

Abstract
The human pyruvate dehydrogenase complex (PDC) is regulated by reversible phosphorylation by four isoforms of pyruvate dehydrogenase kinase (PDK). PDKs phosphorylate serine residues in the dehydrogenase (E1p) component of PDC, but their amino-acid sequences are unrelated to eukaryotic Ser/Thr/Tyr protein kinases. PDK3 binds to the inner lipoyl domains (L2) from the 60-meric transacetylase (E2p) core of PDC, with concomitant stimulated kinase activity. Here, we present crystal structures of the PDK3–L2 complex with and without bound ADP or ATP. These structures disclose that the C-terminal tail from one subunit of PDK3 dimer constitutes an integral part of the lipoyl-binding pocket in the N-terminal domain of the opposing subunit. The two swapped C-terminal tails promote conformational changes in active-site clefts of both PDK3 subunits, resulting in largely disordered ATP lids in the ADP-bound form. Our structural and biochemical data suggest that L2 binding stimulates PDK3 activity by disrupting the ATP lid, which otherwise traps ADP, to remove product inhibition exerted by this nucleotide. We hypothesize that this allosteric mechanism accounts, in part, for E2p-augmented PDK3 activity.
Keywords: bound lipoyl domain, lipoyl-lysine binding pocket, phosphorylation, pyruvate dehydrogenase kinase 3, type 2 diabetes
Introduction
The mammalian pyruvate dehydrogenase complex (PDC) catalyzes the oxidative decarboxylation of pyruvate to give rise to acetyl-CoA, linking glycolysis to the Krebs cycle. PDC is a member of the highly conserved mitochondrial α-ketoacid dehydrogenase complexes comprising the PDC, the branched-chain α-ketoacid dehydrogenase complex (BCKDC), and the α-ketoglutarate dehydrogenase complex (Reed et al, 1985; Roche and Patel, 1989; Reed, 2001). PDC is a 9 × 106-Da macromolecular machine organized around a 60-meric transacetylase (E2p) core. To the E2p core, multiple copies of pyruvate dehydrogenase (E1p), dihydrolipoamide dehydrogenase (E3), E3-binding protein (E3BP), pyruvate dehydrogenase kinase (PDK), and pyruvate dehydrogenase phosphatase (PDP) are attached (Reed, 2001). The co-localization of these components greatly enhances the efficiency of multi-step reactions, with the lipoyl domain of E2p participating in most of the reaction steps. Precursors for subunits of these enzyme components are encoded by nuclear genes and imported into the mitochondrial matrix, where they are processed and assembled into PDC (De Marcucci et al, 1988; Lindsay, 1989).
PDC in eukaryotes is regulated by reversible phosphorylation (Reed et al, 1985; Harris et al, 2001; Holness and Sugden, 2003). The phosphorylation of serine residues in E1p by PDK results in the inactivation of PDC, whereas the dephosphorylation by PDP restores its activity. To date, four PDK (1–4) isoforms in mitochondria have been identified (Popov et al, 1997). Phosphorylation of E1p occurs at three serine residues (S264: site 1, S271: site 2, and S203: site 3) (Yeaman et al, 1978; Teague et al, 1979; Sale and Randle, 1981). Although phosphorylation of each site alone inactivates PDC, site 1 is most rapidly phosphorylated and the phosphorylation of site 3 is the slowest among the three sites (Sale and Randle, 1981; Korotchkina and Patel, 1995). Interestingly, each PDK isoform exhibits different site specificity. Using E1p mutants with single-functional phosphorylation sites, it was shown that all the four isoforms phosphorylate sites 1 and 2, but only PDK1 modifies site 3 (Kolobova et al, 2001; Korotchkina and Patel, 2001). PDK isoforms exhibit tissue-specific expression; PDK1 is detected in the heart, pancreatic islets, and skeletal muscles; PDK2 is expressed in all tissues; PDK3 is present in the testes, kidney, and brain; PDK4 is abundant in the heart, skeletal muscle, kidney, and pancreatic islets (Bowker-Kinley et al, 1998). The expression of PDK2 and PDK4 is induced in starvation and diabetes, which is reversed by insulin treatment (Wu et al, 1998; Harris et al, 2001). Impaired insulin-induced downregulation of PDK4 (due to the lack of insulin or insensitivity to insulin) leads to the overexpression of PDK4 and shuts off glucose oxidation in diabetic animals (Holness and Sugden, 2003; Roche et al, 2003). Therefore, PDK4 is a potential drug _target for the treatment of type II diabetes.
