
Abstract
Healthy kidneys filter ∼160 g/day of glucose (∼30% of daily energy intake) under euglycaemic conditions. To prevent valuable energy from being lost in the urine, the proximal tubule avidly reabsorbs filtered glucose up to a limit of ∼450 g/day. When blood glucose levels increase to the point that the filtered load exceeds this limit, the surplus is excreted in the urine. Thus, the kidney provides a safety valve that can prevent extreme hyperglycaemia as long as glomerular filtration is maintained. Most of the capacity for renal glucose reabsorption is provided by sodium glucose cotransporter (SGLT) 2 in the early proximal tubule. In the absence or with inhibition of SGLT2, the renal reabsorptive capacity for glucose declines to ∼80 g/day (the residual capacity of SGLT1), i.e. the safety valve opens at a lower threshold, which makes it relevant to glucose homeostasis from day-to-day. Several SGLT2 inhibitors are now approved glucose lowering agents for individuals with type 2 diabetes and preserved kidney function. By inducing glucosuria, these drugs improve glycaemic control in all stages of type 2 diabetes, while their risk of causing hypoglycaemia is low because they naturally stop working when the filtered glucose load falls below ∼80 g/day and they do not otherwise interfere with metabolic counterregulation. Through glucosuria, SGLT2 inhibitors reduce body weight and body fat, and shift substrate utilisation from carbohydrates to lipids and, possibly, ketone bodies. Because SGLT2 reabsorbs sodium along with glucose, SGLT2 blockers are natriuretic and antihypertensive. Also, because they work in the proximal tubule, SGLT2 inhibitors increase delivery of fluid and electrolytes to the macula densa, thereby activating tubuloglomerular feedback and increasing tubular back pressure. This mitigates glomerular hyperfiltration, reduces the kidney’s demand for oxygen and lessens albuminuria. For reasons that are less well understood, SGLT2 inhibitors are also uricosuric. These pleiotropic effects of SGLT2 inhibitors are likely to have contributed to the results of the EMPA-REG OUTCOME trial in which the SGLT2 inhibitor, empagliflozin, slowed the progression of chronic kidney disease and reduced major adverse cardiovascular events in high-risk individuals with type 2 diabetes. This review discusses the role of SGLT2 in the physiology and pathophysiology of renal glucose reabsorption and outlines the unexpected logic of inhibiting SGLT2 in the diabetic kidney.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a worldwide public health and economic problem [1]. Good control of blood glucose levels is critical in diabetic patients to delay the progression of the underlying metabolic dysfunction [2] and to reduce the risk of diabetic complications, including nephropathy and cardiovascular disease [3]. Some blood glucose lowering therapies _target molecular pathways in the liver, adipose tissue, skeletal muscle and pancreas, and, thus, manipulate central metabolic mechanisms. Hence, these compounds may be prone to dose-limiting side effects, such as hypoglycaemia and weight gain, impairing their ability to reduce cardiovascular complications [2, 4].
In contrast, sodium glucose cotransporter (SGLT) 2 inhibitors _target renal glucose reabsorption via an insulin-independent mechanism, resulting in the loss of glucose (and, hence, glucose-associated energy) via overspill into the urine, while leaving central metabolic regulation and responsiveness intact. Therefore, by _targeting this process, SGLT2 inhibitors may offer unique benefits as blood glucose lowering agents. This is supported by recent evidence that the use of SGLT2 inhibitors as an adjunct to standard care in type 2 diabetic patients with high cardiovascular risk can have protective effects with regard to clinically relevant renal and cardiovascular outcomes [5, 6].
This review discusses the role of SGLT2 in the physiology and pathophysiology of renal glucose reabsorption, as well as the use of SGLT2 inhibitors as new blood glucose lowering drugs. In particular, we aim to outline the unexpected logic of inhibiting renal glucose transport in the diabetic setting. This includes the counterproductive enhancement of renal glucose reabsorption via SGLT2, as well as the basic mechanisms that link the primary inhibitory effect of these drugs on SGLT2 to the secondary beneficial consequences on metabolism, the kidneys and the cardiovascular system.
SGLT2 mediates the majority of renal glucose reabsorption
The renal glomeruli of healthy normoglycaemic individuals filter about 140–160 g of glucose each day. If not reclaimed by the renal tubules, this would result in a urinary loss of energy substrate equal to ∼30% of the daily energy expenditure. Instead, more than 99% of filtered glucose is reabsorbed in the proximal tubule and returned to the body or used by other renal epithelia (Fig. 1a). By reabsorbing large amounts of filtered glucose, the kidneys contribute to blood glucose homeostasis.

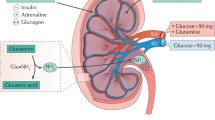
SGLT2-mediated glucose reabsorption in the kidney: consequences of diabetes and links to Na+ reabsorption. (a) SGLT2 and SGLT1 exist in the luminal membrane of the early and late proximal tubule, respectively. SGLT2 reabsorbs ∼97% of filtered glucose in normoglycaemia, while SGLT1 reabsorbs ∼3%. Consequently, between 0–0.2% of glucose is excreted in the urine in normoglycaemia. Contrastingly, SGLT2 inhibition shifts glucose reabsorption downstream, unmasking a significant capacity for SGLT1 to reabsorb ∼50% of filtered glucose. Thus, following SGLT2 inhibition, up to ∼50% of filtered glucose is excreted in normoglycaemia. (b) In health, sodium and glucose uptake via luminal SGLT2 and SGLT1 is followed by basolateral glucose reabsorption via the facilitative glucose transporters, GLUT2 and GLUT1, respectively, and sodium reabsorption via the Na+/K+ pump. (c) Diabetes induces tubular hypertrophy, which is associated with increased SGLT2. Consequently, this may contribute to hyperglycaemia and primary tubular Na+ hyperreabsorption. SGLT2 inhibition counteracts these effects and promotes partial compensation by downstream SGLT1. Preclinical studies propose that SGLT2 inhibition may also attenuate the diabetes-induced increase in renal gluconeogenesis. This figure was modified with permission from the Annual Review of Medicine, Volume 66 © 2015 by Annual Reviews, www.annualreviews.org [26]. Blue arrows, symbols and text relate to SGLT2; green arrows, symbols and text relate to SGLT1; the grey arrow in (a) indicates urinary excretion; black arrows refer to transport mechanisms; in (c), red arrows, symbols and text relate to variables/processes affected by diabetes
Studies in the early 1980s indicated the presence of two different glucose transporters along the apical surface of the proximal tubule [7, 8], leading Wright and colleagues to subsequently clone the responsible Na+/glucose cotransporters, SGLT1 and SGLT2 (for review see [9]). These studies indicated that the majority of tubular glucose uptake occured in the early proximal tubule via SGLT2, leaving SGLT1 to ‘clean up’ most of the remaining luminal glucose in the late proximal tubule (Fig. 1a,b).
