
Abstract
Background:
This randomised, open-label, multicenter phase II study compared progression-free survival (PFS) of S-1 plus oxaliplatin (SOX) with that of S-1 alone in patients with gemcitabine-refractory pancreatic cancer.
Methods:
Patients with confirmed progressive disease following the first-line treatment with a gemcitabine-based regimen were randomised to receive either S-1 (80/100/120 mg day−1 based on body surface area (BSA), orally, days 1–28, every 6 weeks) or SOX (S-1 80/100/120 mg day−1 based on BSA, orally, days 1–14, plus oxaliplatin 100 mg m−2, intravenously, day 1, every 3 weeks). The primary end point was PFS.
Results:
Between January 2009 and July 2010, 271 patients were randomly allocated to either S-1 (n=135) or SOX (n=136). Median PFS for S-1 and SOX were 2.8 and 3.0 months, respectively (hazard ratio (HR)=0.84; 95% confidence interval (CI), 0.65–1.08; stratified log-rank test P=0.18). Median overall survival (OS) was 6.9 vs 7.4 months (HR=1.03; 95% CI, 0.79–1.34; stratified log-rank test P=0.82). The response rate (RR) was 11.5% vs 20.9% (P=0.04). The major grade 3/4 toxicities (S-1 and SOX) were neutropenia (11.4% and 8.1%), thrombocytopenia (4.5% and 10.3%) and anorexia (12.9% and 14.7%).
Conclusions:
Although SOX showed an advantage in RR, it provided no significant improvement in PFS or OS compared with S-1 alone.
Similar content being viewed by others


Main
Pancreatic cancer is associated with an extremely poor prognosis and the eighth leading cause of cancer-related deaths worldwide (Jemal et al, 2011).
Gemcitabine (GEM) has been the standard first-line therapy in patients with advanced pancreatic cancer for a long time. In order to improve the prognosis of such patients, there is an urgent need to establish an effective second-line therapy. Although various second-line therapies have been studied, any phase III data do not support a particular regimen (Petrelli et al, 2010). In Japan, S-1 is commonly used for the treatment of pancreatic cancer patients who failed GEM-based treatment. In the phase II study of S-1 for pancreatic cancer resistant to GEM, the median progression-free survival (PFS) and overall survival (OS) were only 2.0 months and 4.5 months, respectively (Morizane et al, 2009). More effective regimens had been eagerly awaited.
In 2008, the addition of oxaliplatin to 5-FU and leucovorin (5-FU/LV (OFF)) was reported to show significant improvements in OS and PFS in a second-line setting in the CONKO 003 trial (Oettle et al, 2014). The good efficacy of oxaliplatin plus S-1 (SOX) had been previously demonstrated in colorectal and gastric cancer (Yamada et al, 2008; Koizumi et al, 2010), and was, therefore, expected to provide similar efficacy to the OFF treatment. Here, we conducted a randomised phase II trial to evaluate the efficacy and safety of SOX compared with S-1 alone in patients with GEM-refractory pancreatic cancer as a second-line setting.
Materials and methods
Patients
The inclusion criteria were as follows: refractory to GEM, histologically or cytologically confirmed pancreatic adenocarcinoma or adenosquamous carcinoma, a measurable metastatic lesion based on Response Evaluation Criteria in Solid Tumors (RECIST) guidelines version 1.0, age of ⩾20 years, Eastern Cooperative Oncology Group (ECOG) performance status (PS) score of 0 or 1, adequate bone marrow function (haemoglobin level of ⩾9.0 g dl−1, white blood cell count of ⩽12 000 per mm3, neutrophil count of ⩾1500 per mm3, and platelet count of ⩾100 000 per mm3), adequate liver function (total bilirubin ⩽2.0 mg dl−1, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ⩽150 IU l−1), and adequate renal function (serum creatinine (CRE) level of ⩽1.2 mg dl−1). GEM-refractory was defined as follows: (i) patients who developed progression confirmed by the image in the first-line therapy including GEM or (ii) patients who relapsed during the GEM-adjuvant treatment or within 24 weeks after the last GEM administration.
