

Abstract
A major unanswered question in biology and medicine is the mechanism by which the product of the apolipoprotein E ε4 allele, the lipid-binding protein apolipoprotein E4 (ApoE4), plays a pivotal role in processes as disparate as Alzheimer's disease (AD; in which it is the single most important genetic risk factor), atherosclerotic cardiovascular disease, Lewy body dementia, hominid evolution, and inflammation. Using a combination of neural cell lines, skin fibroblasts from AD patients, and ApoE _targeted replacement mouse brains, we show in the present report that ApoE4 undergoes nuclear translocation, binds double-stranded DNA with high affinity (low nanomolar), and functions as a transcription factor. Using chromatin immunoprecipitation and high-throughput DNA sequencing, our results indicate that the ApoE4 DNA binding sites include ∼1700 gene promoter regions. The genes associated with these promoters provide new insight into the mechanism by which AD risk is conferred by ApoE4, because they include genes associated with trophic support, programmed cell death, microtubule disassembly, synaptic function, aging, and insulin resistance, all processes that have been implicated in AD pathogenesis.
SIGNIFICANCE STATEMENT This study shows for the first time that apolipoprotein E4 binds DNA with high affinity and that its binding sites include 1700 promoter regions that include genes associated with neurotrophins, programmed cell death, synaptic function, sirtuins and aging, and insulin resistance, all processes that have been implicated in Alzheimer's disease pathogenesis.
- activity-dependent neuroprotective protein
- Alzheimer's disease
- amyloid precursor protein
- apolipoprotein E
- MAP kinase-activating death domain
- sirtuin
Introduction
The apolipoprotein E ε4 allele (ApoE4; chromosomal locus 19q13) is the single most important genetic risk factor associated with Alzheimer's disease (AD) and also plays important roles in longevity, atherosclerotic cardiovascular disease, evolution, inflammation, and development (Roses, 1996; Bretsky et al., 1999; Ashford and Mortimer, 2002; Mahley et al., 2006; Finch and Morgan, 2007; Finch, 2010). The mechanisms by which these remarkably widespread effects are achieved are not well understood. Although it has been demonstrated that ApoE4 leads to reduced clearance of amyloid-β peptides in the brain (Refolo and Fillit, 2004; Mahley et al., 2006), this effect does not explain the far-reaching observed phenotypic effects of ApoE4. We reported previously that ApoE4 induces isoform-specific signaling events that affect amyloid precursor protein (APP) proteolysis and signaling, caspase activation, downstream kinase activity, and Sirtuin 1 (SirT1) expression. More specifically, our data indicated that ApoE4 interacts with APP with nanomolar affinity, decreases soluble APPα (sAPPα) secretion, and reduces sAPPα/amyloid-β (Aβ) and sAPPα/sAPPβ ratios (Theendakara et al., 2013). Furthermore, there was a significant reduction in ApoE4-mediated SirT1 mRNA and protein levels. Thus, our work along with other reports links ApoE4 to reduced sAPPα and a general shift of APP proteolysis away from the trophic peptides sAPPα and α C-terminal fragment and toward the production of Aβ (Bredesen, 2009; Bredesen and John, 2013; Theendakara et al., 2013) via an effect on SirT1. Because the SirT1 levels (mRNA and protein) were significantly reduced by ApoE4, it was of interest to determine whether ApoE4 interacts with SirT1 directly or indirectly.
Our present data reveal differential effects of ApoE4 versus ApoE3 on SirT1 expression and interaction. Although neither ApoE3 nor ApoE4 directly associated with SirT1 protein, chromatin immunoprecipitation (ChIP) with an ApoE antibody, followed by PCR using SirT1 promoter-specific primers, revealed a ∼250 bp band corresponding to the SirT1 promoter. Furthermore, both ApoE isoforms bound to the SirT1 promoter and repressed the estradiol-induced SirT1–EGFP fluorescence and SirT–luciferase activity.
Therefore, we performed ChIP followed by high-throughput DNA sequencing (ChIP-seq) to identify other ApoE3 or ApoE4 _target genes. Functional annotation of genes associated with ApoE4 binding revealed a network of genes involved in axonal guidance, neural cell death, trophic support, glucose and insulin regulation, inflammation, and neural transmission, among others.
To confirm the role of ApoE4 in the expression of a subset of the _targets, we performed quantitative real-time PCR and identified three genes—MAP kinase-activating death domain (MADD), activity-dependent neuroprotective protein (ADNP), and copper metabolism gene MURR1 (mouse U2af1-rs1region 1) domain 6 (COMMD6)—that may be critical for AD pathogenesis. Because these genes are known to regulate gene transcription by binding to the nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) complex and given the mutual antagonism between SirT1 and the NFκB complex (Hwang et al., 2013; Kauppinen et al., 2013), we investigated the relationship between ApoE and the RelA (v-rel avian reticuloendotheliosis viral oncogene homolog A/nuclear factor NF-κB p65 subunit) subunit of the NFκB complex. Indeed, RelA was found to immunoprecipitate with ApoE3, as well as ApoE4; however, only ApoE4 led to the translocation of RelA into the nucleus, suggesting the possibility that ApoE affects NFκB-mediated gene transcription, as well. Furthermore, ApoE4, but not ApoE3, triggered a significant elevation of two cytokines, interleukin-6 (IL6) and IL8, both of which have been implicated in AD.
Although the relative contribution of the transcriptional effects of ApoE4 compared with its nontranscriptional effects (e.g., Aβ clearance) in AD pathology is not yet completely defined, our present findings demonstrate and dissect direct transcriptional effects of ApoE4, thus offering new insight into the mechanism by which ApoE4 achieves its diverse and robust effects on lifespan, cardiovascular disease, inflammation, and neurodegeneration, and providing a medium-throughput model for therapeutic candidate screening in AD drug discovery.
Materials and Methods
Cell culture conditions and transfection.
All experiments were performed in SHSY5Y human neuroblastoma cells or A172 human glioblastoma cells. Cells were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. Human ApoE3 and ApoE4 cDNA constructs were generously provided by Dr. Yadong Huang (Gladstone Institute of Cardiovascular Disease, University of California, San Francisco, CA). N-terminal FLAG–SirT1 cDNA construct in pECE expression vector was obtained from Addgene. The human SirT1 promoter–EGFP and SirT1 promoter–luciferase constructs were provided generously by Dr. M. Thangaraju (Department of Biochemistry and Molecular Biology, Medical College of Georgia, Georgia Health Sciences University, Augusta, GA). The reporter constructs were generated by cloning the 2.2 kb SirT1 promoter (obtained by PCR using human genomic DNA as the template) into pGEM-T Easy vector, followed by subcloning into pU3R2–EGFP and pGL3 vectors. The RelA GSTp65(1-180) and GSTp65(365-551) constructs were generously provided by Dr. Warner Greene (Gladstone Institute of Virology and Immunology, University of California, San Francisco, CA). All DNA constructs were confirmed by sequence analysis. Transient transfection of cells was performed using a combination of Lipofectamine 2000 and siPORT Amine (Life Technologies). The transfection efficiency, using a combination of these two reagents (1:1), was usually 60–70% and was performed as described previously (Rao et al., 2004).
Antibodies.
The following antibodies were used in the present study: anti-ApoE mouse mAb (E6D7; EMD Millipore/Calbiochem), anti-ApoE rabbit mAb (NB110-55466; Novus Biologicals), human-IL8-IP-WB Antibody Pair (H00003576-PW5; Abnova), anti-IL6 mouse mAb (clone 3G9; Origene), anti-IL6 rabbit mAb (D3K2N; Cell Signaling Technology), anti-NFκB p65 rabbit mAb (C22B4; Cell Signaling Technology), anti-NFκB p65 mouse mAb (L8F6; Cell Signaling Technology), anti-SirT1 rabbit polyclonal antibody (HPA006295; Sigma), anti-SirT1 rabbit polyclonal antibody (NBP1-87038; Novus Biologicals), anti-SirT1 rabbit mAb (D1D7-9475; Cell Signaling Technology), anti-COMMD6 rabbit polyclonal antibody (SC-84239; Santa Cruz Biotechnology), anti-MADD rabbit polyclonal antibody (M-5683; Sigma), and anti-ADNP rabbit polyclonal antibody (HPA-006371; Sigma). All HRP-conjugated secondary antibodies were purchased from GE Healthcare or Santa Cruz Biotechnology.