In accordance with the above, individuals with mutations in the gene for SGLT2, SLC5A2, diagnosed as having familial renal glucosuria, have persistent renal glucosuria, often in the range of 60–120 g/day [10]. More recently, the renal locations of SGLT1 and SGLT2 have been confirmed using validated antibodies in human and rodent kidneys [11–13]. Moreover, studies in Sglt2 (also known as Scl5a2) and Sglt1 (also known as Scl5a1) knockout mice demonstrated that SGLT2 is responsible for all glucose reabsorption in the early proximal tubule [12] and, overall, accounts for ∼97% of renal glucose reabsorption under normoglycaemic conditions, whereas SGLT1 mediates the remaining 2–3% (Fig. 1a) [12, 14, 15].
Renal glucose reabsorption is increased in diabetes
Hyperglycaemia enhances the amount of glucose filtered by the kidneys and increases the capacity for tubular glucose reabsorption by approximately 20%, to around 500–600 g/day in patients with type 2 [16] and type 1 diabetes [17]. Under conditions of normal GFR and blood glucose levels of up to approximately 10–12 mmol/l, the kidneys reabsorb all the filtered glucose; only further increases in tubular glucose load (at blood glucose ≥12 mmol/l) results in a linear increase in glucosuria [16, 17]. This increase in renal glucose reabsorption makes sense from an energy substrate conserving basis, but is counterproductive in diabetes since it contributes to sustained hyperglycaemia (Fig. 1c).
Upregulated SGLT2 enhances glucose reabsorption in diabetes
An increase in glucose transport capacity in animal models of diabetes results from upregulation of SGLT2; using knockout mice as critical negative antibody controls, renal protein levels of SGLT2 are increased by 40–80% in db/db mouse models of type 2 diabetes and Akita mouse models of type 1 diabetes [18, 19]. Pharmacologic inhibition of SGLT2 in normoglycaemic mice also increased renal membrane SGLT2 protein levels [19]. However, these findings are yet to be repeated in primary human tissue samples using validated antibodies.
SGLT2 upregulation may simply reflect overall growth (hypertrophy) of the proximal tubule in diabetes (Fig. 1c), or it may occur in response to feedback from a glucose sensor downstream of the early proximal tubule [20, 21]. In any case, the glucosuric and blood glucose lowering effects of SGLT2 inhibition become more potent in the diabetes setting, where more glucose is filtered and SGLT2 is upregulated [19] (as discussed further in the next section).
SGLT1 levels vary among genetic mouse models of diabetes: they are increased in ob/ob mouse models of type 2 diabetes [22] but reduced in Akita mouse models of type 1 diabetes [18]. Reduced renal SGLT1 levels were also observed in response to genetic or pharmacological SGLT2 inhibition in non-diabetic mice [12, 19]. We speculate that this reduction in renal membrane SGLT1 protein content under conditions of increased glucose delivery may serve to limit glucose reabsorption in the vulnerable late proximal tubule, where excessive glucose uptake may be toxic [19].
SGLT2 inhibitors reduce blood glucose levels in diabetes but keep metabolic counterregulation intact
Although human SGLT2 mutations are uncommon and understudied genetic disorders, they have not been consistently associated with renal complications (e.g. impaired kidney function or urinary tract infections) [9, 10], hence adding to the rationale for developing SGLT2 inhibitors to act as blood glucose lowering drugs.
Specific SGLT2 inhibition
Phlorizin, a flavonoid contained in the bark of various fruit trees, was found to cause glucosuria over 100 years ago [23]. Phlorizin competitively inhibits SGLT2 and SGLT1 and does so with a tenfold higher affinity for the former [9, 24]. However, SGLT1 is the primary pathway for glucose reabsorption in the intestine and is widely expressed throughout the body [25]. Thus oral administration of phlorizin is encumbered by extrarenal side effects, such as diarrhoea. In contrast, SGLT2 appears only to be expressed in the proximal tubule of the kidneys [11, 26] (the proposed expression and function in alpha cells of the pancreas [27] still requires confirmation). Hence, the development of SGLT2-specific phlorizin derivatives (with good oral bioavailability and suitability for once-daily dosing) provided an important breakthrough for the practical use of renal glucose transport _targeting therapies for blood glucose management [28]. Three members of this group of drugs, dapagliflozin (Forxiga [known as Farxiga in the USA]), canagliflozin (Invokana) and empagliflozin (Jardiance), have been approved in the USA and Europe for the treatment of patients with type 2 diabetes.
Enhanced urinary glucose loss
Potent SGLT2 inhibitors induce a similar sustained urinary glucose loss of 40–80 g/day under conditions of normal blood glucose (Fig. 1a) [28–30]. According to a meta-analysis of patients with type 2 diabetes, SGLT2 inhibitors decreased HbA1c levels by 0.5–0.7% after 12 weeks of treatment and this effect persisted for up to 52 weeks of treatment [31]. The higher the blood glucose level and GFR, the more glucose is filtered and reabsorbed and, as a consequence, can be excreted during SGLT2 blockade. Thus, SGLT2 blockers naturally have a greater efficacy when it is desirable for them to be more efficacious [12, 32, 33].