Patients were excluded if they had received previous chemotherapy containing platinum or fluoropyrimidine drugs, previous radiotherapy except for intraoperative radiation therapy, or either blood transfusion, blood products, or hematopoietic growth factor preparations such as granulocyte-colony stimulating factor within 14 days prior to enrolment; if they were receiving treatment with phenytoin, potassium warfarin, or flucytosine at the time of the study; or if they had grade 2 or higher peripheral sensory neuropathy, more than moderate coelomic fluid (pleural effusion, ascites, or pericardial fluid), watery stools, a synchronous cancer with the exception of an early stage tumour (stage I), poorly controlled diabetes, or other serious concomitant diseases.
This study was conducted in accordance with the Declaration of Helsinki principals and Good Clinical Practice (GCP) guidelines. The protocol was approved by the ethics committees of all participating institutions and informed consent was obtained from all patients before their enrolment into the study.
The study is registered with JAPIC Clinical Trial Information (Japic CTI-090685).
Study design
This multicenter, randomised, open-label phase II trial was conducted at 24 centres. Patients were randomly allocated to receive either treatment with SOX or S-1 alone. Randomisation was performed centrally in a 1 : 1 ratio with stratification according to center, PS (0 or 1), and duration of previous GEM treatment (<90 days, 90 to <180 days, or ⩾180 days) using the minimisation method (Pocock and Simon, 1975) independently processed by the Contract Research Organization (AC Medical). The study treatments were started within 8 days from the randomisation and within 29 days from disease progression of the first-line GEM treatment. An Independent Data Monitoring Committee (IDMC) supervised the assessment of efficacy and safety.
The primary end point was PFS and secondary end points were OS, time-to-treatment failure (TTF), response rate (RR), disease control rate (DCR), and safety.
Treatments
Patients allocated to the S-1 arm received S-1 orally twice daily at a dose according to the body surface area (<1.25 m2, 80 mg day−1; 1.25 m2 to 1.5 m2, 100 mg day−1; ⩾1.5 m2, 120 mg day−1) on days 1–28 of every 6-week period. Patients allocated to the SOX arm received oxaliplatin at a dose of 100 mg m−2 as a 2-hour intravenous infusion on day 1 plus S-1 at the same daily-dose of the S-1 alone arm on days 1–14 of every 3-week period. The treatment was continued until disease progression, unacceptable toxicity, or patient refusal.
In both treatment arms, chemotherapy was delayed until recovery from toxicities (neutrophil count of <1500 per mm3, platelet count of <75 000 per mm3, AST or ALT >150 IU l−1, CRE >1.2 mg dl−1, grade 2 or higher diarrhoea or stomatitis, fever suspicious of infection, and grade 3 or higher peripheral sensory neuropathy (only for the SOX arm)). During the administration of S-1, patients who developed neutrophil count of <1000 per mm3, platelet count of <50 000 per mm3, CRE >1.5 mg dl−1, grade 2 or higher diarrhoea or stomatitis, grade 3 rash, or fever suspicious of infection had to withdraw S-1 until they recovered from the toxicities. The doses of oxaliplatin and S-1 could be reduced by 25 mg m−2 and 10–30 mg day−1, respectively, up to two times, but the treatment was discontinued if subsequent reduction was required. In both arms, the dose of each drug was reduced by one level if grade 4 neutropenia, febrile neutropenia, grade 4 thrombocytopenia, thrombocytopenia requiring platelet transfusion, or grade 3 or higher diarrhoea or stomatitis occurred. In the SOX arm, if grade 2, 3, or 4 sensory neuropathy occurred and did not recover before the next administration, oxaliplatin was reduced by one dose level, skipped, or discontinued.