Animals.
Human ApoE3 and ApoE4 transgenic mice (driven by the human APOE promoter) were made in the ApoE-ko background (Xu et al., 1996; Sullivan et al., 1997; Holtzman et al., 1999; Teter et al., 2002; Arold et al., 2012). These mice show equivalent levels of ApoE expression in astrocytes, microglia, and neurons and maintain the human pattern of regulation and regional expression (Xu et al., 1996). Brain sections from the hApoE _targeted replacement (TR) mice were kindly provided by Dr. Gregory Cole (Easton Center for Alzheimer's Disease Research, University of California, Los Angeles, CA) for histochemistry.
Skin fibroblasts.
Skin fibroblasts from AD patients maintained at the National Institute on Aging Cell Culture Repository were obtained from the Coriell Institute. Fibroblasts had a normal karyotype and were genotyped for the ApoE status. Patient 04402, male, 47 years old at the time of sampling, with an ApoE3/E4 profile and clinically affected member of a family with AD, presented with signs of early dementia, inappropriate jocularity, and occasional paraphasias. Patient 21158, female, 69 years old at the time of sampling, ApoE2/E3, was clinically affected with AD.
Cell extracts, subcellular fractionation, immunoprecipitation, and Western blotting.
Whole-cell extracts were prepared as described previously (Rao et al., 2004). Subcellular fractionation was performed as described previously (Rao et al., 2004; Poksay et al., 2011) with some modifications. Cells were resuspended in ice-cold fractionation buffer (in mm: 20 HEPES, pH 7.4, 10 KCl, 250 sucrose, 1.5 MgCl2, 1 EDTA, 1 EGTA, 1 dithiothreitol, and protease inhibitor mixture), and, after cell lysis, nuclei were pelleted by a 10 min, 750 × g spin. The purity of the nuclear fraction was assessed by the presence of poly(ADP-ribose) polymerase (PARP) protein. The supernatant was then centrifuged at 100,000 × g for 30 min. The resulting supernatant contained the soluble cytosolic fraction. Equal amounts of protein (∼100 μg) were loaded for SDS-PAGE and Western blot analysis, as described previously (Rao et al., 2004). Culture media or cell extracts (500 μg of protein) were subjected to immunoprecipitation (IP) as described previously (Theendakara et al., 2013). The immunoprecipitated proteins were subjected to SDS-PAGE and Western blot analysis (Theendakara et al., 2013). Enhanced chemiluminescent detection of the proteins was performed with Pierce ECL detection reagents (Thermo Fisher Scientific).
Quantitative real-time PCR.
To analyze SirT1 mRNA levels, real-time PCR was performed on first-strand cDNAs. Primers for SirT1 were designed using Roche universal probe library system, and the primers were synthesized by Integrated DNA Technology. A172 cells were transfected with ApoE3 or ApoE4 cDNA, and, after 24 h, the cells were trypsinized and collected. Total RNA was isolated using High Pure RNA isolation kit (Roche), and 1 μg of RNA from untransfected and transfected samples were reverse transcribed. The real-time PCR cycling was performed in SYBR Green master mix (Roche) on the Light Cycler 480 (Roche) using 384-well plates. The melting curves of PCR products were monitored to ensure that a single melting curve was obtained. For analysis of the real-time PCR data, signals from each sample were normalized to values obtained for the housekeeping gene GAPDH, which was assayed simultaneously with experimental samples. Relative quantification using the ΔΔCt method was adopted to calculate relative quantity of SirT1 levels (Theendakara et al., 2013). SirT1 mRNA data (δCt values) are expressed as a percentage of untreated control.
SirT1 activity assay.
SirT1 enzyme activity was measured using the Sensolyte green SirT1 assay kit (AnaSpec). In the first step, an acetylated substrate is incubated with the sirtuin-containing samples (cell extracts isolated from untransfected cells or cells transfected with the ApoE constructs). Deacetylation of the substrate sensitizes it to the sirtuin developer, which, in the second step, releases the green fluorophore. Fluorescence produced is proportional to SirT1 activity and can be detected with excitation at 490 nm and emission at 520 nm. SirT1 enzyme activity measured as relative fluorescence units (RFUs) is expressed as percentage of untreated control.
Fluorescence microscopy.
A172 or SHSY5Y cells were transfected with SirT1–pU3R2EGFP reporter construct, followed by transfection of the ApoE isoforms and then cultured in regular medium for 24 h. At 24 h after transfection, cells were treated with or without 17β-estradiol (E2) for 24 h. The expression of GFP was monitored by epifluorescence microscopy. Untreated and E2-treated cells were counted manually by accounting for all the EGFP fluorescent cells. A minimum of 100 cells from several independent experimental fields was evaluated for quantitation of fluorescence. Images were captured on a Nikon Eclipse-800 fluorescence microscope. The effect of ApoE on E2-induced fluorescence was expressed as percentage of untreated, SirT1–pU3R2EGFP-transfected, ApoE-nontransfected control.
Promoter activity.
A172 or SHSY5Y cells were cotransfected with ApoE isoforms and SirT1–pGL3 reporter construct and then cultured in regular medium for 24 h. Luciferase activity was measured in cell lysates using the Promega luciferase assay system (Promega) following the instructions of the manufacturer. Luciferase activity was measured in triplicate, normalized to total protein, and expressed as percentage of untreated control.
Cytokine assay.
We used a Multi-Analyte ELISArray kit (Qiagen) that simultaneously detects 12 cytokines. Repeated measurements resulted in a consistent change in the levels of IL6 and IL8, so we chose to focus only on these two cytokines. Cell culture media harvested from control and ApoE-transfected A172 cells was used in a single-analyte ELISArray kit (Qiagen) for measuring IL6 and IL8. The analysis was performed according to the instructions of the manufacturer. The levels of IL6 (nanograms per milliliters) and IL8 (picograms per milliliters) were quantified from a standard curve and expressed as percentage of untransfected control. Equal amounts of protein from corresponding cell lysates (∼100 μg) were loaded for SDS-PAGE and Western blot analysis as described previously (Theendakara et al., 2013). Blots were probed with antibodies specific for IL6 or IL8.
ChIP and ChIP-seq.
A172 cells were transiently transfected with ApoE3 or ApoE4 cDNA constructs in 10 cm plates. After 24 h, the cells were fixed with 1% formaldehyde, lysed, and then sonicated. ChIP was performed using a ChIP assay kit (EMD Millipore). Human ApoE antibody that recognizes both isoforms of ApoE was used for immunoprecipitating the DNA. For ChIP-seq, purified DNA obtained after ChIP was submitted to Arraystar (http://www.arraystar.com/) for library construction, sequencing, and basic data analyses. Illumina genomic adaptors were used to make the ChIP-seq library. The library was then sequenced on an Illumina HiSeq 2000 following the TruSeq Rapid SBS protocol. The stages of image analysis and base calling were performed using Off-Line Basecaller software (OLB version 1.8). Clean reads were aligned to the human genome [University of California Santa Cruz (UCSC) HG19] using BOWTIE software (version 2.1.0). Aligned reads were used for peak calling of the ChIP regions using MACS version 1.4.0. Statistically significant ChIP-enriched regions (peaks) were identified by comparison with a Poisson background model using a p value threshold of 10−5. The peaks in samples were annotated to the nearest gene using the newest UCSC RefSeq database.
Immunohistochemistry.
