
Abstract
The reduction of intracellular 1,4,5-inositol trisphosphate (IP3) levels stimulates autophagy, whereas the enhancement of IP3 levels inhibits autophagy induced by nutrient depletion. Here, we show that knockdown of the IP3 receptor (IP3R) with small interfering RNAs and pharmacological IP3R blockade is a strong stimulus for the induction of autophagy. The IP3R is known to reside in the membranes of the endoplasmic reticulum (ER) as well as within ER–mitochondrial contact sites, and IP3R blockade triggered the autophagy of both ER and mitochondria, as exactly observed in starvation-induced autophagy. ER stressors such as tunicamycin and thapsigargin also induced autophagy of ER and, to less extent, of mitochondria. Autophagy triggered by starvation or IP3R blockade was inhibited by Bcl-2 and Bcl-XL specifically _targeted to ER but not Bcl-2 or Bcl-XL proteins _targeted to mitochondria. In contrast, ER stress-induced autophagy was not inhibited by Bcl-2 and Bcl-XL. Autophagy promoted by IP3R inhibition could not be attributed to a modulation of steady-state Ca2+ levels in the ER or in the cytosol, yet involved the obligate contribution of Beclin-1, autophagy-related gene (Atg)5, Atg10, Atg12 and hVps34. Altogether, these results strongly suggest that IP3R exerts a major role in the physiological control of autophagy.
Similar content being viewed by others

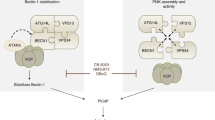

Main
The first step of macroautophagy consists in the gradual envelopment of cytoplasmic material (cytosol and/or organelles) in the phagophore, a cistern that finally sequesters cytoplasmic material in autophagosomes (also called autophagic vacuoles (AVs)) lined by two membranes. Autophagosomes then undergo a progressive maturation by fusion with endosomes and/or lysosomes. This latter step creates autolysosomes in which the inner membrane as well as the luminal content of the AVs is degraded by lysosomal enzymes. The process of autophagy is controlled by a series of evolutionary conserved genes, the atg genes, whose products are essential for specific steps of the autophagic process.1, 2
One of the strongest triggers of autophagy is nutrient stress.3, 4 In response to starvation, cells degrade nonessential components thereby generating nutrients for meeting the cell's energetic demand as well as for vital biosynthetic reactions. In such circumstances, when autophagy is an adaptation, inhibition of autophagy has a negative impact on cell survival. For example, mice lacking the atg5 gene survive without any major developmental defect until birth, yet succumb to the stressful neonatal period unless puppies are force-fed with milk within the first hours after birth.5 Similarly, human cell lines cultured in nutrient-free media mount a cytoprotective autophagic response. Suppression of autophagy by chemical inhibitors or knockdown of essential genes thus sensitize cells to starvation-induced cell death.6, 7 Autophagy inhibition also sensitizes cells to the depletion of obligatory growth (or survival) factors resulting in a decrease of nutrient import through the plasma membrane. For example, the knockdown of Atg5, Atg10 or Atg12 (which are all involved in a ubiquitin-like conjugation system required for autophagosome formation) prevents the generation of AV and sensitizes cells to cell death induced by withdrawal of essential growth factors.8 Similarly, knockdown of Atg6/Beclin-1 (whose major physiological partner is the class III phosphatidylinositol 3- kinase Vps34) inhibits autophagy and sensitizes to cell death induced by starvation.6, 8 Beclin-1 also interacts with Bcl-2,9 which inhibits autophagy,10 perhaps by preventing the interaction between Beclin-1 and Vps34.11
One recent study suggested that myo-inositol-1,4,5-trisphosphate (IP3) could regulate autophagy because inhibition of inositol monophosphatase by lithium or L-690,330 stimulates autophagy through the depletion of inositol and IP3.12 IP3 is a second messenger produced primarily in response to the stimulation of G-protein-coupled receptor or receptor tyrosine kinases. IP3 acts on the IP3 receptor (IP3R), a mostly endoplasmic reticulum (ER)-sessile Ca2+ release channel that integrates signals from numerous small molecules and proteins including nucleotides, kinases and phosphatases, as well as nonenzyme proteins.13 IP3R is known to play a major role within the Ca2+ microdomains that transmit Ca2+ spikes generated by the ER to mitochondria.14, 15 IP3R is also regulated by Bcl-2 16, 17 and Bcl-XL,18 which affect Ca2+ fluxes through IP3R by direct molecular interactions, by influencing its regulatory phosphorylation and/or by modulating its response to IP3. Through these regulatory mechanisms, IP3R modulates diverse cellular functions, which include, but are not limited to, contraction/excitation, gene expression, cellular growth and apoptosis.13, 19
On the basis of these premises, we decided to investigate the contribution of IP3R to the regulation of autophagy. Here, we report that the depletion of IP3R or its inhibition triggers rapid autophagy affecting both ER and mitochondria. IP3R functions as a Bcl-2-regulated repressor of autophagy.