Assessments
Complete blood counts, blood chemical tests, and physical examinations were performed at least once a week for 12 weeks, and every 3 weeks thereafter. Computed tomography scans were performed and tumour markers (CEA and CA19-9) were measured every 4 weeks. Tumour responses were extramurally reviewed by Independent Review Committee in accordance with the RECIST version 1.0 guidelines. Safety was assessed using the Common Terminology Criteria for Adverse Events version 3.0.
Statistical analysis
The safety analysis set included the patients who received at least one dose of study drugs and had no major GCP violations. The full analysis set (FAS) included the patients who met the eligibility criteria in the safety analysis set. Efficacy was evaluated in the FAS population. PFS was defined as the time from randomisation to the first event of progressive disease or death because of any cause. If no such event occurred in a patient, data for that patient was censored on the day of the last imaging confirmation. OS was defined as the time from randomisation to death from any cause. In the absence of the event, data was censored on the last day of survival confirmation. TTF was the time from enrolment to the first event of discontinuation of treatment, progressive disease, or death because of any cause.
This trial was designed to detect an hazard ratio (HR) of 0.667 with an increase in median PFS from 2 to 3 months. In order to detect the assumed differences with a power of 85% and a two-sided 5% type 1 error, a minimum of 220 events were required for primary analysis. Considering patients’ dropout, the sample size was set at 240.
Regarding patient background, imbalances among the groups were examined using the χ2 test or Fisher’s exact test for categorical variables. The stratified log-rank test was used to assess the differences between groups in PFS and OS. The stratification factors were PS (0 or 1), and duration of previous GEM treatment (<90 days, 90 to <180 days or ⩾180 days). The stratified Cox proportional hazards model was used to estimate HRs and corresponding 95% confidence intervals (CI) for PFS, OS, and TTF. The point estimate of the median PFS, OS, and TTF and the 95% CI were calculated using the Kaplan–Meier method. The exploratory subgroup analysis was performed with stratified Cox proportional hazards model. All analyses were done with SAS (version 9.1.3).
Results
Patient characteristics
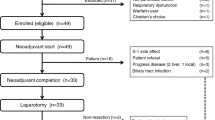
From January 2009 to July 2010, a total of 271 patients from 24 Japanese centres were enroled, and 135 patients were allocated to the S-1 arm and 136 patients allocated to the SOX arm. The cut-off date for analysis was 31 March 2011.
In the S-1 arm, three patients were excluded from the safety analysis set; two patients did not receive S-1 treatment and one patient received commercial S-1 instead of the provided study drug. Two patients were excluded from the FAS because they were ineligible; one patient was not proven to have adenocarcinoma or adenosquamous carcinoma, and another patient had massive fluid retention. In the SOX arm, two patients were also excluded from the FAS because of the same reasons mentioned above (Figure 1). As a result, a total of 268 patients (S-1 arm: 132 patients, SOX arm: 136 patients) were assessed for safety and 264 patients (S-1 arm: 130 patients, SOX arm: 134 patients) were assessed for efficacy. Both arms were well balanced in patients’ characteristics (Table 1). Approximately 70% of patients had PS 0 or liver metastasis in both arms. Patients whose primary tumour was resected accounted for about 20% in both arms.

CONSORT diagram.