Immunohistochemical experiments were performed on 7-month-old or 18-month-old ApoE3/3 and ApoE4/4 TR mouse brain. Animals were perfused with HEPES buffer containing complete protease inhibitor (Roche), and hemibrains were submersion fixed in 4% paraformaldehyde for 3 d. The tissues were then cryoprotected in increasing concentrations of sucrose in PBS (10, 20, and 30%) over 3 d at 4οC. They were then snap frozen in liquid nitrogen, cryosectioned at 12 μm, and mounted on slides. For labeling, sections from approximately bregma 0 (the level at which the septum and striatum are visible) or bregma 2.0 (hippocampus and thalamus are visible) were used; they were rehydrated in PBS, and endogenous peroxidase activity was quenched with 3% H2O2 in PBS for 10 min. After washing in PBS, nonspecific antibody binding was blocked with 5% normal goat serum (NGS) in PBS for 1 h, followed by incubation with primary antibodies at a dilution of 1:300 in PBS with 0.1% Tween 20 (PBST) and 5% NGS overnight at 4°C. Sections were washed three times for 5 min each in PBS and then incubated with biotinylated anti-rabbit secondary antibody (Vector Laboratories) at 1:400 in PBST with 5% NGS for 2 h at room temperature. After washing, sections were incubated using the VECTASTAIN ABC kit (Vector Laboratories), washed, and developed for 10 min with the Vector DAB substrate kit. After rinsing in Milli-Q filtered water, sections were briefly counterstained with hematoxylin, processed into xylene, mounted with Permount (Thermo Fisher Scientific), and coverslipped. Labeled sections were imaged using an Olympus BX51 microscope/camera and Olympus Microsuite Five imaging software at 40× magnification.
Surface plasmon resonance.
Nine DNA segments ranging from eight to 30 bases of the ∼250 bp SirT1 promoter and their complements were obtained from Eurofins MWG Operon. Double-stranded sequences were prepared by mixing each single-strand segment and its complement in a 1:1 ratio and incubating for 60 min at 20°C. The surfaces of all four flow cells (FC1, FC2, FC3, FC4) of a carboxymethylated-dextran (CM-5) chip were washed sequentially with 50 mm NaOH, 1 mm HCl, 0.05% H3PO4, and 10 mm sodium phosphate, pH 6.0, in parallel using a flow rate of 30 μl/min for 1 min using a Biacore T-100 (GE Healthcare). Avidin (Thermo Fisher Scientific) was diluted to 1 mg/ml in PBS (20 mm phosphate and 137 mm sodium chloride, pH 7.4) and then to 40 μg/ml in 10 mm sodium acetate, pH 5.5. The avidin was immobilized via amine coupling using 10 mm phosphate, pH 6.0. The final response unit (RU) values were 2475 ± 30. Thioredoxin (trx)-fusion proteins containing ApoE3 (trx–ApoE3) and ApoE4 (trx–ApoE4) were expressed and purified as described previously (Theendakara et al., 2013). The ApoE fusion proteins and trx (Sigma) were biotinylated using the EZ-Link Sulfo-NHS-SS-Biotin biotinylation kit following the instructions to produce one to two biotins per molecule. After biotinylation, the biotinylated proteins were dialyzed extensively [3 h at 1:1000 dilution 20 mm phosphate, pH 7.4, 137 mm sodium chloride, 0.005% Tween 20 (PBST) for three changes]. The chip had only avidin in flow cell one, biotinylated trx in flow cell two with a final RU of 102, biotinylated trx–ApoE3 in flow cell three with a final RU of 100, and biotinylated trx–ApoE4 in flow cell four with a final RU of 100. After loading with biotinylated proteins, the chip was washed with alternating washes of 100 μl of 20 mm Tris, pH 8.0, 2 m NaCl, and 100 μl of 20 mm sodium acetate, pH 4.5, and 100 μl of 3× PBST.
The oligonucleotides (100 μm) were reconstituted in 10 mm Tris, pH 7.0, and 1 mm EDTA. The flow rate for the experiments was 60 μl/min. Buffer controls were made to measure the bulk effect caused by the buffer mismatch with flow buffer. The buffer controls indicated that the linear drift in the sensograms during the flow stage was attributable to bulk effects. The sensograms were derived using double subtraction between the ApoE-containing flow cell and the trx-containing control flow cell and between analyte and buffer control. The resultant curves were fitted to a 1:1 binding model using Prism (GraphPad Software). The bulk effects were modeled as a linear contribution with time during the flow stage and then an exponential decay just after the cessation of flow.
Statistical analysis.
All statistics used either Student's unpaired t test or one-way ANOVA, followed by Newman–Keuls multiple-comparison test using the scientific statistic software Prism (version 4.0c; GraphPad Software). Results were considered statistically significant at p < 0.05. Data were expressed as mean ± SEM.
Results
ApoE4-mediated reduction in ADAM10 expression and pharmacological reversal of SirT1 expression and activity
Recent studies point to SirT1 as an inducer of transcription of ADAM10. Mechanistically, SirT1 increases sAPPα by increasing α-secretase activity through activation of the α-secretase gene ADAM10 (Qin et al., 2006; Donmez, 2013). Our previous report demonstrated that ApoE4, but not ApoE3, significantly reduced sAPPα secretion, SirT1 mRNA, and protein (Theendakara et al., 2013). To investigate what role ApoE has on ADAM10 expression, we performed quantitative real-time PCR in A172 human glioblastoma cells transfected with ApoE cDNA constructs. Our results indicated a significant downregulation of ADAM10 mRNA by ApoE4 (Fig. 1A) compared with ApoE3 (data not shown). The ApoE4-mediated reduction in ADAM10 may have contributed to the observed reduction in the levels of neuroprotective sAPPα (Theendakara et al., 2013).
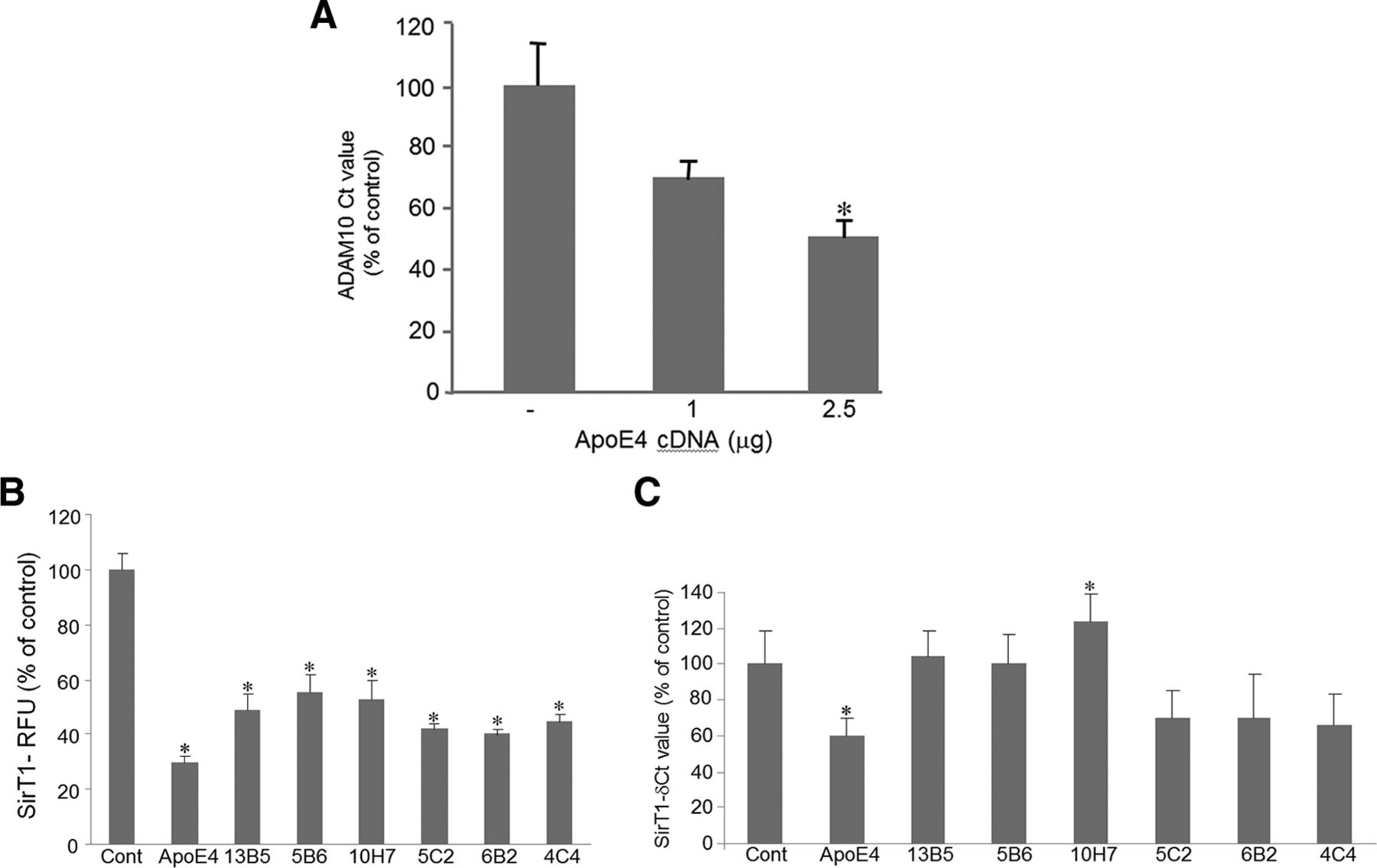
ApoE4-mediated reduction in ADAM10 expression and pharmacological reversal of SirT1 expression and activity. A, Human glioblastoma A172 cells were transfected with ApoE4 cDNA construct, and, 24 h later, cell pellets were collected and subjected to RNA isolation and PCR. The real-time PCR was performed as described in Materials and Methods. ADAM10 mRNA data (δCt values expressed as percentage of untransfected control) are from three experiments performed in triplicate, *p < 0.05. B, C, Pharmacological reversal of ApoE4-mediated reduction in SirT1 levels by a subset of compounds from a CNS-focused library. A172 cells were transfected with ApoE4 cDNA construct and 24 h later were treated with various compounds at a final concentration of 2 μm. After an additional 24 h, cell lysates were prepared and assayed for SirT1 mRNA (B) and SirT1 enzyme activity (C) as described in Materials and Methods. Data are from three independent experiments performed in triplicate, *p < 0.05 compared with ApoE4-transfected and untreated cells. Cont, Control.