Results and Discussion
Starvation-induced autophagy concerns both ER and mitochondria
Upon 12 h of culture in the absence of serum and nutrients (starvation), HeLa cells manifest the relocalization of LC3-GFP into AVs and this effect is inhibited by addition of the cell-permeable IP3 precursor myo-inositol (Figure 1a and b). Under these conditions, little cell death (as determined with the vital dye propidium iodine (PI)) occurred after serum withdrawal and the reduction of the mitochondrial transmembrane potential (ΔΨm) induced by starvation was inhibited by myo-inositol (Figure 1c). Starvation-induced LC3-GFP aggregates partially colocalized with the ER marker calreticulin and with the mitochondrial matrix marker HSP60, indicating that this regimen caused autophagy of both ER and mitochondria (Figure 1d–f).

Autophagy induced by starvation. (a–c) LC3-GFP redistribution inhibited by myo-inositol. HeLa cells transiently transfected with LC3-GFP were subjected to nutrient depletion in the presence or absence of myo-inositol for 12 h and representative pictures of Hoechst 33342-counterstained cells were taken (a) and the percentage of adherent cells exhibiting a clear LC3-GFP relocalization into cytoplasmic vacuoles was determined (b). The frequency of adherent and nonadherent cells with a low ΔΨm was determined by DiOC6(3) staining (and counterstaining with PI) (c). Results are means±S.D., of three independent experiments. (d–f). Organelle-specific autophagy triggered by starvation. Cells subjected to LC3-GFP transfection and nutrient depletion as in a–c were fixed, permeabilized and stained for calreticulin (d) or HSP60 (e) to determine the colocalization of LC3-GFP and ER (d) or mitochondrial (e). The right panels in d and e indicate the profiles of colocalization within the area of interest (labeled by arrows), as determined by quantitative confocal microscopy. (f) Mean level of colocalization of LC3-GFP with calreticulin or HSP60, as determined for 50 cells (X±S.E.M.)
ER stress but not mitochondrial stress induce autophagy
As starvation can induce an ER stress response20, 21 as well as mitochondrial stress,22 we investigated whether direct induction of organelle-specific stress would stimulate autophagy. Thapsigargin (which selectively inhibits the Ca2+–ATPase responsible for Ca2+ accumulation by the ER, SERCA), tunicamycin (which blocks N-linked protein glycosylation), brefeldin A (which inhibits ER–golgi transport) are well known for their capacity to induce ER stress21, 23 and all of them induce rapid LC3-GFP aggregation in AVs (Figure 2a and b), well before they cause cell death and ΔΨm dissipation (Figure 2c). In contrast, induction of mitochondrial stress by antimycin A (which inhibits complex III of the respiratory chain), betulinic acid (which can cause direct permeabilization of mitochondrial membranes)24 or cadmium (a heavy metal that induces oxidative stress and permeability transition)25 did not stimulate autophagy under conditions in which all three agents caused a significant ΔΨm loss (Supplementary Figure S1). These data suggest that ER stress is a stronger inducer of autophagy than mitochondrial stress.