Efficacy
The analysis of PFS was based on 256 events among 264 patients (97.0%) with a median follow-up time of 12.6 months. Median PFS was 2.8 months (95% CI, 1.9–3.5) in the S-1 arm and 3.0 months (95% CI, 2.8–3.7) in the SOX arm (Figure 2), and there was no significant difference between the arms (HR=0.84; 95% CI, 0.65–1.08, stratified log-rank test P=0.18). The analysis of OS was based on 232 deaths among the 264 patients (87.9%). Median OS was 6.9 months (95% CI, 5.8–9.0) in the S-1 arm and 7.4 months (95% CI, 6.2–8.6) in the SOX arm (Figure 3), and there was no significant difference between the arms (HR=1.03, 95% CI, 0.79–1.34, stratified log-rank test P=0.82). The RR was 11.5% (95% CI, 6.6–18.3) in the S-1 arm and 20.9% (95% CI, 14.4–28.8) in the SOX arm (P=0.04). The DCR was 53.8% (95% CI, 44.9–62.6) in the S-1 arm and 60.4% (95% CI, 51.6–68.8) in the SOX arm (P=0.28). With respect to tumour markers, serum CRE levels decreased in 31% of patients in the S-1 arm and in 24% of patients in the SOX arm. Serum CRE 19-9 levels decreased in 54% of patient of the S-1 arm and in 55% of patients in the SOX arm. There was no obvious difference in tumour marker decrease between the S-1 arm and the SOX arm. No clear correlation was also seen between tumour response and tumour maker decrease. After the study treatment, 75 patients in the S-1 arm and 72 patients in the SOX arm received the third-line therapies, and those contents were similar in the two arms.

Kaplan–Meier estimates of progression-free survival (PFS). The median PFS was 2.8 months (95% CI, 1.9–3.5) in the S-1 arm and 3.0 months (95% CI, 2.8–3.7) in the SOX arm.

Kaplan–Meier estimates of overall survival (OS). The median OS was 6.9 months (95% CI, 5.8–9.0) in the S-1 arm and 7.4 months (95% CI, 6.2–8.6) in the SOX arm.
In the subgroup analysis, the SOX arm showed longer PFS in patients with age <65 years (HR, 0.68; 95% CI, 0.47–0.99; Figure 4). The SOX arm also showed longer OS in patients with age <65 years (HR, 0.68; 95% CI, 0.46–1.00), whereas the S-1 arm showed longer OS in patients with age >65 years (HR, 1.58; 95% CI, 1.06–2.36) (Figure 5).

Subgroup analyses of progression-free survival. *Some patients had overlapped locations.

Subgroup analyses of overall survival. *Some patients had overlapped locations.
Safety
The major toxicities are summarised in Table 2. Anorexia, decreased serum sodium, lymphopenia, and neutropenia were the most common events in both arms. The incidence of anaemia, stomatitis, and pigmentation were 10% or higher in the S-1 arm than in the SOX arm. The incidence of thrombocytopenia, peripheral sensory neuropathy, anorexia and vomiting were 10% or higher in the SOX arm than in the S-1 arm. There was no difference between the arms in grade 3 or worse adverse events. One treatment-related death owing to bacterial peritonitis was observed in the SOX arm.
Treatment
The median treatment duration was 73 days (range 4–840 days) in the S-1 arm and 78 days (range 3–455 days) in the SOX arm. The major reasons for discontinuation of the treatment were disease progression (S-1 arm: 113 patients, SOX arm: 96 patients), adverse events (S-1 arm: 14 patients, SOX arm: 22 patients), and consent withdrawal (S-1 arm: 1 patient, SOX arm: 10 patients). As for the treatment discontinuation relating to adverse events, neutropenia, and thrombocytopenia were more frequently seen in the SOX arm than the S-1 arm. The major causes for consent withdrawal in the SOX arm were non-haematological toxicities such as fatigue and vomiting. There was no patient who withdrew consent owing to neurotoxicity. The median relative dose intensity (RDI) and the median total dose of S-1 were 89.7% and 6000 mg m−2 in the S-1 arm and 84.3% and 5010 mg m−2 in the SOX arm, respectively. The median RDI of oxaliplatin was 95.4% and the median total dose was 400 mg m−2 in the SOX arm.