Preserved efficacy in progressive insulin resistance
Lowering blood glucose levels and body weight (by any means) ameliorates the function of beta cells and the insulin sensitivity status of patients with type 2 diabetes, thereby further improving blood glucose control. As indicated by preclinical [34–36] and clinical studies [32, 37], this also applies to SGLT2 inhibition. The mechanism of action of SGLT2 inhibitors is independent of insulin secretion or action. As a consequence, their efficacy does not decline with progressive beta cell dysfunction and/or insulin resistance, and SGLT2 inhibitors act synergistically with other blood glucose lowering agents [28].
Low hypoglycaemia risk
Since the glucosuric effect of SGLT2 inhibitors is tied to the filtered load of glucose, they become ineffective at further lowering blood glucose once the filtered load reaches ≤80 g/day (a load which can be handled by SGLT1). The residual capacity of SGLT1 relates to the question: how do normal, euglycaemic humans/animals reabsorb ∼97% of filtered glucose via SGLT2, yet maintain a fractional glucose reabsorption of around 50% when SGLT2 is inhibited pharmacologically [38–40] or deleted by genetic mutation [10, 12]? Studies in mice using pharmacologic and/or genetic inhibition of SGLT1 and SGLT2 demonstrate that an increase in SGLT1-mediated transport fully accounts for residual glucose reabsorption during SGLT2 inhibition [14]. These findings are consistent with a high maximal glucose transport rate of human SGLT1 [24]. In other words, SGLT2 inhibition enhances the glucose load to the late proximal tubule where it unmasks a significant transport capacity of SGLT1 for glucose that limits glucosuria and hypoglycaemia risk (Fig. 1a, c).
Additionally, as expected for blood glucose lowering agents that leave the metabolic counterregulation intact, SGLT2 inhibitors increase plasma glucagon concentrations and endogenous hepatic glucose production (gluconeogenesis) in patients with type 2 diabetes [32, 37]. In addition, SGLT2 inhibition enhances lipolysis and shifts substrate utilisation from carbohydrates to lipids [32], contributing to a reduction in fat mass and body weight [41]. This physiological response to excessive renal glucose excretion prevents episodes of hypoglycaemia and secures glucose availability for glucose-dependent organs, such as the brain, while maintaining body energy supplies. As a consequence, hypoglycaemia was only noted when SGLT2 inhibitors have been used in combination with other drugs, but not as a monotherapy [31].
Beneficial impact of SGLT2 inhibitors on renal and cardiovascular outcomes in patients with type 2 diabetes and high cardiovascular risk
The EMPA-REG OUTCOME trial assessed the long-term effects of empagliflozin on renal and cardiovascular outcomes in patients with type 2 diabetes, a high cardiovascular risk and an estimated GFR (eGFR) of ≥30 ml min−1 (1.73 m2)−1 of body surface area [5, 6]. In this study, alongside empagliflozin, around 80% of patients were treated with an angiotensin converting enzyme (ACE) inhibitor or angiotensin II type 1 receptor (AT1) antagonist as part of their standard care. The study found that, in addition to standard care, empagliflozin reduced the rate of incident or worsening nephropathy (defined as progression to macroalbuminuria, doubling of serum creatinine level, initiation of renal replacement therapy or death from renal disease), with a relative risk reduction of 39%. Moreover, empagliflozin reduced the rate of doubling serum creatinine and the initiation of renal replacement therapy, with relative risk reductions of 44% and 55%, respectively [6]. Empagliflozin treatment also reduced the risk of death from cardiovascular disease, hospitalisation for heart failure and death from any cause, with relative risk reductions of 38%, 35% and 32%, respectively, but did not alter the risk of nonfatal myocardial infarction or nonfatal stroke [5]. Thus, in patients with type 2 diabetes and high cardiovascular risk, the addition of empagliflozin to standard care delayed the progression of kidney disease and lowered rates of clinically relevant renal and cardiovascular events.
In terms of side effects, the adverse-event profile of empagliflozin in patients with normal and impaired kidney function at baseline was similar to that reported in the total trial population. The main side effect of SGLT2 inhibitors was an increased risk of genitourinary infections [42]. Therefore, these drugs appear to be cardioprotective and preventative of renal disease in patients with type 2 diabetes and high cardiovascular risk, while having only minor side effects.
How does SGLT2 inhibition protect the renal and cardiovascular systems?
One possible explanation for the cardioprotective and renal protective effect of SGLT2 inhibition is the associated pleiotropic effects; i.e. the induction of multiple processes that have a synergistic beneficial influence on the renal and cardiovascular systems. For example, in the EMPA-REG OUTCOME trial, empagliflozin was associated with small improvements in blood glucose control and small reductions in body weight, waist circumference, uric acid levels, and systolic and diastolic blood pressure, with no increase in heart rate [5, 6]. Hence, important renal and cardiovascular risk factors, including hyperglycaemia, hypertension, obesity and hyperuricaemia, are improved via SGLT2 inhibition.
Lowering of hyperglycaemia
The small effect of SGLT2 inhibition on blood glucose control alone seems unlikely to induce the rapidly observed beneficial effects, which occur within a few months. In support of this theory, other blood glucose lowering agents do not show this cardiovascular benefit in such a short amount of time, or even at all. However, these agents may have simultaneous countervailing effects that offset the benefits of reduced blood glucose levels, such as increased obesity or hypoglycaemia.
By reducing hyperglycaemia, SGLT2 inhibitors have the potential to reduce glucotoxicity in the kidney and in extrarenal organs [43, 44]. For example, studies in rodent models of diabetes have shown that SGLT2 inhibition can reduce renal growth, inflammation and injury, secondary to a potent blood glucose lowering effect [18, 19, 22, 45–48] (Fig. 2).