To determine whether the ApoE4-mediated reduction in SirT1 mRNA or SirT1 enzyme activity could be reversed by therapeutic candidates, A172 human glioblastoma cells were transfected with ApoE4 cDNA construct and, 24 h later, treated with various drugs (2 μm) from a CNS-focused library (∼10,000 compounds). ApoE4-mediated reduction in SirT1 enzyme activity (Fig. 1B) and mRNA (Fig. 1C) was reversed (significantly, in some cases) by a subset of the drugs, including 13B5 (riboflavin), 5B6 (clonidine), and 10H7 (alaproclate; Fig. 1B,C).
ApoE binds to the SirT1 promoter
The abovementioned results prompted us to examine whether there was any direct association of the ApoE isoforms with SirT1. A172 cells were transiently transfected with the FLAG–SirT1 construct together with ApoE isoforms. IP of FLAG–SirT1 from the lysates did not reveal any association of SirT1 with ApoE3 or ApoE4 (Fig. 2A). Because we were unable to identify any protein–protein interaction between ApoE and SirT1 and to further explore the ApoE-mediated reduction in SirT1 expression, we evaluated the possibility that ApoE binds to the SirT1 promoter and regulates its activity. The 2.2 kb human SirT1–pEGFP and SirT1–luciferase reporter vectors together with ApoE constructs were used for transfecting cells. Because E2 (17-β-estradiol) binds to its specific receptor, estrogen receptor α, and induces the expression of SirT1, we used E2 (10 nm) as a positive regulator of SirT1 expression. SirT1 expression was very low in untreated A172 cells (Fig. 2B, top left); however, the addition of 10 nm E2 significantly increased the SirT1 promoter-specific EGFP fluorescence (Fig. 2B, top right, C). Although E2-induced SirT1–EGFP fluorescence was reduced in cells transfected with ApoE3 (Fig. 2B, bottom left, C), it was significantly suppressed only by ApoE4 (Fig. 2B, bottom right, C).

ApoE isoforms do not associate with SirT1 protein but bind to SirT1 promoter. A, After transfection of human glioblastoma A172 cells with ApoE isoforms and FLAG–SirT1 constructs (1:1), cell extracts were subjected to IP with an anti-FLAG antibody, followed by SDS-PAGE and Western blotting (WB) to detect SirT1 and ApoE. The bottom panel represents endogenous GAPDH as a loading control before the pull-down. B, A172 cells were transfected with the SirT1–pU3R2EGFP reporter construct followed by transfection of the ApoE isoforms (1:1). At 24 h after transfection, cells were treated with or without E2 (10 nm) for 24 h as a positive regulator of SirT1 expression. The expression of EGFP was later monitored by epifluorescence. C, Untreated and E2-treated cells were counted manually by accounting for all of the EGFP fluorescent cells. A minimum of 100 cells from several independent experimental fields was evaluated for quantitation of fluorescence. E2-induced fluorescence was expressed as a percentage of untreated, SirT1–pU3R2EGFP-transfected, ApoE-nontransfected control, *p < 0.05. D, Human neuroblastoma SHSY5Y cells were cotransfected with the SirT1–pGL3 reporter construct and ApoE isoforms (1:1). After 24 h, cell lysates were prepared and luciferase activity was measured in triplicate. Data (mean ± SE) are from three independent experiments, *p < 0.05. E, F, ApoE isoforms bind to the SirT1 promoter. E, After transfection of SHSY5Y cells with ApoE4, ChIP was performed as described in Materials and Methods. Genomic DNA present in the immunoprecipitate was examined using the SirT1 promoter-specific primers by PCR. F, ChIP was performed on skin fibroblasts isolated from two AD patients as described in Materials and Methods. Patient 04402 had an ApoE3/E4 profile, and patient 21158 had an ApoE2/E3 profile. Genomic DNA present in the immunoprecipitate was examined using the SirT1 promoter-specific primers by PCR. C, Control.
Similarly, the basal SirT1–luciferase activity was significantly reduced by ∼40% in SHSY5Y human neuroblastoma cells cotransfected with an expression construct for ApoE3 and by ∼60% in cells cotransfected with the ApoE4 construct (Fig. 2D), both of which were statistically significant. However, the reduction in SirT1–luciferase activity by ApoE4 was significantly greater compared with ApoE3 (Fig. 2D, ApoE3 vs ApoE4, *p < 0.05). Thus, both isoforms of ApoE affect SirT1 expression at the transcriptional level, with ApoE4 being more effective than ApoE3.
To investigate the transcriptional effect of ApoE on SirT1 expression, we performed ChIP. SHSY5Y cells transfected with ApoE4 cDNA were lysed, and IP was performed with an antibody that recognizes all three isoforms of ApoE. The genomic DNA present in the immunoprecipitates was analyzed by PCR using SirT1 promoter-specific primers. A ∼250 bp band corresponding to the SirT1 promoter was present only in the cell fraction that received the ApoE antibody (Fig. 2E), suggesting that ApoE4 interacts with the SirT1 promoter and thus may regulate its activity transcriptionally.
Because the abovementioned results reflect interactions between Sirt1 and ApoE4 that came from transfection and an overexpression system, we repeated these observations in human skin fibroblasts derived from AD patients. Fibroblasts from two patients (patient 04402 ApoE3/E4 and patient 21158 ApoE2/E3) were lysed, and IP was performed as mentioned above. The genomic DNA present in the immunoprecipitates was analyzed by PCR using SirT1 promoter-specific primers. A ∼250 bp band corresponding to the SirT1 promoter was detected only in the samples that received the ApoE antibody (Fig. 2F). Our rationale was to determine whether an ApoE–SirT1 interaction occurs in an in vivo setting as well, and, in both cases (ApoE 2/3 and ApoE 3/4), this interaction was observed, suggesting a physiological interaction of ApoE with the SirT1 promoter.