Dose reduction of S-1 was observed in 16 patients (12.1%) in the S-1 arm and 24 patients (17.6%) in the SOX arm. Dose reduction of oxaliplatin was observed in 28 patients (20.6%). Dose reduction of both drugs was observed in 14 patients (10.3%). Postponement of drug administration was observed in 58 patients (43.9%) in the S-1 arm and 76 patients (55.9%) in the SOX arm. Dose interruption of S-1 was observed in 59 patients (44.7%) in the S-1 arm and 38 patients (27.9%) in the SOX arm.
Discussion
This phase II study could not demonstrate the relevant benefit by adding oxaliplatin to S-1 in PFS and OS, although some increased response was gained. In the first-line therapy, GEM alone treatment is considered for the majority of patients. Recently, two regimens, FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin combination) and GEM plus nab-paclitaxel, have become the new standard options for advanced pancreatic cancer (Conroy et al, 2011; Von Hoff et al, 2013). Fluoropyrimidine-based regimens could be used as the second-line chemotherapy in patients who failed in GEM alone or GEM plus nab-paclitaxel treatment, although their benefit has not been proven yet (Boeck et al, 2007; Gebbia et al, 2007; Kulke et al, 2007; Xiong et al, 2008; Morizane et al, 2009; Novarino et al, 2009). In the second line setting for pancreatic cancer, patients with poor PS were enroled in some studies (Gebbia et al, 2007; Xiong et al, 2008). However, we are not sure if SOX therapy fits such patients because only patients with good PS were enroled in this study.
In this trial, the primary end point was PFS, and we assessed response every 4 weeks to detect small difference between the two arms (expected median PFS; S-1 2.0 months vs SOX 3.0 months). However, the primary end point was not met owing to unexpectedly good PFS in the S-1 arm. The subgroup analysis showed that SOX improved both OS and PFS in the group of patients <65 years. In the SOX arm, the RDI of S-1 was 87.6% for the group of patients <65 years and 79.0% for the group ⩾65 years. One possible explanation is that anorexia or fatigue requiring a dose reduction of S-1 was more frequently observed in the group of aged ⩾65 years than that of aged <65 years. The SOX regimen might be an effective therapy for particularly non-elderly patients with GEM-refractory pancreatic cancer.
Many trials with 5-fluorouracil and platinum combination have been reported in the second-line treatment for advanced pancreatic cancer. Recently, a comprehensive analysis of published 12 trials data (n=450) for that combination was performed (Rahma et al, 2013). It provided a median PFS of 2.9 months and OS of 5.7 months for the combination of 5-fluorouracil and platinum agents. These are similar to those reported in the OFF arm of CONKO-003 trial (median PFS 2.9 month and median OS 5.9 months) and in the SOX arm of our study (median PFS 3.0 month and median OS 7.4 months). There was no big difference between the median total dose of oxaliplatin reported in the OFF arm (340 mg m−2) and that obtained in the SOX arm of this study (400 mg m−2). Although cross-trial comparison should be carefully interpreted because of the different inclusion/exclusion criteria, different drugs (oral and intravenous), and schedules, these data indicate that the benefit by this kind of combination was limited.
Toxicities were generally mild-to-moderate in both arms, and similar to those reported in the previous trials using 5-fluorouracil and oxaliplatin. Grade 3 peripheral sensory neuropathy, the most critical toxicity of oxaliplatin, was low (2.9%). In the treatment for metastatic colorectal cancer, oxaliplatin-induced grade 3 neuropathy was reported to occur in around 30% of the patients treated with cumulative doses of oxaliplatin ranging from 765 mg m−2 to 1020 mg m−2 (Kalofonos et al, 2005; Van Cutsem et al, 2006; Argyriou et al, 2007; Land et al, 2007; Otsu et al, 2014). In this trial, the median cumulative dose of oxaliplatin was 400 mg m−2, and the less cumulative dose of oxaliplatin would lead to less frequency of grade 3 neuropathy.
In conclusion, SOX showed no significant improvement in PFS and OS as compared with S-1 in the second-line chemotherapy for patients with GEM-refractory pancreatic cancer.