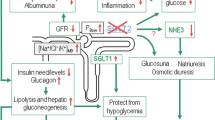
The pleiotropic effects of SGLT2 inhibition: the potential for kidney and cardiovascular protection in diabetes. SGLT2 inhibition may provide its cardioprotective and renal protective effects via several pleiotropic mechanisms: (1) SGLT2 inhibition attenuates primary proximal tubular hyperreabsorption in the kidney in diabetes, increasing/restoring the tubuloglomerular feedback signal at the macula densa ([Na+/Cl−/K+]MD) and hydrostatic pressure in Bowman’s space (PBow). This reduces glomerular hyperfiltration, beneficially affecting albumin filtration and tubular transport work and, thus, renal oxygen consumption; (2) by lowering blood glucose levels, SGLT2 inhibitors can reduce kidney growth, albuminuria and inflammation; (3) SGLT2 inhibitors have a modest osmotic diuretic, natriuretic and uricosuric effect, which can reduce extracellular volume (ECV), blood pressure, serum uric acid levels and body weight. These changes may have beneficial effects on both the renal and cardiovascular systems; (4) SGLT2 may be functionally linked to NHE3, such that SGLT2 inhibition may also inhibit NHE3 in the proximal tubule, with implications on the natriuretic, GFR and blood pressure effect; (5) SGLT2 inhibition reduces insulin levels and the need for therapeutic and/or endogenous insulin, and increases glucagon levels. As a consequence, lipolysis and hepatic gluconeogenesis are elevated. These metabolic adaptations reduce fat tissue/body weight and hypoglycaemia risk, and result in mild ketosis, potentially having beneficial effects on both the renal and cardiovascular systems; (6) SGLT2 inhibition may also enhance renal HIF content, which may have renal protective effects. White text boxes indicate affected variables; grey text boxes indicate processes that link SGLT2 inhibition to the reduction in GFR. Green arrows demonstrate consequences; red arrows indicate changes in associated variables (increase/decrease)
Reduction in blood pressure
Lowering blood pressure is an effective strategy for delaying the progression to more advanced stages of nephropathy in patients with type 1 and type 2 diabetes [49, 50]. Consistent with preclinical data and the EMPA-REG OUTCOME trial, a meta-analysis of type 2 diabetes patients treated with SGLT2 inhibitors showed a consistent decrease in systolic blood pressure of 3–6 mmHg [31]. Importantly, these reductions in blood pressure are comparable with those induced by established blood pressure lowering agents and expected to have similar vascular protective effects, particularly in high-risk patients [53].
This effect of SGLT2 inhibitors on blood pressure is partly due to their direct natriuretic effect and was not fully explained by reductions in body weight (see subsequent section) [43, 51, 52]. SGLT2 inhibition induces a modest glucose-based osmotic diuresis (100–470 ml/day) and a small natriuretic effect. This natriuretic effect is sustained until the resulting decline in extracellular volume invokes compensatory mechanisms that equilibrate urinary fluid and sodium excretion with dietary sodium intake.
The reduction in blood pressure and associated modest reductions in plasma volume [54] upon SGLT2 blockade may quickly reduce cardiac pre- and afterload, potentially contributing to the rapid beneficial cardiac effects observed with SGLT2 inhibitor use, particularly in heart failure patients [5] (Fig. 2).
Weight loss
In patients with type 2 diabetes, including those in the EMPA-REG OUTCOME trial, the glucosuric effect of SGLT2 inhibition was consistently associated with a 2–3 kg lower body weight. While fluid loss may contribute to initial weight loss, the majority of steady-state weight loss with SGLT2 inhibitor treatment appears to result from fat loss [55, 56]. In obese rodents, the reduction in body weight in response to SGLT2 inhibition was associated with increased lipolysis and fatty acid oxidation and, consequently, lower body fat [57, 58]. This is consistent with the shift in substrate utilisation from carbohydrates to lipids [32], as previously mentioned (Fig. 2). Reduced visceral and subcutaneous fat has also been reported in type 2 diabetes patients treated with SGLT2 inhibitors [41, 55].
Inhibition of hyperuricaemia
SGLT2 blockade may also induce beneficial renal and cardiovascular effects by lowering plasma uric acid levels [59]. While the mechanism of uric acid transport and the relevance of this effect remains to be determined, studies in healthy individuals and patients with type 2 diabetes indicate that increases in tubular glucose delivery may explain the uricosuric effect of SGLT2 inhibitors [60, 61] (Fig. 2). Based on in vitro transport studies in Xenopus oocytes, it has been hypothesised that glucose in the lumen of the proximal tubule may facilitate intracellular urate exchange via the urate transporter, GLUT9 isoform 2 (encoded by the SLC2A9b isoform), thereby enhancing urinary urate excretion [61]. Evidently, further research is required to elucidate the precise mechanisms and consequence of this SGLT2-induced effect.
SGLT2 inhibition reduces diabetic glomerular hyperfiltration
Diabetes is a major cause of chronic kidney disease (CKD), and hyperglycaemia and CKD are major risk factors for cardiovascular and overall mortality. Thus, the improvement of renal outcomes in response to SGLT2 inhibition [6] may contribute to their observed cardiovascular benefits [5]. In a subset of patients, glomerular hyperfiltration occurs at the onset of type 1 or type 2 diabetes and can increase the risk for subsequent development of diabetic nephropathy [62]. Since <1% of filtered sodium is excreted in urine to maintain sodium balance on a normal diet, GFR is the primary determinant of renal sodium reabsorption and transport work, and thus of renal oxygen consumption. As a consequence, reducing glomerular hyperfiltration is expected to reduce the transport work and oxygen consumption of the kidney in diabetic individuals, particularly in the proximal convoluted tubule [63].
Hyperfiltration in diabetes
Several studies have been published in support of the ‘tubular hypothesis’, according to which nuances of glomerular filtration in diabetes, such as hyperfiltration, are explained by prior changes in tubular reabsorption (for review see [64]). Moderate hyperglycaemia induces a ‘primary’ increase in proximal tubular reabsorption by providing substrate for SGLTs or by causing the tubule to undergo hypertrophy (Figs 1c and 3f). This primary increase in proximal reabsorption reduces NaCl and fluid delivery to the downstream macula densa, causing GFR to increase via normal physiologic actions of tubuloglomerular feedback (Fig. 3f). The tubuloglomerular feedback mechanism works on the single nephron level and adjusts the tone of the afferent arteriole, and thereby GFR, to stabilise the NaCl and fluid load to the macula densa. This contributes to the autoregulation of GFR and facilitates the fine regulation of NaCl and fluid balance in the distal nephron, downstream of the macula densa, by neurohumoral control (including sympathetic tone and aldosterone and vasopressin regulation). A primary increase in proximal reabsorption also reduces distal tubular flow rate, which can increase GFR by lowering tubular back pressure, i.e. the hydrostatic pressure in the Bowman’s space (Fig. 3f).