Interaction of ApoE and SirT1 promoter by surface plasmon resonance
We also confirmed the interaction of both ApoE isoforms and the SirT1 promoter by surface plasmon resonance (SPR) by measuring the binding of recombinant ApoE isoforms with single-stranded, single-stranded complement, and double-stranded DNA segments of the larger ∼250 bp SirT1 promoter sequence. Of the nine different segments of the promoter tested (Tables 1, 2), both ApoE isoforms bound avidly only to a 30-base double-stranded segment, cagcctccgcccgccacgtgacccgtagtg, of the SirT1 promoter. Both ApoE3 and ApoE4 bound with an estimated KD of 3 nm (Fig. 3A,B). Addition of 34 nm ApoE3 (Fig. 3C) or ApoE4 (Fig. 3D) significantly decreased the binding to the double-stranded SirT1 promoter sequence, consistent with a high-affinity specific interaction between the double-stranded segment of the SirT1 promoter sequence and the anchored ApoE isoforms (Table 2). Neither ApoE3 nor ApoE4 showed significant binding to the single-stranded DNA sequence of the segment or its complement or a double-stranded scrambled sequence with the same base composition as the segment (Fig. 3E; Tables 1, 2). Eight other DNA sequences were tested from the ∼250 bp SirT1 promoter sequence, and all showed similar sensograms to the scrambled peptide (Table 1), indicating no significant binding.
Summary of SPR experiments to screen for ApoE-binding sites within the SirT1 promoter sequence
Kinetic parameters of ApoE binding to a segment of DNA from the ∼250 bp SirT1 promoter sequence

SPR analysis of the binding of ApoE isoforms to the SirT1 promoter region. A, B, Binding of ApoE3 (A) or ApoE4 (B) to the double-stranded oligonucleotide of the SirT1 promoter. The oligonucleotide cagcctccgcccgccacgtgacccgtagtg and its complement were mixed at a 1:1 ratio and incubated at 20°C for at least 1 h before the experiment. All the sensograms were fitted to a 1:1 binding model using Prism. The effective KD was calculated with a single-site binding model. C, D, Competition between ApoE in solution and ApoE anchored to the flow cell for binding to the 30 bp SirT1 promoter segment by SPR. Competition with ApoE in solution reduced the binding to the chip by >50% for both ApoE3 (C) or ApoE4 (D), confirming the magnitude of the estimated KD. E, Sensograms generated using a scrambled sequence drawn from the sequence of the 30 bp segment of the SirT1 promoter that bound ApoE. Neither ApoE3 (Table 1) nor ApoE4 (E) showed significant binding to a scrambled sequence.
Subcellular localization of ApoE
Our previous report (Theendakara et al., 2013) together with the abovementioned results suggested that ApoE may enter the nucleus and bind to SirT1 DNA. Thus, the results prompted us to examine the subcellular localization of ApoE. Because SirT1 is also known to shuttle to the cytoplasm and affect neuronal differentiation and neurite outgrowth (Herskovits and Guarente, 2014), we also examined the cellular distribution of SirT1. Cell lysates from untransfected and ApoE-transfected cells were fractionated into nuclear and soluble (cytosolic) fractions and analyzed by Western blotting. The quality of the fractionation experiments was controlled by assessing the distribution of PARP, a nuclear protein. Although both ApoE3 and ApoE4 predominantly existed in the cytosolic fraction, a subpopulation of both isoforms was present in the nuclear fraction as well (Fig. 4A,B). SirT1 is predominantly a nuclear protein (Fig. 4), and its expression was significantly reduced in ApoE3- or ApoE4-transfected cells (Fig. 4A,C) as we reported previously (Theendakara et al., 2013). However, the subcellular distribution of SirT1 was found to be ApoE isoform specific: only ApoE4 expression led to the redistribution of a subpopulation of SirT1 into the cytosolic fraction, with some remaining in the nuclear fraction as well. ApoE4 triggered nearly a sixfold increase in the levels of cytosolic SirT1, although this did not reach statistical significance (Fig. 4C). ApoE3 did not trigger any SirT1 redistribution. Thus, the presence of ApoE3 and ApoE4 in the nucleus is compatible with their binding to the SirT1 promoter and regulating SirT1 transcription; in addition, ApoE4 mediates the redistribution of SirT1 to the cytosol.

Subcellular localization of ApoE isoforms and SirT1. A, Subcellular fractions (SCF) were derived as described in Materials and Methods. C, Cytosolic; N, nuclear. The purity of the nuclear fraction (N) was assessed by the presence of PARP protein. Equal amounts of protein (∼100 μg) from A172 cells were loaded for SDS-PAGE and Western blot analyses. Membranes were probed with anti-ApoE or anti-SirT1 antibody. B, C, Band densities of SirT1 or ApoE is expressed as a percentage of control cytosolic fraction. Data (mean ± SE) are from three independent experiments, *p < 0.05, **p < 0.01.
Genome-wide mapping of ApoE binding
Because ApoE3 and ApoE4 affect SirT1 expression at the transcriptional level and to explore further the ApoE–SirT1 promoter interaction, we performed ChIP-seq to assess ApoE3 and ApoE4 binding on a genome-wide scale. The start and end sites of the peak corresponding to the SirT1 gene (Fig. 5A) from the ChIP-seq encompasses an ∼950 bp region that includes the following: (1) the primer pair sequences that was used for the ChIP; (2) the DNA fragment, cagcctccgcccgccacgtgacccgtagtg, that bound to ApoE on SPR (Fig. 3A,B); (3) the sequence, ccgccacgtga, within the ApoE binding region that binds other transcription factors, including upstream transcription factor 1 (USF-1) and retinoic acid receptor α (RARα); (4) at least two sequences, aagttgagaa and tgggtggcgggag, within the ∼250 bp promoter sequence and in the vicinity of the ApoE binding region that overlap with regions on the ApoD promoter (Do Carmo et al., 2002; Levros et al., 2013); and (5) the sequence, ccacaaagagg, that may be a putative serum responsive element as well as other notable features described below.

A, Analysis of the ∼250 bp SirT1 promoter sequence using the web server LASAGNA-Search version 2.0. The start and end site of the peak corresponding to the SirT1 gene from the ChIP-seq encompasses an ∼950 bp region that includes the following: (1) the primer pair sequences that was used for the ChIP (green); (2) the DNA fragment cagcctccgcccgccacgtgacccgtagtg that bound to ApoE on SPR (red); (3) the sequence ccgccacgtga that is predicted to bind to two other transcription factors, namely RARα and USF-1 (bold red and in italics); (4) a putative RelA binding site, acgtggaaattccca, ∼200 bp upstream of the ApoE binding region (blue); (5) two sequences, aagttgagaa and tgggtggcgggag, within the ∼250 bp promoter sequence and in the vicinity of the ApoE binding region and ggcggaccaaaa adjacent to the RelA binding site, all of which match with regions on the ApoD promoter (orange); and (6) ccacaaagagg within the ∼250 bp promoter sequence that may be a putative serum response element (brown). B, ChIP-seq was performed to explore ApoE binding on a genome-wide scale. Statistically significant ChIP-enriched regions (peaks) were identified when compared with a Poisson background model using a p value threshold of 10−5. The peaks located within −2 to +2 kb around the corresponding gene TSS were subgrouped into five classes on the basis of their distances to UCSC RefSeq gene and included promoters, upstream, introns, exons, and intergenic peaks.
Statistically significant ChIP-enriched regions (peaks) were identified by comparison with a Poisson background model, using a p value threshold of 10−5. The peaks in samples were annotated by the nearest gene using the newest UCSC RefSeq database and were found to be located within −2 to +2 kb around the corresponding transcriptional start site (TSS). The peaks were subgrouped into five classes on the basis of their distances to the UCSC RefSeq gene (http://genome.ucsc.edu/cgi-bin/hgGateway) and included promoters, upstream, introns, exons, and intergenic peaks (Fig. 5B). Promoters were defined as 2000 bp upstream and downstream from the TSS. Peaks whose centers were located in these promoter regions were defined as promoter peaks. Of a total of 3080 promoter peaks examined, ∼1700 were found to be associated with ApoE4 but not ApoE3, and National Center for Biotechnology Information (NCBI) gene database and simple PubMed-based literature searches were used to identify genes exclusive to ApoE4 with potential involvement in AD. This approach led to the identification of 76 genes (Table 3) and included among them SirT1, which was independently re-identified in this assay.