PDKs form dimers as the biologically functional unit (Bowker-Kinley et al, 1998; Baker et al, 2000; Korotchkina and Patel, 2001). PDK dimers are recruited to the PDC by preferentially binding to the inner lipoyl (L2) domain of the E2p core (Liu et al, 1995). Binding of L2 to PDKs requires the covalently attached lipoyl group at the lysine 173 of L2 (Radke et al, 1993). Reduction or acetylation of the lipoyl group significantly increases the affinity of L2 for PDKs compared to L2 containing an oxidized lipoyl group (Baker et al, 2000; Roche et al, 2003). The activity of PDKs is stimulated upon binding to E2p, but the response of different isoforms to isolated L2 varies. PDK3 is robustly activated by E2p, and the majority of this activation can be achieved by isolated L2 (Baker et al, 2000; Roche et al, 2003). In contrast, PDK2 activity is augmented by binding to E2p (Baker et al, 2000) or a di-domain (Tuganova and Popov, 2005) consisting of both the L2- and E1p-binding domains, but not binding to L2 alone. Individual isoforms exhibit different binding affinities for L2 with PDK3>PDK1=PDK2>PDK4 (Tuganova et al, 2002). The mammalian PDC contains substoichiometric number of PDK dimers relative to the 20–30 E1p molecules bound to the E2p core (Radke et al, 1993). For efficient phosphorylation of E1p molecules by PDKs, a hand-over-hand model for kinase movements has been proposed (Liu et al, 1995; Roche et al, 2003). According to this model, the PDK dimer moves along the surface of the E2p 60-meric core by repeated dissociation and association, in a relay fashion, with L2 domains from different constitutive E2p monomers. This mechanism presumably enables the limited copies of PDK molecules to phosphorylate the 20–30 copies of E1p molecules bound to the E2p scaffold.
Mitochondrial protein kinases comprising PDKs and the related BCKD kinase (BCK) constitute a novel family of protein kinases, in which motifs that are normally present in eukaryotic Ser/Thr/Tyr kinases (Hanks et al, 1988) were not found. Structural studies of rat PDK2 and rat BCK have revealed that these kinases consist of two distinct domains (Machius et al, 2001; Steussy et al, 2001). The C-terminal domain (or K domain in BCK) is an α/β structure with a five-stranded β-sheet, and this fold is shared between the members of the GHKL ATPase/kinase superfamily (http://scop.mrc-lmb.cam.ac.uk/scop/data/scop.b.e.bff.b.html) that includes DNA Gyrase B (Wigley et al, 1991), Hsp90 (Prodromou et al, 1997), histidine kinases including CheA (Bilwes et al, 1999) and EnvZ (Tanaka et al, 1998), as well as MutL (Ban and Yang, 1998). Members of this superfamily share four conserved motifs (N-, G1-, G2-, and G3-boxes), which build a unique ATP-binding fold (Alex and Simon, 1994; Bergerat et al, 1997; Smirnova et al, 1998; Dutta and Inouye, 2000). This fold includes a common structural element known as the ‘ATP lid', whose conformational change is coupled to both ATP hydrolysis and protein–protein interactions (Wigley et al, 1991; Ban et al, 1999; Machius et al, 2001). The ATP lid is conserved in the PDK2 and BCK structures. In the PDK2–ADP structure, the ATP lid is partially ordered, whereas in BCK the lid is more ordered in the ADP-bound form than in the ATP-bound structure. The N-terminal domain of PDK2 (or B domain in BCK) consists of a four-helix bundle that resembles the histidine phosphoryl-transfer (HPt) domains of the two-component systems. The HPt domains contain histidine residues for forming phospho-histidine intermediates in a phospho-relay system. However, the phosphoryl-transfer mechanism in mitochondrial protein kinases is distinct and does not involve a histidine residue in the four-helix bundle domain (Davie et al, 1995; Tovar-Mendez et al, 2002).
The structural basis for the upregulation of PDKs through binding to E2p is unknown. To address this problem, we produced a stable PDK3–L2 complex, based on the high affinity of PDK3 for the L2 domain, and determined the crystal structures of this complex. Our structural data show a novel mode of L2 binding to PDK3, which induces significant conformational changes in PDK3. We show that these conformational changes markedly reduce the affinity of PDK3 for ADP to mitigate product inhibition by this nucleotide. We suggest that this allosteric mechanism explains, in part, the stimulation of PDK3 activity by E2p binding.