Autophagy induced by organelle-specific stress. (a–c) Autophagy induced by ER stress. LC3-GFP-transfected cells were treated with thapsigargin, tunicamycin or brefeldin A and the frequency of cells exhibiting the vacuolar redistribution of LC3-GFP was determined among adherent cells in b (for representative pictures taken at 6 h see a). The percentage of cells with a reduced ΔΨm was determined by DiOC6(3) staining (and counterstaining with PI). (c) The inset in c confirms the induction of ER stress, as detected by measuring the expression level of GADD34. Results are means±S.D. of three independent experiments. (d, e) Transmission electron microscopy of thapsigargin-induced autophagy after 6 h. Note the presence of clearly distinguishable rough ER in some AV. The number of AV of type 1 or 2 is quantified in d on a per cell basis (X±S.E.M., n=50). (f–h) Organelle-specific autophagy triggered by starvation. Cells subjected to LC3-GFP transfection and nutrient depletion as in f, g were fixed, permeabilized and stained for calreticulin (f) or HSP60 (g). The right panels in f and g depict profiles of colocalization. The percentage of colocalization (X±S.E.M.) was quantified for 50 cells for each condition (h)
ER stress-induced autophagy affects ER and mitochondria
ER stress triggered the emergence of bona fide AVs, as defined by cytoplasmic structures lined by two membranes that can be detected by transmission electron microscopy (Figure 2d and e). The LC3-GFP-positive vesicles induced by thapsigargin or tunicamycin colocalized with the ER marker calreticulin, but also colocalized in part with the mitochondrial matrix marker HSP60, suggesting that ER stress causes autophagy of both ER and mitochondria (Figure 2f–h). ER stress-induced autophagy followed a classical autophagic pathway to the extent that the knockdown of essential atg genes such as atg6/beclin-1, atg5, atg10, atg12 as well as the knockdown of the mammalian class III phosphatidylinositol 3-kinase vps34 inhibited autophagy induced by thapsigargin, tunicamycin or brefeldin A (Figure 3). Altogether, these data suggest that ER stress effectively trigger autophagy that follows a canonical pathway.

Implication of essential atg genes in ER stress-induced autophagy. Cells were transfected with the indicated siRNAs and protein downregulation was determined by immunoblot for two different siRNAs _targeting Beclin-1 (48 h after transfection) and for one siRNA _targeting hVps34 (a). One day after siRNA transfection, cells were retransfected with LC3-GFP (optionally together with Beclin-1) and the cells were subjected to ER stress the day after for 6 h, followed by assessment of the cytoplasmic aggregation of LC3-GFP. Results are means±S.D. of three experiments (b)
IP3 depletion and IP3R inhibition stimulates autophagy of both ER and mitochondria
The inositol monophosphatase inhibitors L-690,330 and lithium both induced autophagy (Figure 4a and b) under conditions in which neither agent induced toxic effects on the viability or bioenergetic state of cells (Figure 4c). Both L-690,330 and lithium triggered significant autophagy of both ER and mitochondria (Figure 4d–f). Very similar results were obtained by inhibition of IP3R. Knockdown of the IP3R I and III isoforms caused a strong, nonsynergistic induction of autophagy, as measured by LC3-GFP relocalization (Figure 5a and b). Similarly, xestospongin B, a chemical antagonist of IP3R,26 caused LC3-GFP aggregation in cytoplasmic vacuoles (Figure 5c). Stimulation of autophagy was more pronounced for xestospongin B than for xestospongin C, which has a lower affinity for IP3R27 (Figure 5d). Moreover, the xestospongin B effect was rapid (⩽2 h, Figure 5e) and did not involve any major toxicity on mitochondria (Figure 5f) nor on the ER (as estimated by measuring the ER stress indicator GADD34; inset in Figure 5f). Xestospongin B-induced autophagy required atg6/beclin-1, atg5, atg10, atg12 and vps34 (Figure 5g), indicating that it followed an orthodox pathway. Under the conditions in which xestospongin B was employed in this study, the luminal ER Ca2+ and the baseline cytosolic Ca2+ concentrations were not modulated (Figure 6a and b), although xestospongin clearly blunted the histamine-induced IP3/IP3R-dependent ER Ca2+ efflux, as an internal control of its efficacy (Figure 6c). Moreover, depletion of extracellular Ca2+ did not affect the xestospongin B-induced autophagy (Figure 6d), suggesting that the xestospongin B effects were not mediated by gross perturbations of intracellular Ca2+ fluxes. Ultrastructural analysis of xestospongin B-elicited AV indicated that they frequently contained degenerating mitochondria or ER (Figure 7a). Accordingly, LC3-GFP aggregates elicited by xestospongin B colocalized with either calreticulin or HSP60 (Figure 7b–d). Thus, IP3R blockade caused the same pattern of autophagy as that induced by starvation.