NCBI gene database and simple PubMed-based literature searches were used to identify genes exclusive to ApoE4 with potential involvement in AD
Several of the 76 identified genes (Table 3) have been implicated in AD and could be grouped broadly into the following classes: (1) axon guidance; (2) neuronal signaling; (3) diabetes, glucose metabolism, and energy homeostasis; (4) inflammation (specifically NFκB), tumor necrosis factor (TNF), and cytokine signaling; and (5) neurotrophins and neuronal cell death. The findings reinforce the role of ApoE4 in several key areas that have been shown to contribute to AD pathophysiology. Based on the genomic findings, we then used a combination of techniques to confirm the involvement of three potential candidates in AD.
MADD is a component of the TNF receptor 1 (TNFR1) signaling complex, linking TNFR1 activation with downstream MAP kinase activity. MADD expression has been shown to be reduced in the hippocampus in areas of AD pathology compared with normal controls. Similarly, in AD brain homogenates, MADD protein and mRNA expression were significantly reduced in AD compared with controls, with a concomitant increase in TNF receptor-associated death domain, TNFR1, and activated c-Jun N-terminal protein kinase (Del Villar and Miller, 2004).
ADNP plays an essential role in brain development and autophagy and is expressed predominantly in the cerebellum, hippocampus, and cerebral cortex (Fernandez-Montesinos et al., 2010). In mouse models of AD, ADNP is highly expressed during early or mild stages of AD. ADNP may have an important neuroprotective role in response to neuronal damage during the onset and progression of AD. A peptide derived from ADNP is currently in clinical development for the treatment of neurodegenerative disorders (Quintana et al., 2006).
COMMD6 belongs to the COMMD family of proteins that share a common COMM domain at the C terminus, which facilitates protein–protein interactions. COMMD6 associates with the RelA subunit of NFκB complex and inhibits NFκB-mediated transcriptional activation in the nucleus (de Bie et al., 2006).
To investigate what role ApoE has on the expression of these genes, we performed quantitative real-time PCR and also assessed protein levels in lysates from A172 cells transfected with ApoE3 or ApoE4 (Fig. 6). For all three genes, ApoE4 effected a statistically significant reduction at the protein level (Fig. 6A,B). These reductions were also reflected at the mRNA level, although for ADNP the mRNA reduction did not reach statistical significance, and, for all three, there was some (albeit lesser for ADNP and COMMD6) reduction in mRNA induced by ApoE3 as well (Fig. 6C). These results suggest that ApoE4 exerts transcriptionally repressive effects at four promoters (MADD, ADNP, COMMD6, and SirT1) and leave open the possibility that posttranscriptional effects may also contribute to the marked reduction in protein level.

Expression of a subset of proteins whose promoters bind ApoE. A, After transfection of A172 cells with ApoE isoforms, cell pellets were collected and analyzed by SDS-PAGE and Western blot to detect MADD, ADNP, and COMMD6. B, Band densities of the immunoblots are expressed as a percentage of untransfected control. Data (mean ± SE) are from three independent experiments, *p < 0.05. C, After transfection of A172 cells with ApoE isoforms, cell pellets were collected for RNA isolation and PCR. The real-time PCR cycling was performed as described in Materials and Methods. Data (δCt values expressed as a percentage of untransfected control) are from three experiments performed in triplicate, *p < 0.05. D, E, Expression of candidate proteins in ApoE3/3 and ApoE4/4 TR mouse brain by immunohistochemistry. D, Immunohistochemistry was performed on septal sections from 7-month-old ApoE3/3 and ApoE4/4 TR mice. Immunohistochemistry was performed as described in Materials and Methods. E, Immunohistochemistry was performed on mouse brain dentate gyrus from ∼18-month-old ApoE3/3 and ApoE4/4 TR mice as described in Materials and Methods. C, Control.
The expression of these proteins was also studied by immunohistochemistry on 7-month-old ApoE3/3 and ApoE4/4 TR mouse brain sections. Immunohistochemical labeling of MADD and ADNP revealed greater expression of these proteins in the septal region of brains from ApoE3 TR mice compared with ApoE4 TR mice (Fig. 6D). COMMD6 appeared to be comparatively higher in ApoE3 mice, although labeling was not as intense for this protein.
Similarly, immunohistochemical labeling of MADD in the dentate gyrus of 18-month-old ApoE4 TR mice showed clear reduction in expression compared with ApoE3 TR mice. No difference in ADNP expression was observed in ApoE3 and ApoE4 TR mice, although COMMD6 expression did not reach the level of detection (Fig. 6E).
Differential effects of ApoE on RelA binding, distribution, and associated gene expression
Two of the genes described above, namely MADD and COMMD6, are known to regulate transcription by binding to the NFκB complex or the RelA subunit of the complex (Del Villar and Miller, 2004; de Bie et al., 2006). Similarly, the 8 aa neuroprotective peptide NAP, derived from ADNP, downregulates key inflammatory cytokines, including TNF, IL16, and IL12 in macrophages, indicating its role in immune regulation and neuroprotection (Quintana et al., 2006; Fernandez-Montesinos et al., 2010). Additionally, recent studies have described a mutual antagonism between RelA and SirT1 in favor of inflammation through downregulation of SirT1 (Hwang et al., 2013; Kauppinen et al., 2013). Furthermore, the analysis of the ∼250 bp SirT1 promoter sequence using the web server LASAGNA-Search version 2.0 revealed an NFκB–RelA binding site, acgtggaaattccca, ∼200 bp upstream of the ApoE binding region (Fig. 5A). This is in addition to the sequence ccgccacgtga within the ApoE binding region that served as a binding site for other transcription factors, including USF-1 and RARα (Du et al., 1998; Chen et al., 2005a; Isotalo et al., 2012; Kawahara et al., 2014).
The abovementioned analysis coupled with the finding that ApoE4 downregulates SirT1, MADD, ADNP, and COMMD6 levels led us to investigate specifically the role of ApoE on RelA binding, expression, and subcellular localization. Cell lysates from untransfected and ApoE-transfected cells were fractionated into nuclear and soluble (cytosolic) fractions and analyzed by Western blotting. Expression of ApoE4 was associated with a redistribution of the majority of RelA from the cytosol to the nucleus, whereas the expression of ApoE3 had no effect on RelA distribution, with the vast majority remaining cytosolic (Fig. 7A). This marked effect of ApoE4 on the RelA intracellular distribution raised the question of whether these two proteins may interact directly. IP of endogenous RelA from SHSY5Y cell lysates obtained after transient transfection of ApoE isoforms provided evidence of association between ApoE isoforms and RelA (Fig. 7B). Thus, although RelA coimmunoprecipitated with both ApoE3 and ApoE4, only ApoE4 led to the translocation of RelA into the nucleus.

Subcellular localization and ApoE binding properties of RelA. A, Subcellular fractionation (SCF) was performed as described in Materials and Methods, N, Nuclear; C, cytosolic. Equal amounts of protein (∼100 μg) from A172 cells were loaded for SDS-PAGE and Western blot analyses. Membranes were probed with anti-ApoE or anti-RelA antibody. PARP served as a standard marker protein for nuclei (N). B, ApoE and RelA interaction. After transfection of A172 cells with ApoE isoforms, cell extracts were subjected to IP with an anti-RelA antibody, followed by SDS-PAGE and Western blotting (WB) to detect RelA or the ApoE isoforms. The bottom represents endogenous GAPDH as a loading control before the pull-down. C–F, SPR analysis of the binding of ApoE isoforms to GST–RelA. C, Biacore analysis of RelA consensus sequence. Sensograms shows the results of pumping the double-stranded RelA _target DNA sequence through the biotinylated ApoE4 flow cell. No significant binding was observed up to 5 μm to either ApoE3 (data not shown) or ApoE4 for the single-stranded, double-stranded, or complement RelA consensus sequence. D, E, Binding of GST–RelA(365–551) in solution to biotinylated ApoE3 (D) or ApoE4 (E) attached to the SPR chip. For both ApoE3 and ApoE4, the sensograms were generated with 0.28, 0.55, 1.1, and 2.2 μm GST–RelA(365–551). F, Binding of GST–RelA(1–180) in solution to biotinylated ApoE4 attached to the SPR chip. No significant binding was observed for GST–RelA(1–180) up to 1 μm for ApoE4 (F) or ApoE3 (data not shown).