Results and discussion
L2 binding markedly stimulates the kinase activity of PDK3
There are inconsistencies in the literature regarding the magnitude of L2 domain-stimulated PDK3 activity. The study by the Roche group showed that the L2 domain and E2p stimulate human PDK3 activity by 12.8- and 17-fold, respectively (Baker et al, 2000). These authors concluded that L2 binding is the major determinant for stimulated PDK3 activity and that the small additional activation by E2p over L2 is imparted by the co-localization of the kinase and substrate E1p to E2p. In contrast, the Popov group showed that PDK3 activity is activated by 3- and 38-fold, respectively, by the L2 domain and E2p (Tuganova et al, 2002).
To resolve this discrepancy, the kinase activity of human PDK3 was measured by E1p phosphorylation in the absence and presence of a lipoylated L2 construct (residues 126–233, designated as L2), with an intrinsic C-terminal linker (residues 209–233) identical to that used by the Roche group. A maltose-binding protein (MBP) was N-terminally linked to the kinase to produce a soluble MBP-fused PDK3 (MBP–PDK3) fusion protein. The presence of the MBP tag has no effect on the enzymatic properties of the related rat BCK (Davie et al, 1995; Wynn et al, 2000). As shown in Figure 1A and B, at saturating L2 concentrations (20–30 μM), PDK3 activity is stimulated by 11.9-fold, whereas at the 40 nM concentration of E2p used previously (Tuganova et al, 2002), PDK3 activity is maximally stimulated by 16.4-fold. Thus, the fold increases of PDK3 activity stimulated by L2 and E2p in the present study are in good agreement with those reported previously by the Roche group. The significantly lower E2p concentration required for maximally stimulated PDK3 activity presumably results, in part, from the multiple copies of L2 domain present in the 60-meric E2p core.
Figure 1.
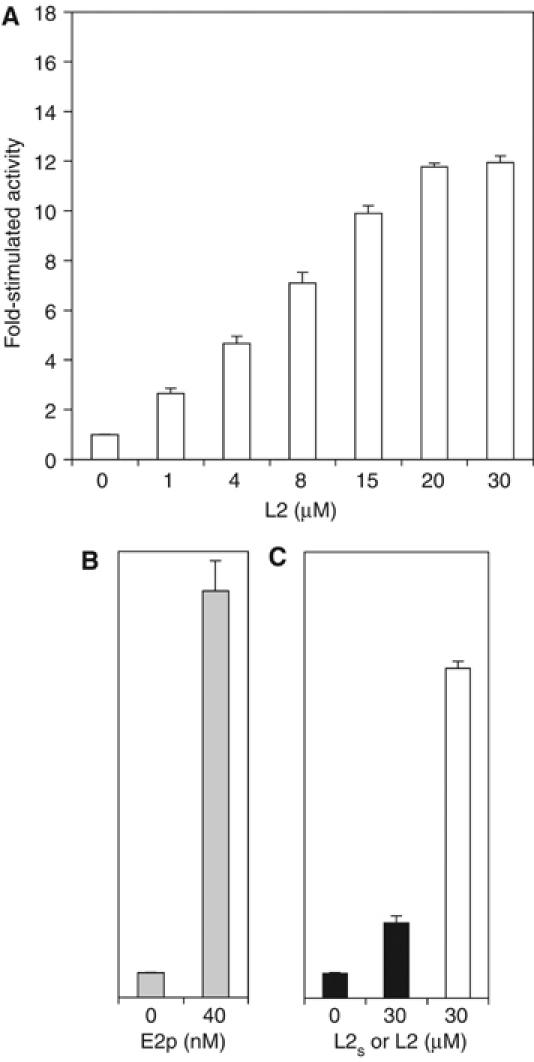
Stimulation of PDK3 kinase activity by the L2 domain and E2p. The kinase activity of MBP–PDK3 (45 nM) was measured by E1p phosphorylation in three independent experiments: (A) in the absence and presence of increasing concentrations (1–30 μM) of the lipoylated L2 domain (residues 126–233, designated as L2) (open bars), (B) in the absence and presence of 40 nM lipoylated E2p/E3BP 60-meric core (shaded bars), and (C) in the absence and presence of L2 (open bar) and a lipoylated L2s construct (residues 126–214) with a shorter C-terminal linker than L2 (solid bars), as described in ‘Materials and methods'. The basal (one fold) PDK3 activities in (A), (B), and (C) are similar at 21 nmol 32P incorporated/min/mg PDK3. To calculate the molar concentration of E2p, a molecular mass of 3 458 544 Da for lipoylated E2p/E3BP was used, according to a molecular ratio of E2p:E3BP=48:12 in the 60-meric E2p/E3BP core (Hiromasa et al, 2004). The ordinates in (B) and (C) are the same as in (A).