Induction of autophagy by inositol monophosphate inhibitors. Cells were treated with L-690,330, lithium and/or myo-inositol for 12 h, followed by determination of the cytoplasmic redistribution of LC3-GFP (a, b) or the quantitation of the ΔΨm (c). Results are means±S.D. of three experiments. In addition, the colocalizaton of LC3-GFP and calreticulin (d, f) or HSP60 (e, f) was determined (n=50 in f)

Autophagy triggered by IP3R knockdown. HeLa cells were transfected with siRNA specific for distinct IP3R isoforms. The protein expression level was controlled 48 h after transfection (a). In addition, the impact on LC3-GFP relocalization (X±S.D., n=3) was determined at different times posttransfection (b). (c–e) LC3-GFP aggregation induced by IP3R inhibitors. LC3-GFP-expressing cells were treated with the indicated doses of xestospongin B or C and the degree of cells with vacuolar redistribution of LC3-GFP was measured (standard dose for xestospongin B is 2 μ M in c and e; the treatment is 6 h in c and d). (f) Effect of IP3R blockade on the ΔΨm, as determined by cytofluorometry and on GADD34 expression as determined by immunoblot (insert). (g) Implication of essential atg genes in autophagy induced by IP3R inhibition. Cells were transfected with the indicated siRNAs (day 0), then with LC3-GFP (optionally together with Beclin-1, on day 1), treated with xestospongin B (day 2) for 6 h and scored for redistribution of LC3-GFP

Ca2+ independence of xestospongin-induced autophagy. (a) XeB inhibits IP3-induced Ca2+ release but does not change steady [Ca2+]er, ER Ca2+ leak and basal [Ca2+]c [Ca2+]er was measured in erAEQmut-transfected HeLa cells ([Ca2+]er was 475±25 μ M in controls, 481±32 in XeB-treated cells; n=7). XeB (1 μ M) was added to the cells 2 h before the experiment. Cells were kept in KRB–EGTA (600 μ M) solution, then ER was Ca2+ loaded by the addition of 1 mM CaCl2 (in KRB) to the cells. ER Ca2+ leak was estimated by the addition of the SERCA blocker tert-butylhydroquinone (tBHQ). (b) Basal [Ca2+]c was measured in cells loaded with fura-2 as described in the Materials and Methods section (controls: 46.2±4.5 nM; after XeB treatment: 42.2±5.5 nM, n=35). After the control period, tBHQ (in KRB 1 mM CaCl2) was added to the cells in order to estimate Ca2+ leak into the cytosol and the activity of Ca2+ extrusion through the plasma membrane. (c) Histamine-induced Ca2+ release was measured in cytAEQ-transfected cells (lower right panel). As an internal control of the efficacy of xestospongin B as a IP3R antagonist, histamine-induced (IP3/IP3R-dependent) Ca2+ variations in the ER were measured in the absence or presence of 2 μ M xestospongin. The peak [Ca2+]c response was 3.3±0.6 μ M in controls, 0.87±0.4 μ M after XeB treatment (n=9). Note that xestospongin did not affect the baseline ER Ca2+ concentration. (d) Effect of Ca2+ depletion on xestospongin-induced autophagy. The concentration of extracellular Ca2+ was modulated before addition of xestospongin B for 6 h and determination of LC3-GFP aggregation. Results are means±S.D. of three experiments

Organelle-specific autophagy after IP3R inhibition. (a) Transmission electron microscopy of xestospongin B-induced autophagy after 6 h (I). Note the presence of mitochondria and rough ER in some AV (II). The number of type 1 or type 2 AV is quantified in B (X±S.E.M., n=50) (III). (b–d) Organelle-specific autophagy triggered by IP3R inhibition. LC3-GFP-expressing cells exposed to xestospongin B were stained for calreticulin (b) or HSP60 (c), both labeled in red. The degree of colocalization (X±S.E.M.) was measured for 50 cells (d)
ER-_targeted Bcl-2 and Bcl-XL differentially inhibit autophagy
IP3R has been shown to interact physically with Bcl-2 within the ER.16, 17 Moreover, the interaction of IP3 with the ANT interactome is strongly influenced by pro-apoptotic signaling.15 Xestospongin B strongly affected the coimmunoprecipitation of Bcl-2 and IP3R, depending on the subcellular localization of Bcl-2. The coimmunoprecipitation of IP3R with Bcl-2 _targeted to the ER (Bcl-2 cytochrome b5 (Cb5)) was attenuated by xestospongin B, whereas that of IP3R with wild-type (WT) Bcl-2 or with Bcl-2 _targeted to mitochondria (Bcl-2 Acta) was increased (Figure 8a). Although these results do not provide a molecular explanation for xestospongin B-induced autophagy, they point to an organelle-specific interaction between IP3R and Bcl-2, which