SGLT2 inhibition lowers diabetic glomerular hyperfiltration. (a) In vivo micropuncture studies were performed in non-diabetic and streptozotocin-induced diabetic rats, both with superficial glomeruli [65]. Measurements were performed under control conditions or following application of phlorizin directly into the early proximal tubule, i.e. systemic blood glucose levels were not changed. Small amounts of blue dye were injected into the Bowman’s space to determine the configuration of the nephron, including the early proximal tubular loop and the early distal tubule located near the macula densa. The distal tubule is stained with blue dye in the figure. The macula densa is just upstream of the early distal tubule but does not project to the kidney surface. Tubular fluid was collected from the early distal tubule, i.e. just downstream of the macula densa, to determine: (1) the strength of the tubuloglomerular feedback signal at the macula densa ([Na+/Cl−/K+]MD); and (2) single nephron glomerular filtration rate (SNGFR) by inulin clearance. Additionally, the Bowman’s space was punctured to determine the hydrostatic pressure (PBow). BS, Bowman’s space; EPT, early proximal tubule; EDT, early distal tubule; SG, superficial glomerulus. Scale bar, 100 μm. ‘V-shaped’ lines indicate the micropuncture capillaries and their position; arrows indicate the withdrawal or application of fluid or pressure measurement. (b–e) Basal measurements (B) revealed that whole kidney glomerular hyperfiltration in diabetes was associated with reductions in (b) [Na+] and [Cl−] at the macula densa ([Na+/Cl−]MD), (c) [K+] at the macula densa ([K+]MD) and (d) in PBow, and (e) an increase in SNGFR (nl/min). Phlorizin (P) did not greatly affect any of these measures in non-diabetic rats, but normalised [Na+/Cl−/K+]MD, PBow and SNGFR in diabetes [65]. In (b–e), white circles, non-diabetic; black circles, diabetic. (f) Diabetes induces a primary hyperreabsorption in the proximal tubule, which causes glomerular hyperfiltration via tubuloglomerular feedback (reduction in [Na+/Cl−/K+]MD levels) and reductions in tubular back pressure (PBow). SGLT2 contributes to hyperreabsorption (this process is further enhanced by tubular growth in diabetes) and, consequently, SGLT2 inhibition mitigates hyperreabsorption in diabetes, inhibiting glomerular hyperfiltration. In (f), yellow boxes indicate the main consequences of diabetes; grey boxes indicate mechanisms that link hyperreabsorption to hyperfiltration; black arrows indicate changes in variable (increase/decrease); red arrows indicate the enhanced reabsorption
SGLT2 inhibitors suppress hyperfiltration
SGLT2 inhibition attenuates primary proximal tubule hyperreabsorption in diabetes and thereby lowers glomerular hyperfiltration (Fig. 3f). This has been demonstrated in micropuncture studies in rats using local application of phlorizin into the Bowman’s space of individual nephrons (Fig. 3a–e) [65] and by acute or chronic systemic application of selective SGLT2 inhibitors [66]. Hyperfiltration has been suppressed on the whole-kidney level in mouse models of diabetes by pharmacologic or genetic inhibition of SGLT2 [18, 19]. In each case diabetic hyperfiltration suppression was independent of effects on blood glucose [18, 65, 66], but was associated with an increase in NaCl concentration at the macula densa [65, 66] and in hydrostatic pressure in the Bowman’s space [65] (Figs 2 and 3).
Recent studies have also confirmed a GFR lowering effect of SGLT2 inhibitors in humans; in an 8-week study, the SGLT2 inhibitor empagliflozin decreased GFR by 19% in type 1 diabetic patients with baseline hyperfiltration. This effect was also independent of lowering blood glucose levels [33]. The SGLT2 inhibitor canagliflozin also initially lowered eGFR in patients with type 2 diabetes (basal eGFR ≥55 ml min−1 [1.73 m2]−1). Following this initial dip, eGFR declined at a slower rate in the canagliflozin-treated group such that it was better preserved after 2 years of follow-up and associated with reduced urinary albumin:creatinine ratio, as compared with a control group that had been treated with glimepiride to achieve similar blood glucose control [67].
SGLT2 inhibitors in diabetic patients with CKD
The nephrons that survive in advanced stages of CKD are assumed to hyperfilter as a way of compensating for the loss of other nephrons. If so, then the remaining nephrons maintain a high tubular glucose load. As a consequence, the acute GFR lowering effect of SGLT2 inhibition should be preserved in CKD, even if the effect on overall glucose homeostasis were attenuated. In accordance, the SGLT2 inhibitor canagliflozin modestly reduced eGFR, together with proteinuria, within 3 weeks in patients with type 2 diabetes and basal eGFR values between 30–50 ml min−1 [1.73 m2]−1 (stage 3 CKD) [68]. Empagliflozin also induced a small decline in eGFR in patients with type 2 diabetes and stage 2 CKD and stage 3 CKD. This effect was maintained at 52 weeks of treatment and was fully reversible after a 3-week wash-out period. The decline in eGFR was associated with reduced urine albumin:creatinine ratio [69]. Reductions in single nephron glomerular hyperfiltration by SGLT2 inhibition in CKD and, thereby, the oxygen-consuming transport workload, may help to preserve the integrity of remaining nephrons and overall kidney function in the long term (Fig. 2). This has also been proposed for angiotensin II blockade [70]; the EMPA-REG OUTCOME trial provides evidence that the combination of SGLT2 inhibitor plus angiotensin II blockade provides an additive nephroprotective effect in type 2 diabetic patients with high cardiovascular risk, including those with initial GFRs of at least 30 ml min−1 [1.73 m2]−1 of body surface area [6]. Currently SGLT2 inhibitors are approved for use in patients with type 2 diabetes, with dapagliflozin being approved for those with a GFR >60 ml/min, and canagliflozin and empagliflozin for GFRs >45 ml/min.
Perspectives
The pleiotropic impact of SGLT2 inhibition creates a favourable environment for beneficial and synergistic effects on the renal and cardiovascular systems (Figs. 2 and 3). However, we have to be open-minded with regard to additional beneficial mechanisms.