References
Argyriou AA, Polychronopoulos P, Iconomou G, Koutras A, Makatsoris T, Gerolymos MK, Gourzis P, Assimakopoulos K, Kalofonos HP, Chroni E (2007) Incidence and characteristics of peripheral neuropathy during oxaliplatin-based chemotherapy for metastatic colon cancer. Acta Oncol 46: 1131–1137.
Boeck S, Wilkowski R, Bruns CJ, Issels RD, Schulz C, Moosmann N, Laessig D, Haas M, Golf A, Heinemann V (2007) Oral capecitabine in gemcitabine-pretreated patients with advanced pancreatic cancer. Oncology 73: 221–227.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, Fouchardiere C, Bennouna J, Bachet J-B, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Chem M, Ducreux M (2011) FOLFIRINOX versus gemcitabine for metastatic pancreas cancer. N Engl J Med 364: 1817–1825.
Gebbia V, Maiello E, Giuliani F, Borsellino N, Caruso M, Di Maggio G, Ferraù F, Bordonaro R, Verderame F, Tralongo P, Di Cristina L, Agueli R, Russo P, Colucci G (2007) Second-line chemotherapy in advanced pancreatic carcinoma: a multicenter survey of the Gruppo Oncologico Italia Meridionale on the activity and safety of the FOLFOX4 regimen in clinical practice. Ann Oncol 6: vi124–vi127.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61: 69–90.
Kalofonos HP, Aravantinos G, Kosmidis P, Papakostas P, Economopoulos T, Dimopoulos M, Skarlos D, Bamias A, Pectasides D, Chalkidou S, Karina M, Koutras A, Samantas E, Bacoyiannis C, Samelis GF, Basdanis G, Kalfarentzos F, Fountzilas G (2005) Irinotecan or oxaliplatin combined with leucovorin and 5-fluorouracil as first-line treatment in advanced colorectal cancer: a multicenter, randomized, phase II study. Ann Oncol 16: 869–877.
Koizumi W, Takiuchi H, Yamada Y, Boku N, Fuse N, Muro K, Komatsu Y, Tsuburaya A (2010) Phase II study of oxaliplatin plus S-1 as first-line treatment for advanced gastric cancer (G-SOX study). Ann Oncol 21: 1001–1005.
Kulke MH, Blaszkowsky LS, Ryan DP, Clark JW, Meyerhardt JA, Zhu AX, Enzinger PC, Kwak EL, Muzikansky A, Lawrence C, Fuchs CS (2007) Capecitabine plus erlotinib in gemcitabine-refractory advanced pancreatic cancer. J Clin Oncol 25: 4787–4792.
Land SR, Kopec JA, Cecchini RS, Ganz PA, Wieand HS, Colangelo LH, Murphy K, Kuebler JP, Seay TE, Needles BM, Bearden JD 3rd, Colman LK, Lanier KS, Pajon ER Jr, Cella D, Smith RE, O'Connell MJ, Costantino JP, Wolmark N (2007) Neurotoxicity from oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: NSABP C-07. J Clin Oncol 25: 2205–2211.
Morizane C, Okusaka T, Furuse J, Ishii H, Ueno H, Ikeda M, Nakachi K, Najima M, Ogura T, Suzuki E (2009) A phase II study of S-1 in gemcitabine-refractory metastatic pancreatic cancer. Cancer Chemother Pharmacol 63: 313–319.
Novarino A, Satolli MA, Chiappino I, Giacobino A, Bellone G, Rahimi F, Milanesi E, Bertetto O, Ciuffreda L (2009) Oxaliplatin, 5-fluorouracil, and leucovorin as second-line treatment for advanced pancreatic cancer. Am J Clin Oncol 32: 44–48.