can be modulated by xestospongin B. Transfection-enforced overexpression of Bcl-2 Cb5 inhibited autophagy induced by starvation, L-690,330, lithium or xestospongin B. In contrast, WT Bcl-2 (which is mostly mitochondrial) or Bcl-2 Acta failed to inhibit autophagy (Figure 8b), although they were expressed at a similar level as Bcl-2 Cb5 (inset in Figure 8b). Very similar results were obtained for the Bcl-2 analogue Bcl-XL. ER-_targeted Bcl-XL Cb5 (but not WT Bcl-XL and mitochondrion-_targeted Bcl-XL Acta) inhibited autophagy induced by starvation, L-690,330, lithium or xestospongin B (Figure 8c). Of note, none of the Bcl-2 or Bcl-XL constructs inhibited autophagy by thapsigargin or tunicamycin (Figure 8b and c), again suggesting that ER stress-induced and IP3R-regulated autophagy differ in mechanistic terms.

Impact of Bcl-2 and Bcl-XL on autophagy induced by IP3R inhibition. (a) Relocalization of Bcl-2 after IP3R association. Cells expressing WT Bcl-2, mitochondrion-_targeted Bcl-2 (Acta) or ER-_targeted Bcl-2 (Cb5) were treated with xestospongin B, followed by immunoprecipation of Bcl-2 and immunochemical detection of IP3R-III or Bcl-2. (b, c) Impact of WT, mitochondrion-_targeted (Acta) or ER-_targeted (Cb5) Bcl-2 (b) or Bcl-XL (c) on autophagy induced by IP3R inhibition, ER stress or inositol monophosphatase inhibition. Cells were transfected with LC3-GFP together with the indicated constructs, and 24 h later the cells were exposed to different stimuli for 12 h. Then, LC3-GFP redistribution (X±S.D., n=3) was determined by fluorescence microscopy. Asterisks indicate significant (P<0.01) inhibitory effects of ER-_targeted Bcl-2 and Bcl-XL. The inserts in b and c confirm the expression of WT, mitochondrion-_targeted (Acta) or ER-_targeted (Cb5) Bcl-2 and Bcl-XL, respectively
Concluding Remarks
In this article we provide arguments in favor of the implication of the IP3R in the regulation of autophagy. First, the elevation of IP3 levels using cell-permeable myo-inositol inhibited autophagy induced by starvation (Figure 1b). Similarly, the pharmacological depletion of IP3 sufficed to induce autophagy in human cells (Figure 4), as shown previously for murine and hamster cell lines.12 Second, depletion of IP3R by small interfering RNAs (siRNAs) or pharmacological blockade of IP3R (Figure 5) was highly efficient in inducing autophagy, which was similar to that induced by starvation (Figure 1) or IP3 depletion (Figure 4) to the extent that it induced the autophagic sequestration of both ER and mitochondria (Figure 7). Pharmacological inhibition of IP3R induced autophagy as quickly and as efficiently as ER stress-inducing agents including thapsigargin. At first glance, it appears paradoxical that IP3R antagonists (that should increase the luminal ER Ca2+) and the SERCA agonist thapsigargin (that should deplete luminal ER Ca2+ stores) both induced autophagy. One explanation would be to assume that any kind of perturbation in ER homeostasis would induce autophagy. However, xestospongins do not trigger the unfolded protein response in the ER (as this is the case for thapsigargin), whereas a variety of mechanistically unrelated ER stressors (including brefeldin A and tunicamycin) triggered autophagy. We failed to establish a link between Ca2+ signaling and IP3R-modulated autophagy. Thus, under conditions in which xestospongin B induced autophagy, no change in cytosolic or ER Ca2+ levels could be measured (Figure 6a–c), and withdrawal of external Ca2+ (Figure 6d) or chelation of intracellular Ca2+ (data not shown) failed to suppress xestospongin B-triggered autophagy. Rather, it appears that xestospongin B affects the protein–protein interaction in which IP3R engages, including the interaction with Bcl-2 in the ER (Figure 8a). Specifically ER-_targeted Bcl-2 (Figure 8b) or Bcl-XL (Figure 8c) strongly inhibited the induction of autophagy by starvations, several agents that reduce IP3 levels and the IP3R antagonist xestospongin B. Under the same experimental setting, however, ER-_targeted Bcl-2 (Figure 8b) or Bcl-XL (Figure 8c) failed to reduce autophagy induced by ER stressors including thapsigargin. These data strongly support the notion that induction of autophagy by inhibition of the IP3/IP3R signaling cascade is mechanistically different from autophagy induced by ER stress. It remains a conundrum through which molecular mechanisms ER stress can activate the autophagic response.