We also evaluated the interaction of ApoE and the putative RelA-binding region, acgtggaaattccca, on the SirT1 promoter by SPR. Neither ApoE isoform displayed binding to the RelA _target DNA sequence (Fig. 7C; Table 1). Because both ApoE3 and ApoE4 coimmunoprecipitated with RelA, we evaluated this interaction further by SPR using protein fragments of RelA. GST–RelA(365-551) fusion protein bound strongly to both ApoE isoforms, with an estimated KD of 30 nm (Fig. 7D,E). No significant binding was observed with ApoE3 or ApoE4 and GST–RelA(1-180) (Fig. 7F) at similar concentrations, suggesting the specificity of the binding of both ApoE isoforms to the C-terminal region of RelA.
The interaction of ApoE isoforms with RelA raised the question of whether the presence of ApoE affects NFκB-dependent gene transcription. To understand this better, we assessed the levels of several cytokines after transfecting A172 cells with ApoE3 or ApoE4. Additionally, cell lysates were also examined for cytokine expression by Western blotting. We focused on IL6 and IL8 because these two cytokines showed consistent results. Transfection of ApoE4 resulted in a significant elevation in IL6 levels compared with ApoE3 (Fig. 8A). Although there was a threefold increase in IL8 levels in ApoE4-transfected cells compared with ApoE3, this did not reach statistical significance. Furthermore, transfection of ApoE4 resulted in a significant elevation in IL6 and IL8 expression by Western blot analysis compared with ApoE3 (Fig. 8B). By analogy to previous studies that have shown the role of NFκB–RelA complex in modulating IL6 and IL8 levels through the transcriptional modulator TIP60 (Matsusaka et al., 1993; Kim et al., 2012), it is possible that the ApoE4–RelA complex transcriptionally activates the expression of inflammatory cytokines, including IL6 and IL8, similarly to RelA/TIP60.

Effect of ApoE isoforms on cytokine release. A, After transfection of A172 cells with ApoE isoforms, media were collected and IL6 and IL8 were measured as described in Materials and Methods. The levels of IL6 (nanograms per milliliters) and IL8 (picograms per milliliters) were quantified from a standard curve and expressed as percentage of untransfected control. Data (mean ± SE) are from three experiments performed in triplicate, *p < 0.05. B, Equal amounts of protein (∼100 μg) from untreated or treated A172 cells were loaded for SDS-PAGE and Western blot analyses. Membranes were probed with anti-IL6 or anti-IL8 antibody. C, Band density of blot is expressed as a percentage of untransfected control. Data (mean ± SE) are from three independent experiments, *p < 0.05. C, Control.
Discussion
The apolipoprotein E ε4 allele is the single most important genetic risk factor associated with AD. Recently, we showed that ApoE4 expression is associated with a marked reduction in SirT1 levels, in both cultured neural cells and the brains of patients with AD (Theendakara et al., 2013). Furthermore, in addition to the overall reduction in SirT1 protein expression, our present results showed a subpopulation of SirT1 in the cytosolic fraction only in ApoE4-transfected cells, suggesting that ApoE4 (but not ApoE3) causes the redistribution of SirT1 into the cytosolic fraction. This effect may be attributable, at least in part, to cellular stress, including physiological and pathological stimuli triggered by ApoE4. Because SirT1 has been implicated in neuroprotection, this effect of ApoE4 may be important from both mechanistic and therapeutic development standpoints. SirT1 has been shown to be a transcriptional activator of the α-secretase ADAM10 gene, resulting in increased levels of the neuroprotective sAPPα peptide (Lee et al., 2014). Although SirT1 is believed to be a nuclear protein, its presence in the cytosolic compartment has also been reported (Herskovits and Guarente, 2014). Although the significance of the cytoplasmic localization of SirT1 is not clear, it is possible that cytosolic SirT1 may modulate cytoplasmic proteins or trophic mediators that are involved in nerve growth and differentiation. A recent study has shown that cytosolic SirT1 interacts with the Ras GTPase superfamily and regulates axonal growth (Law et al., 2009). Interestingly, although both ApoE3 and ApoE4 bound to SirT1 promoter with similar binding affinities, only ApoE4 triggered the redistribution of SirT1 to the cytosol. We do not know that the same KD of ApoE3 and ApoE4 for DNA has any relevance for differential nuclear localization because that is an event that happens after the ApoE enters the nucleus. Similarly, it is also not clear whether DNA binding plays a significant role in stabilizing the nuclear population of ApoE and SirT1. Differential SirT1 nuclear localization could also be influenced by other events in the cytoplasm or the nucleus that control the rate of the entry and exit of ApoE from the nucleus.
Although both ApoE3 and ApoE4 predominantly existed in the cytosolic fraction, a subpopulation of both isoforms was present in the nuclear fraction as well. The mechanism by which ApoE isoforms escape the secretory pathway and translocate to the cytosol or the nucleus is currently unclear. ApoE4 is known to trigger endoplasmic reticulum (ER) stress and cell death (Zhong et al., 2009; Cash et al., 2012; Theendakara et al., 2013), and several reports have suggested that ER stress can result in mislocalization of proteins, resulting in their redistribution to the cytosol and nucleus (Rao and Bredesen, 2004; Drori et al., 2010; Miesbauer et al., 2010). Cancer cells are particularly prone to ER stress and mislocalization of proteins, with ER stress contributing to either survival or apoptosis of cancer cells, depending on the setting (Wang and Kaufman, 2014). Interestingly, several reports have described a positive correlation of cytosolic/nuclear ApoE immunoreactivity and clinical aggressiveness in prostate and ovarian cancers (Venanzoni et al., 2003; Chen et al., 2005b). From the cytosol, both ApoE isoforms could be transported into the nucleus by binding to specific nuclear proteins through their weak polybasic domains that could serve as potential nuclear signaling sequences (NLSs; Kim et al., 2008; Levros et al., 2013). Additionally, ApoE4 might also be ferried into the nucleus through its interaction with RelA as explained in a later section.
Using ChIP assay and SPR, we showed that ApoE not only binds to the SirT1 promoter with high affinity but also represses promoter activity, suggesting a plausible role for ApoE4 in the nucleus as a transcriptional repressor in at least some settings. This conclusion is supported by the finding that three other genes—MADD, ADNP, and COMMD6—whose promoters also interact with ApoE4 based on ChIP-seq data all show reduced transcription in the presence of ApoE4 (and, to a lesser extent, in the presence of ApoE3). However, we have not yet analyzed the interaction of ApoE with all of the gene promoters to fully substantiate the role of ApoE as a transcriptional repressor. Thus, although the direct effects of ApoE4 on transcription of the small number of genes studied to date reveal only repressive effects, we cannot yet exclude the possibility that ApoE may in some cases turn out, with or without additional complex members (e.g., RelA), to function as a transcriptional activator.
In a report on the astrocyte transcriptome, Simpson et al. (2011) identified 237 genes that were differentially regulated in association with ApoE4. Of these 237 genes, ApoE4 triggered directly or indirectly the downregulation of 164 genes, whereas the remainder was upregulated. These 237 genes were categorized into six functional groups and included genes associated with signaling pathways, the cytoskeleton, DNA damage, metabolism, transcription, and the immune response (Simpson et al., 2011). Some of these groups matched with the functional groups that we categorized from our ChIP-seq studies and within these groups, we noticed few similar genes as well.