The kidney is a complex organ with many interacting parts. We believe that we understand how SGLT2 blockade invokes tubuloglomerular feedback and tubular back pressure to suppress hyperfiltration (Fig. 2). We imagine that SGLT2 blockade also reduces glomerular capillary pressure through the tubuloglomerular feedback mechanism; however, we have not yet measured glomerular capillary pressure under these conditions. Moreover, alterations in physical and metabolic stresses in other parts of the kidney upon manipulation of glucose transport in the early proximal tubule remain to be defined.
Renal hypoxia
SGLT2 inhibition shifts transport to segments further downstream of the early proximal tubule. It is possible for this to exacerbate renal medullary hypoxia, although such an effect would be counteracted by reductions in blood glucose and GFR [63, 71]. A recent study on renal ischaemia reperfusion injury indicated that the commencement of SGLT2 inhibitor (dapagliflozin) application 1 day before surgery attenuated serum creatinine increases, tubular injury and markers of apoptosis after 24 h, and that this was associated with increased renal levels of the transcription factor, hypoxia-inducible factor (HIF)1-α [72]. Notably, this study was performed in non-diabetic mice, indicating the potential for renoprotective effects of SGLT2 inhibition independent of blood glucose control. Renal hypoxia is known to predominantly induce HIF1-α and upregulate the tissue protective _target gene HMOX1 (encoding haem oxygenase 1) in tubular cells [73]. Notably, Hmox1 mRNA expression was also increased in the kidneys of non-diabetic mice lacking SGLT2 [18]. Thus, while the effect on HIF1-α would be consistent with enhancements in medullary tubular hypoxia in response to SGLT2 inhibition, it remains to be determined whether the increase of HIF1-α actually contributes to the nephroprotective effects of SGLT2 inhibition (Fig. 2).
Moreover, the proximal tubule is thought to make little use of reabsorbed glucose under normal conditions but acute kidney injury may induce a relevant glycolytic shift in the outer medullary proximal tubules [74, 75], an effect that may involve tubular upregulation of HIF1-α [76]. Whether the increased delivery of glucose to this region of the kidney via SGLT2 inhibition is detrimental or beneficial in conditions of acute kidney injury remains to be determined. In comparison, the hypoxia-induced formation of erythropoietin in peritubular interstitial fibroblast-like cells of the cortico-medullary border is associated with increased levels of HIF2-α in these cells [73, 77, 78]. The effect of SGLT2 blockade on HIF-2 remains to be determined and may provide knowledge that is relevant for the cardiovascular system.
SGLT2 inhibition and natriuresis
In the brush border of the early proximal tubule, SGLT2 is co-expressed with the Na+/H+-exchanger 3 (NHE3), which reabsorbs about 30% of filtered sodium. Recent studies provide evidence that SGLT2 may be functionally linked to NHE3 [79–81] such that SGLT2 inhibition may also inhibit NHE3 in the proximal tubule (Fig. 2). Whereas such an interaction could be relevant for the natriuretic effect of SGLT2 inhibition and its effect on blood pressure and GFR, more studies are required to further establish this interaction and the relevance of these outcomes.
SGLT2 inhibition and gluconeogenesis
The kidneys produce between 15–55 g of glucose per day, with this process being upregulated in diabetes [82]. Studies in Akita mouse models of type 1 diabetes confirmed that renal mRNA expression of phosphoenolpyruvate carboxykinase, the main regulator of gluconeogensis, was upregulated; however this effect was attenuated by SGLT2 inhibition [19] (Fig. 1c). Further studies are needed to determine whether this interaction may be explanatory of the blood glucose lowering effect of SGLT2 inhibitors that is independent of a glucosuric effect (allowing this effect to be maintained in CKD). It is also important to determine whether similar findings would be observed in humans.
SGLT2 inhibition and ketoacidosis
In May 2015 the Food and Drug Administration (FDA) released a warning that SGLT2 inhibitors might increase the risk of ketoacidosis [83], with cases reported in patients with type 1 diabetes and some (although fewer) in type 2 diabetes. Potential mechanisms include a reduction in insulin and higher glucagon levels in response to SGLT2 inhibition, which can lead to increased lipolysis and ketogenesis. The EMPA-REG OUTCOME trial found no difference in rates of ketoacidosis in type 2 diabetic patients treated with the SGLT2 inhibitor empagliflozin vs placebo over the course of 3 years [5]. Nevertheless, dedicated studies are required to further define the clinical relevance and potential mechanisms of SGLT2-associated ketoacidosis. Notably, the development of mild ketosis has been hypothesised to contribute to the beneficial effects of SGLT2 inhibition on cardiac and renal outcomes because of the use of ketone bodies as energy substrates, as well as their potential interactions with G-protein coupled receptors and other signalling pathways [84–86] (Fig. 2).
Ongoing trials
In addition to the EMPA-REG OUTCOME trial there are several studies currently under way, investigating the effects of SGLT2 inhibition on kidney and cardiovascular outcomes. These include the Dapagliflozin Effect on CardiovascuLAR Events (DECLARE; ClinicalTrial.gov registration no.NCT01730534) study, the CANagliflozin cardioVascular Assessment Study (CANVAS; ClinicalTrial.gov registration no. NCT01032629) and the Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE; ClinicalTrial.gov registration no. NCT02065791) (for review see [87]). Their results are expected to further refine our understanding of the therapeutic potential and safety of SGLT2 inhibition.
Summary
SGLT2 inhibitors are important blood glucose lowering agents for use in diabetes treatment. They primarily function by altering glucose reabsorption via SGLT2 in the proximal tubule. However, they also improve body weight and composition, uric acid levels and blood pressure in patients. Furthermore, they lower glomerular hyperfiltration which reduces the kidney’s demand for oxygen and lessens albuminuria. Since they do not alter metabolic regulation of glucose and are not effective when filtered glucose levels are below 80 g/day, these drugs are not associated with hypoglycaemia. Their use has been approved in diabetic patients with normal and preserved renal function. The pleiotropic and synergistic effects of SGLT2 inhibitors are likely to contribute to the cardio- and renoprotective effects recently reported with their use, including in patients with modestly impaired renal function. Further research is required to fully establish their therapeutic value in individuals with diabetes and to explore their potential in non-diabetic renal and cardiovascular disease.