In conclusion, our results strongly support a signaling pathway in which IP3 and IP3R act to suppress autophagy. Thus, interruption of this pathway by depletion/inhibition of IP3 or IP3R can unleash the autophagic response. These findings underscore the cardinal importance of IP3R in the integration of cell fate-determining signals and their conversion into biological responses including cell growth, motility, differentiation, apoptosis and, as shown here, autophagy.
Materials and Methods
Cell lines, culture conditions and treatment
Rat-1 fibroblasts transfected with an empty control vector (CMV) or with a plasmid-encoding Bcl-2, either in WT configuration or fused to peptides that allow the _targeting to mitochondria (Acta) or to ER (Cb5) were described previously.28 Rat-1 fibroblasts as well as HeLa cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic (HEPES) buffer, 100 units/ml penicillin G sodium and 100 μg/ml streptomycin sulfate at 37°C under 5% CO2. All media and supplements for cell culture were purchased from Gibco-Invitrogen (Carlsbad, CA, USA). For serum and amino-acid starvation, cells were cultured in serum-free Earle's Balanced Salt Solution medium (Sigma).6 Cells (30–50 × 103)were seeded in 12- or 24-well plates and grown for 24 h before treatment with antimycin A (from 0.06 to 0.5 μ M; Sigma-Aldrich, St Louis, MO, USA), betulinic acid (from 1.25 to 10 μ M; Sigma-Aldrich), brefeldin A (20 μ M; Sigma-Aldrich), cadmium (from 2.5 to 20 μ M; Sigma-Aldrich), L-690330 (100 μ M; Tocris), lithium chloride (10 mM; Sigma-Aldrich), myo-inositol (Bt3(1,3,5)P3/AM; 10 μ M; Calbiochem), thapsigargin (3 μ M; Calbiochem), tunicamycin (2.5 μ M; Sigma-Aldrich), xestospongin B (from 0.08 to 10 μ M; extracted from the marine sponge Xestospongia exigua),26 or X. C (from 0.08 to 10 μ M; Calbiochem) for 6–24 h.
Plasmids, transfection and RNA interference
Cells were cultured in six-well plates and transfected at 80% confluence with Oligofectamine reagent (Invitrogen), in the presence of 100 nM of siRNAs specific for human Beclin-1 and other atg genes,6, 7 IP3-RI and IP3-RIII (Oakes SA et al., PNAS 2005), hVps34,29, 30 a siRNA _targeting the unrelated protein emerin.31 siRNA effects were controlled by immunoblots with suitable antibodies specific for Beclin-1 (Santa Cruz) or Vps34 (Zymed). Transient transfections with plasmids were performed with Lipofectamine 2000 reagent (Invitrogen) and cells were used 24 h after transfection unless specified differently. Cells were transfected with empty vector alone or together with LC3-GFP,7 in the presence or absence of WT Beclin-1, WT Bcl-2, ER-_targeted Bcl-2 (Bcl-2 Cb5), mitochondrion-_targeted Bcl-2 (Bcl-2 Acta),28 WT Bcl-XL, ER-_targeted Bcl-XL (Bcl-XL Cb5) or mitochondrion-_targeted Bcl-XL (Bcl-XL acta). The cDNA encoding amino acids 1–210 of Bcl-XL were fused either to the sequence encoding the C-terminus of Acta or the Cb5 hydrophobic tail for mitochondrial or ER _targeting, respectively. Correct subcellular localization of the mutants expressed in cells was verified by immunofluorescence microscopy as described previously,28 using rabbit anti-Bcl-XL anti-sera followed by either a monoclonal antibody to the ER protein calreticulin or to an inner mitochondrial membrane protein (2G2, ExAlpha Biologicals).