Although the presence of ApoE in the nucleus has already been demonstrated in several cell types and tissues (Panin et al., 2000; Chen et al., 2005b; Levros et al., 2013), its underlying role in the nucleus remains unclear. A recent study demonstrated the entry of ApoE and ApoD into the nucleus under stress conditions, with ApoE binding constitutively to the ApoD promoter and repressing it in vivo (Levros et al., 2013). ApoE and ApoD promoters share regions, including the serum response elements characterized by two C residues, followed by a run of six residues that can be either A or T and then followed by an additional two G residues (Do Carmo et al., 2002). We noticed within the ∼250 bp SirT1 promoter region the sequence ccacaaagagg that may be a putative serum response element. Furthermore, we also identified three sequences on the SirT1 promoter (aagttgagaa and tgggtggcgggag within the ∼250 bp promoter sequence and in the vicinity of the ApoE binding region and ggcggaccaaaa adjacent to the RelA binding site), all of which overlap with regions on the ApoD promoter. All of these results indicate that the ∼250 bp SirT1 promoter region may be an important binding site for transcription factors, including ApoE.
Our studies also clearly demonstrate the role of ApoE as a DNA binding protein and transcriptional regulator of multiple genes. The estimated binding affinity of ApoE3 and ApoE4 is in the range of known transcription factors and is ∼10 nm (Majka and Speck, 2007; Jantz and Berg, 2010). Both ApoE isoforms do not have high sequence similarity to the classical DNA binding proteins but do contain the helix–loop–helix secondary structure, as well as a high arginine content, found in many DNA-binding proteins. Binding of arginine residues to phosphate is very strong, and DNA-binding proteins use arginine to make critical interactions with the DNA backbone (Rohs et al., 2010). Because of its high arginine content, ApoE was originally named the arginine-rich protein (Danielsson et al., 1978), and these Arg residues might conceivably facilitate ApoE–DNA interaction.
The SPR experiments that we reported were performed with partially lipidated ApoE that exists as a mixture of monomeric-, tetrameric-, and octomeric-sized complexes in rapid equilibrium (Perugini et al., 2000). ApoE3 tends to form more of the larger oligomers than ApoE4, and this effect can be seen by comparing Figure 3, A and B. The difference in the maximal RUs achieved for the same concentration of ApoE3 and ApoE4 indicates that the average ApoE3 oligomer binds more DNA than the average ApoE4 oligomer binding to the chip. Although it is true that ApoE3 and ApoE4 are known to change structures depending on how much lipid is associated with the protein (Hatters et al., 2006; Peters-Libeu et al., 2006), it is likely that several different conformations of both ApoE3 and ApoE4 were present in our system. Because the kinetics of binding of ApoE3 or ApoE4 to DNA was not significantly different, our data are consistent with the notion that the DNA binding site on ApoE is not influenced by either the sequence variation or the oligomerization state but by a different distribution of oligomers.
In our study, a fraction of both ApoE3 and ApoE4 underwent nuclear translocation and binding to the SirT1 promoter. Using ChIP-seq analysis, we identified ∼1700 promoter peaks that were found to be unique to ApoE4, of which 76 have been implicated previously in AD. The 76 genes (Table 3) included SirT1, which was independently re-identified in this assay. These genes could be grouped broadly into the following classes: (1) axon guidance; (2) neuronal signaling; (3) diabetes, glucose metabolism, and energy homeostasis; (4) inflammation; and (5) neurotrophins and neuronal cell death. Some of these functional groups were similar to the gene clusters in the astrocyte transcriptome, and also included a few similar genes (Simpson et al., 2011).
Three of the genes that we identified from the ChIP-seq, namely MADD, ADNP, and COMMD6 (Del Villar and Miller, 2004; Quintana et al., 2006; de Bie et al., 2006; Fernandez-Montesinos et al., 2010), were evaluated further, and all were observed to be repressed. There was a significant reduction in mRNA and protein levels of all three genes only in ApoE4-transfected cells compared with ApoE3-transfected cells or untransfected cells. Additionally, immunohistochemical labeling of MADD, ADNP, and COMMD6 revealed greater expression of these proteins in the septal region of brains from 7-month-old ApoE3 TR mice compared with ApoE4 TR mice. Although MADD and COMMD6 regulate transcription by binding to the NFκB complex or RelA, respectively, ADNP functions as both an anti-apoptotic and anti-inflammatory gene. Thus, the protein products of all three genes affect the inflammatory response, and, in each case, repression by ApoE4 would be expected to trigger inflammation and cell death.
The involvement of these three genes in inflammation and the fact that there is a mutual antagonism between RelA and SirT1 (Kauppinen et al., 2013) led us to investigate the relationship between ApoE and RelA. Although RelA bound to both ApoE3 and ApoE4, interestingly, only ApoE4 binding led to the translocation of RelA into the nucleus; thus, this may also be one of the mechanisms by which ApoE4 enters the nucleus, especially if the ApoE4–RelA interaction led to the exposure of the RelA NLS. It is not yet clear whether ApoE4 affects the NFκB–inhibitor of nuclear factor-κB interaction or uses some other mechanism to trigger the nuclear translocation of RelA. It is also not yet clear whether ApoE4 modulates the transcriptional effects of NFκB directly. In any case, however, ApoE4 induces a proinflammatory state that is mediated at least in part by NFκB, and this includes a significant elevation of proinflammatory cytokines IL6 and IL8, which are also associated with the pathological changes found in AD (Finch and Morgan, 2007; Finch, 2010).
The network of genes whose promoters bind ApoE4 suggests a fundamental cellular reprogramming, including downregulation of MADD, ADNP, and COMMD6 and a consequent elevation of IL6 and IL8 by ApoE4 (among many other effects), all of which could participate in triggering the inflammatory and apoptotic pathways associated with AD pathophysiology. Our results regarding ApoE–SirT1 promoter interaction in human fibroblasts isolated from AD patients and the downregulation of a subset of genes specifically in the ApoE4/4 TR young and old mouse brains together with our previous observation of a significant reduction in SirT1 levels in ApoE4-positive postmortem human AD brains (Theendakara et al., 2013), all suggest a physiological role of ApoE as a transcription factor and most likely as a transcriptional repressor.
Thus, our work provides new insight into the mechanism by which ApoE4 may achieve its diverse and robust effects on lifespan, cardiovascular disease, inflammation, and neurodegeneration. Our work also offers a medium-throughput model for screening drug candidates that will not only influence the onset or progression of AD by _targeting the interaction between ApoE4–SirT1 and ApoE4–RelA but also modulate lifespan by rescuing SirT1 levels potentially affecting the normal aging process and mediating other metabolic events.
Footnotes
↵*D.E.B. and R.V.R. are senior co-authors.
The work was supported in part by National Institutes of Health Grant AG034427 (D.E.B.), the Joseph Drown Foundation (D.E.B.), Buck-Impact Circle Funds (R.V.R.), the Marin Community Foundation (D.E.B. and R.V.R.), the Stephen D. Bechtel Jr. Foundation (D.E.B. and R.V.R.), and the John and Bonnie Strauss Foundation (D.E.B. and R.V.R.). We thank Dr. Yadong Huang (Gladstone Institute of Neurological Disease, University of California, San Francisco, CA) for providing the ApoE plasmids, Dr. M. Thangaraju (Department of Biochemistry and Molecular Biology, Medical College of Georgia, Georgia Health Sciences University, Augusta, GA) for the human SirT1 promoter–EGFP and SirT1 promoter–luciferase constructs, Dr. Warner Greene (Gladstone Institute of Virology and Immunology, University of California, San Francisco, CA) for the RelA GSTp65(1-180) and GSTp65(365-551) constructs, Dr. Gregory Cole (Easton Center for Alzheimer's Disease Research, University of California, Los Angeles, CA) for providing brain sections from hApoE TR mice, Dr. Brian Kennedy (Buck Institute for Research on Aging, Novato, CA) for helpful discussions, and Rowena Abulencia for administrative assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: Dale E. Bredesen, Easton Laboratories for Neurodegenerative Disease Research, University of California Los Angeles, Los Angeles, CA 90025, dbredesen{at}mednet.ucla.edu; or Rammohan V. Rao, Buck Institute for Research on Aging, 8001 Redwood Boulevard, Novato, CA 94945, rrao{at}buckinstitute.org




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}