Oettle H, Riess H, Stieler JM, Heil G, Schwaner I, Seraphin J, Görner M, Mölle M, Greten TF, Lakner V, Bischoff S, Sinn M, Dörken B, Pelzer U (2014) Second-line oxaliplatin, folnic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from CONKO-003 trial. J Clin Oncol 32: 2423–2429.
Otsu S, Hirashima Y, Nishikawa K, Sakashita H, Morinaga R, Watanabe K, Shirao K (2014) Neurological toxicity in metastatic colorectal cancer patients treated with modified FOLFOX6 plus bevacizumab. Jpn Clin Med 5: 19–23.
Petrelli F, Borgonovo K, Ghilardi M, Cabiddu M, Barni S (2010) What else in gemcitabine-pretreated advanced pancreatic cancer? An update of second line therapies. Rev Recent Clin Trials 5: 43–56.
Pocock SJ, Simon R (1975) Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics 31: 103–115.
Rahma OE, Duffy A, Liewehr DJ, Steinberg SM, Greten TF (2013) Second-line treatment in advanced pancreatic cancer: a comprehensive analysis of published clinical trials. Ann Oncol 24: 1972–1979.
Van Cutsem E, Nordlinger B, Adam R, Köhne CH, Pozzo C, Poston G, Ychou M, Rougier P (2006) Towards a pan-European consensus on the treatment of patients with colorectal liver metastases. Eur J Cancer 42: 2212–2221.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Cutsem EV, Wei X, Iglesias J, Renschler MF (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369: 1691–1703.
Xiong HQ, Varadhachary GR, Blais JC, Hess KR, Abbruzzese JL, Wolff RA (2008) Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 113: 2046–2052.
Yamada Y, Tahara M, Miya T, Satoh T, Shirao K, Shimada Y, Ohtsu A, Sasaki Y, Tanigawara Y (2008) Phase I/II study of oxaliplatin with oral S-1 as first-line therapy for patients with metastatic colorectal cancer. Br J Cancer 98: 1034–1038.
Acknowledgements
This study was supported by Yakult Honsha Co., Ltd. We thank all patients, clinicians, and support staff who participated in this study. We are grateful to Yuh Sakata, Fumitaka Nagamura, and Satoshi Morita for their helpful advice as members of the IDMC, and Atsushi Sato, Kunihisa Miyakawa and Kouki Yoshikawa as members of the Independent Review Committee. We also thank Yuki Tanaka for his helpful advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Hiroyuki Isayama received honoraria from Yakult Honsha Co., Ltd. and Taiho Pharmaceutical; Chikuma Hamada has received consulting fees from Yakult Honsha Co., Ltd.. The remaining authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ohkawa, S., Okusaka, T., Isayama, H. et al. Randomised phase II trial of S-1 plus oxaliplatin vs S-1 in patients with gemcitabine-refractory pancreatic cancer. Br J Cancer 112, 1428–1434 (2015). https://doi.org/10.1038/bjc.2015.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.103
Keywords
This article is cited by
-
FOLFOX regimen after failure of fluorouracil and leucovorin plus nanoliposomal-irinotecan therapy for advanced pancreatic cancer: a retrospective observational study
BMC Cancer (2023)
-
Clinical significance and functional role of adhesion G-protein-coupled receptors in human pancreatic ductal adenocarcinoma
British Journal of Cancer (2023)
-
Phase II clinical trial of gemcitabine plus oxaliplatin in patients with metastatic pancreatic adenocarcinoma with a family history of pancreatic/breast/ovarian/prostate cancer or personal history of breast/ovarian/prostate cancer (FABRIC study)
International Journal of Clinical Oncology (2020)
-
Establishment and characterization of 6 novel patient-derived primary pancreatic ductal adenocarcinoma cell lines from Korean pancreatic cancer patients
Cancer Cell International (2017)
-
Therapeutic efficacy and safety of S-1-based combination therapy compare with S-1 monotherapy following gemcitabine failure in pancreatic cancer: a meta-analysis
Scientific Reports (2016)