Flow cytometry
The following fluorochromes were employed by cytofluorometry: 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3), 40 nM) for quantification or the ΔΨm (Molecular Probes) and PI (1 μg/ml, Sigma-Aldrich) for determination of cell viability.6 Cells were trypsinized and labeled with the fluorochromes at 37°C, followed by cytofluorometric analysis with a fluorescence-activated cell sorter scan (Becton Dickinson, San Jose, CA, USA). Data were statistically evaluated using Cell Quest software (Becton Dickinson).
Light microscopy and immunofluorescence
Cells were fixed with paraformaldehyde (4%, w/v) for LC3-GFP and immunofluorescence assays.6 Cells were stained for the detection of HSP60 (mAb from Sigma-Aldrich) or calreticulin32 (pAb from Stressgen) revealed by goat anti-mouse or anti-rabbit immunoglobulin Alexa fluor conjugates (Molecular Probes). Nuclei were labeled with 10 μg/ml Hoechst 33342 (Molecular Probes-Invitrogen). Fluorescence microscopy was analyzed with a Leica IRE2 equipped with a DC300F camera. Confocal microscopy was performed with a LSM 510 Zeiss microscope equipped with a × 63 objective. To determine the percentage of colocalization, images were loaded into Image J software.
Electron microscopy
Cells were fixed for 1 h at 4°C in 1.6% glutaraldehyde in 0.1 M Sörensen phosphate buffer (pH 7.3), washed, fixed again in aqueous 2% osmium tetroxide and finally embedded in Epon. Electron microscopy was performed with a Zeiss EM 902 transmission electron microscope, at 90 kV, on ultrathin sections (80 nm), stained with lead citrate and uranyl acetate.
Dynamic in vivo measurements with _targeted aequorin probes and fura-2
cytAEQ, erAEQmut-expressing cells, were reconstituted with coelenterazine, transferred to the perfusion chamber, light signal was collected in a purpose-built luminometer and calibrated into [Ca2+] values as described previously.33 All aequorin measurements were carried out in KRB containing 1 mM CaCl2 (KRB/Ca2+, Krebs-Ringer modified buffer: 135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.4 mM K2HPO4, 1 mM CaCl2, 5.5 mM glucose, 20 mM HEPES, pH 7.4). For [Ca2+]er measurements, erAEQmut-transfected cells were reconstituted with coelenterazine n, following ER Ca2+ depletion in a solution containing 0 [Ca2+], 600 μ M ethylene glycol bis(β-aminoethylether)-N,N,N’,N’,-tetraacetic acid (EGTA), 1 μ M ionomycin, as described.34 Kinetic imaging of [Ca2+]c transients in fura-2-loaded cells was performed as described.34
Immunoblots and immunoprecipitation
HeLa cells (1 × 106 cells) were washed with cold phosphate-buffered saline (PBS) at 4°C and lysed as described previously.7 Forty micrograms of protein were loaded on a 5–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membrane (Millipore). The membrane was incubated for 1 h in PBS–Tween 20 (0.05%) containing 5% bovine serum albumin. Primary antibody Anti-Beclin-1 (Santacruz), anti-GADD34 (Abcam), anti-IP3-RI (Calbiochem), anti-IP3-RIII (BD Biosciences) and anti-hVps34 (Zymed) were incubated overnight at 4°C and revealed with appropriate horseradish peroxidase-labeled secondary antibodies (SouthernBiotech, Birmingham, AL, USA) and the SuperSignal West Pico chemoluminiscent substrate (Pierce). Anti-glyceraldehyde-3-phosphate dehydrogenase (Chemicon) was used to control equal loading. Rat-1 fibroblasts (8 × 106 cells) were collected, lysed, and immunoprecipitation was performed as described16 using anti-Bcl-2 (SantaCruz) antibody and Protein G Sepharose (GE Healthcare) followed by western blotting with anti-IP3-RIII antibody.