Abbreviations
- CKD:
-
Chronic kidney disease
- eGFR:
-
Estimated GFR
- HIF:
-
Hypoxia-inducible factor
- NHE3:
-
Na+/H+-exchanger 3
- SGLT:
-
Sodium glucose cotransporter
References
International Diabetes Federation (2013) www.idf.org/diabetesatlas. Accessed 15 Aug 2015
DeFronzo RA (2009) Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58:773–795
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Gerstein HC, Miller ME, Byington RP et al (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358:2545–2559
Zinman B, Wanner C, Lachin JM et al (2015) Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373:2117–2128
Wanner C, Inzucchi SE, Lachin JM et al (2016) Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 375:323–334
Barfuss DW, Schafer JA (1981) Differences in active and passive glucose transport along the proximal nephron. Am J Physiol 241:F322–F332
Turner RJ, Moran A (1982) Heterogeneity of sodium-dependent D-glucose transport sites along the proximal tubule: evidence from vesicle studies. Am J Physiol 242:F406–F414
Wright EM, Loo DD, Hirayama BA (2011) Biology of human sodium glucose transporters. Physiol Rev 91:733–794
Santer R, Calado J (2010) Familial renal glucosuria and SGLT2: from a mendelian trait to a therapeutic _target. Clin J Am Soc Nephrol 5:133–141
Vrhovac I, Balen ED, Klessen D et al (2015) Localizations of Na-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467:1881–1898
Vallon V, Platt KA, Cunard R et al (2011) SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22:104–112
Sabolic I, Vrhovac I, Eror DB et al (2012) Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am J Physiol Cell Physiol 302:C1174–C1188
Rieg T, Masuda T, Gerasimova M et al (2014) Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Phys Renal Phys 306:F188–F193
Gorboulev V, Schurmann A, Vallon V et al (2012) Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61:187–196
Farber SJ, Berger EY, Earle DP (1951) Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest 30:125–129
Mogensen CE (1971) Maximum tubular reabsorption capacity for glucose and renal hemodynamcis during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand J Clin Lab Invest 28:101–109
Vallon V, Rose M, Gerasimova M et al (2013) Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304:F156–F167
Vallon V, Gerasimova M, Rose MA et al (2014) SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 306:F194–F204
Vallon V (2011) The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300:R1009–R1022
Seyer-Hansen K (1987) Renal hypertrophy in experimental diabetes: some functional aspects. J Diabet Complications 1:7–10
Gembardt F, Bartaun C, Jarzebska N et al (2014) The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am J Physiol Ren Physiol 307:F317–F325
Ehrenkranz JR, Lewis NG, Kahn CR, Roth J (2005) Phlorizin: a review. Diabetes Metab Res Rev 21:31–38
Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM (2011) Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 300:C14–C21
Song P, Onishi A, Koepsell H, Vallon V (2016) Sodium glucose cotransporter SGLT1 as a therapeutic _target in diabetes mellitus. Expert Opin Ther _targets 20:1109–1125
Vallon V (2015) The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu Rev Med 66:255–270
Bonner C, Kerr-Conte J, Gmyr V et al (2015) Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 21:512–517
Washburn WN, Poucher SM (2013) Differentiating sodium-glucose co-transporter-2 inhibitors in development for the treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs 22:463–486
Abdul-Ghani MA, DeFronzo RA (2014) Lowering plasma glucose concentration by inhibiting renal sodium-glucose co-transport. J Intern Med 276:352–363
Hasan FM, Alsahli M, Gerich JE (2014) SGLT2 inhibitors in the treatment of type 2 diabetes. Diabetes Res Clin Pract 104:297–322
Monami M, Nardini C, Mannucci E (2014) Efficacy and safety of sodium glucose co-transport-2 inhibitors in type 2 diabetes: a meta-analysis of randomized clinical trials. Diabetes Obes Metab 16:457–466
Ferrannini E, Muscelli E, Frascerra S et al (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124:499–508
Cherney DZ, Perkins BA, Soleymanlou N et al (2014) Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129:587–597
Jurczak MJ, Lee HY, Birkenfeld AL et al (2011) SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 60:890–898
Hansen HH, Jelsing J, Hansen CF et al (2014) The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves beta-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J Pharmacol Exp Ther 350:657–664
Macdonald FR, Peel JE, Jones HB et al (2010) The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes Metab 12:1004–1012
Merovci A, Solis-Herrera C, Daniele G et al (2014) Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest 124:509–514
Komoroski B, Vachharajani N, Boulton D et al (2009) Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther 85:520–526
Heise T, Seewaldt-Becker E, Macha S et al (2013) Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks’ treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab 15:613–621
Sha S, Devineni D, Ghosh A et al (2011) Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab 13:669–672
Bolinder J, Ljunggren O, Kullberg J et al (2012) Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab 97:1020–1031
Geerlings S, Fonseca V, Castro-Diaz D, List J, Parikh S (2014) Genital and urinary tract infections in diabetes: impact of pharmacologically-induced glucosuria. Diabetes Res Clin Pract 103:373–381
Guthrie RM (2013) Sodium-glucose co-transporter 2 inhibitors and the potential for cardiovascular risk reduction in patients with type 2 diabetes mellitus. Postgrad Med 125:21–32
Basile JN (2013) The potential of sodium glucose cotransporter 2 (SGLT2) inhibitors to reduce cardiovascular risk in patients with type 2 diabetes (T2DM). J Diabetes Complicat 27:280–286
Nagata T, Fukuzawa T, Takeda M et al (2013) Tofogliflozin, a novel sodium-glucose co-transporter 2 inhibitor, improves renal and pancreatic function in db/db mice. Br J Pharmacol 170:519–531
Terami N, Ogawa D, Tachibana H et al (2014) Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS One 9:e100777
Gangadharan KM, Gross S, Mudaliar H et al (2014) Inhibition of kidney proximal tubular glucose reabsorption does not prevent against diabetic nephropathy in type 1 diabetic eNOS knockout mice. PLoS One 9:e108994
Kojima N, Williams JM, Takahashi T, Miyata N, Roman RJ (2013) Effects of a new SGLT2 inhibitor, luseogliflozin, on diabetic nephropathy in T2DN rats. J Pharmacol Exp Ther 345:464–472
Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T (2005) Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care 28:164–176
American Diabetes Association (2014) Standards of Medical Care in Diabetes - 2014. Diabetes Care 37:S14–S80
Baker WL, Smyth LR, Riche DM, Bourret EM, Chamberlin KW, White WB (2014) Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: a systematic review and meta-analysis. J Am Soc Hypertens 8:262–275
Oliva RV, Bakris GL (2014) Blood pressure effects of sodium-glucose co-transport 2 (SGLT2) inhibitors. J Am Soc Hypertens 8:330–339
Foote C, Perkovic V, Neal B (2012) Effects of SGLT2 inhibitors on cardiovascular outcomes. Diab Vasc Dis Res 9:117–123
Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J (2013) Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab 15:853–862
Bolinder J, Ljunggren O, Johansson L et al (2014) Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab 16:159–169
Cefalu WT, Leiter LA, Yoon KH et al (2013) Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet 382:941–950
Yokono M, Takasu T, Hayashizaki Y et al (2014) SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol 727:66–74
Suzuki M, Takeda M, Kito A et al (2014) Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr Diabetes 4, e125
Kanbay M, Jensen T, Solak Y et al (2016) Uric acid in metabolic syndrome: from an innocent bystander to a central player. Eur J Intern Med 29:3–8
Lytvyn Y, Skrtic M, Yang GK, Yip PM, Perkins BA, Cherney DZ (2015) Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am J Physiol Renal Physiol 308:F77–F83
Chino Y, Samukawa Y, Sakai S et al (2014) SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos 35:391–404
Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG (2009) Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 52:691–697
Layton AT, Vallon V, Edwards A (2016) Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am J Physiol Ren Physiol 310:F1269–F1283
Vallon V, Thomson SC (2012) Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74:351–375
Vallon V, Richter K, Blantz RC, Thomson S, Osswald H (1999) Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10:2569–2576
Thomson SC, Rieg T, Miracle C et al (2012) Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 302:R75–R83
Heerspink HJ, Desai M, Jardine M, Balis D, Meininger G, Perkovic V (2016) Canagliflozin slows progression of renal function decline independently of glycemic effects. J Am Soc Nephrol. doi:10.1681/ASN.2016030278
Yale JF, Bakris G, Cariou B et al (2013) Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab 15:463–473
Barnett AH, Mithal A, Manassie J et al (2014) Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2:369–384
Holtkamp FA, de Zeeuw D, Thomas MC et al (2011) An acute fall in estimated glomerular filtration rate during treatment with losartan predicts a slower decrease in long-term renal function. Kidney Int 80:282–287
Neill O, Fasching A, Pihl L, Patinha D, Franzen S, Palm F (2015) Acute SGLT inhibition normalizes oxygen tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am J Physiol Renal Physiol 309:F227–F234
Chang YK, Choi H, Jeong JY et al (2016) Dapagliflozin, SGLT2 inhibitor, attenuates renal ischemia-reperfusion injury. PLoS One 11:e0158810
Rosenberger C, Mandriota S, Jurgensen JS et al (2002) Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 13:1721–1732
Ash SR, Cuppage FE (1970) Shift toward anaerobic glycolysis in the regenerating rat kidney. Am J Pathol 60:385–402
Lan R, Geng H, Singha PK et al (2016) Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27:3356–3367
Farsijani NM, Liu Q, Kobayashi H et al (2016) Renal epithelium regulates erythropoiesis via HIF-dependent suppression of erythropoietin. J Clin Invest 126:1425–1437
Koury ST, Bondurant MC, Koury MJ (1988) Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood 71:524–527
Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M (2008) Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 111:5223–5232
Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G (2014) functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 25:2028–2039
Coady MJ, El TA, Santer R et al (2016) MAP17 Is a necessary activator of renal Na+/glucose cotransporter SGLT2. J Am Soc Nephrol. doi:10.1681/ASN.2015111282
Fu Y, Gerasimova M, Mayoux E, Masuda T, Vallon V (2014) SGLT2 inhibitor empagliflozin increases renal NHE3 phosphorylation in diabetic Akita mice: possible implications for the prevention of glomerular hyperfiltration. Diabetes 63(supplement 1):A132
Gerich JE (2010) Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med 27:136–142
Food and Drug Administration (2015) www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Accessed 10 Aug 2015
Mudaliar S, Alloju S, Henry RR (2016) Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME Study? A unifying hypothesis. Diabetes Care 39:1115–1122
Newman JC, Verdin E (2014) beta-hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract 106:173–181
Ferrannini E, Mark M, Mayoux E (2016) CV Protection in the EMPA-REG OUTCOME Trial: a “thrifty substrate” hypothesis. Diabetes Care 39:1108–1114
Inzucchi SE, Zinman B, Wanner C et al (2015) SGLT-2 inhibitors and cardiovascular risk: proposed pathways and review of ongoing outcome trials. Diab Vasc Dis Res 12:90–100
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors work was supported by the National Institutes of Health (R01DK56248, R01DK106102, P30DK079337), the Department of Veterans Affairs and investigator-initiated research grants by Bristol-Myers Squibb and Boehringer Ingelheim.
Duality of interest
Over the last 36 months, VV has served as a consultant and received honoraria from Boehringer Ingelheim, Intarcia Therapeutics, Astra-Zeneca, Janssen Pharmaceutical, Eli Lilly and Merck, and received grant support for investigator-initiated research from Boehringer Ingelheim, Astra-Zeneca, Fresenius, Janssen and Bayer. Over the past 36 months, SCT has received research support from Merck and Pfizer. The study sponsors were not involved in the design of the study; the collection, analysis and interpretation of data; writing the report; or the decision to submit the report for publication.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 0.98 mb)
Rights and permissions
About this article

Cite this article
Vallon, V., Thomson, S.C. _targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60, 215–225 (2017). https://doi.org/10.1007/s00125-016-4157-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4157-3