Abbreviations
- Atg:
-
autophagy-related gene
- AV:
-
autophagic vacuole
- ER:
-
endoplasmic reticulum
- IP3:
-
1,4,5-inositol trisphosphate
- IP3R:
-
IP3 receptor
- ΔΨm:
-
mitochondrial transmembrane potential
- PI:
-
propidium iodine
References
Shintani T, Klionsky DJ . Autophagy in health and disease: a double-edged sword. Science 2004; 306: 990–995.
Yorimitsu T, Klionsky DJ . Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12 (Suppl 2): 1542–1552.
Levine B, Yuan J . Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115: 2679–2688.
Lum JJ, DeBerardinis RJ, Thompson CB . Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005; 6: 439–448.
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y . In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101–1111.
Boya P, Gonzalez-Polo R-A, Casares N, Perfettini J-L, Dessen P, Larochette N et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–1040.
Gonzalez-Polo R-A, Boya P, Paulau A-L, Jalil A-A, Larochette N, Souquere S et al. The apoptosis/autophagy paradox. Accumulation of autophagic vacuoles triggers apoptosis. J Cell Sci 2005; 118: 3091–3102.
Lum JJ, Bauer DE, Kong M, Harris HM, Li C, Lindsten T et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120: 237–248.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672–676.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927–939.
Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A et al. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol 2005; 15: 1513–1517.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170: 1101–1111.
Patterson RL, Boehning D, Snyder SH . Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem 2004; 73: 437–465.
Bianchi K, Rimessi A, Prandini A, Szabadkai G, Rizzuto R . Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta 2004; 1742: 119–131.
Verrier F, Deniaud A, Lebras M, Metivier D, Kroemer G, Mignotte B et al. Dynamic evolution of the adenine nucleotide translocase interactome during chemotherapy-induced apoptosis. Oncogene 2004; 23: 8049–8064.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 2004; 166: 193–203.
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA 2005; 102: 105–110.
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB et al. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 2005; 7: 1021–1028.
Boehning D, van Rossum DB, Patterson RL, Snyder SH . A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc Natl Acad Sci USA 2005; 102: 1466–1471.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003; 11: 619–633.
Schroder M, Kaufman RJ . The mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–789.
Green DR, Kroemer G . The pathophysiology of mitochondrial cell death. Science 2004; 305: 626–629.
Ferri KF, Kroemer GK . Organelle-specific initiation of cell death pathways. Nat Cell Biol 2001; 3: E255–E263.
Fulda S, Scaffidi C, Susin SA, Krammer PH, Kroemer G, Peter ME et al. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J Biol Chem 1998; 273: 33942–33948.
Zoratti M, Szabò I . The mitochondrial permeability transition. Biochem Biophys Acta – Rev Biomembr 1995; 1241: 139–176.
Jaimovich E, Mattei C, Liberona JL, Cardenas C, Estrada M, Barbier J et al. Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108-15) cells. FEBS Lett 2005; 579: 2051–2057.
Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF et al. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 1997; 19: 723–733.
Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW . Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 1996; 15: 4130–4141.
Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA 2005; 102: 14238–14243.
Byfield MP, Murray JT, Backer JM . hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem 2005; 280: 33076–33082.
Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K . Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 2001; 114: 4557–4565.
Obeid M, Tesniere A, Ghiringhelli F, Firmia GM, Apetoh L, Perfettini JL et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61.
Chiesa A, Rapizzi E, Tosello V, Pinton P, de Virgilio M, Fogarty KE, et al. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem J 2001; 355: 1–12.
Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R . Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 2004; 8: 59–68.
Acknowledgements
We thank Dr Abdelali Jalil for confocal microscopy and the members of our laboratory for continuous discussion. We also thank the International Collaboration Program ECOS-CONICYT, grant C04B03 (to GK and SL). A Criollo is a recipient of a PhD fellowship from CONICYT, Chile. GK is supported by a special grant of the Ligue Nationale contre le Cancer (équipe labelisée), European Commission (RIGHT), Cancéropôle Ile-de-France and Institut National contre le Cancer (INCa).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Criollo, A., Maiuri, M., Tasdemir, E. et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 14, 1029–1039 (2007). https://doi.org/10.1038/sj.cdd.4402099
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402099