In a separate experiment, PDK3 activity was measured in the absence and presence of an L2 construct (designated as L2s) containing the shorter C-terminal linker (residues 209–214) studied by the Popov group or the above L2 construct used in crystallography. With these two L2 constructs, we obtained three- and 13.3-fold stimulation, respectively, of PDK3 activity (Figure 1C). Our results, therefore, establish that the significantly smaller L2-stimulated PDK3 activity reported by the Popov group is due to the shorter L2 C-terminal linker they used. It follows that the length of the C-terminal linker is critical for the augmentation of PDK3 activity by the L2 domain. In BCK, we have shown that a long C-terminal linker (residues 85–99) of its cognate lipoyl domain is also essential for efficient binding to this kinase (Chuang et al, 2002). However, other than stabilization effects on L2 (Gong et al, 2000), the role of the C-terminal linker in PDK3 activation is not clear, since this region does not appear to interact with PDK3 in the present PDK3–L2 structure (see below).
Overall structure of the PDK3–L2 complex
We determined three crystal structures of the PDK3–L2 complex (L2 residues 126–233), either without (apo) or with bound nucleotides (ATP or ADP) (Table I). These structures are virtually identical (RMSD: less than 0.55 Å for over 370 Cα atoms), except for the ATP lid (residues 305–327). The refined PDK3 structure is similar to the published PDK2–ADP structure in the absence of L2 (RMSD: 1.07 Å for 321 Cα atoms); however, there are small, yet significant, conformational differences apparently caused by L2 binding. The structure of the cognate BCK is also similar to PDK3 (RMSD: 1.4 Å for 277 Cα atoms), despite low sequence identity (23%) between these two kinases.
Table 1. Data collection and refinement statistics of PDK3–L2 structures.
| apo | +ADP | +ATP | |
|---|---|---|---|
| Data collectiona | |||
| Space group | P6522 | P6522 | P6522 |
| Unit cell (Å) | |||
| a=b | 120.81 | 120.75 | 120.90 |
| c | 238.59 | 239.15 | 240.09 |
| Wavelength (Å) | 1.54 | 1.0 | 1.0 |
| Resolution (Å) | 2.60 | 2.48 | 2.63 |
| Measurements | 316129 | 422583 | 231967 |
| Unique reflections | 32529 | 37324 | 31079 |
| Completeness (%) | 100 (100) | 99.8 (100) | 98.6 (97.2) |
| R-merge (%)b | 5.3 (54.7) | 3.8 (54.6) | 5.9 (54.2) |
| 〈I〉/〈σ(I)〉 | 28.7 (3.9) | 54.1 (3.5) | 30.0 (2.1) |
| Multiplicity | 9.7 (8.7) | 11.3 (9.1) | 7.5 (5.1) |
| Refinementa | |||
| No. of reflections (work/test) | 30805/1645 | 35738/1494 | 29477/1566 |
| No. of atoms (Mean B value (Å2)) | |||
| Protein | 3803 (62.3) | 3803 (64.2) | 3849 (66.6) |
| Solvents | 53 (46.6) | 67 (52.5) | 56 (52.3) |
| Nucleotide | — | 27 (74.4) | 31 (45.7) |
| Hetero compound | 12 (72.5) | 14 (59.0) | 14 (66.7) |
| R-work (%)c | 21.0 (33.8) | 21.0 (38.9) | 20.4 (40.7) |
| R-free (%)c | 24.8 (38.7) | 23.4 (39.9) | 23.0 (47.8) |
| RMSD | |||
| Bond length (Å) | 0.016 | 0.019 | 0.015 |
| Bond angle (°) | 1.700 | 1.685 | 1.731 |
| Ramachandran plot | |||
| Most favored (%) | 90.4 | 91.4 | 90.3 |
| Allowed (%) | 9.6 | 8.6 | 9.7 |
| Disallowed (%) | 0 | 0 | 0 |
| aValues in parentheses refer to data in the highest resolution shell unless otherwise indicated. | |||
| bR-merge=∑hkl ∑j∣Ij–〈I〉∣/∑hkl ∑j Ij, where 〈I〉 is the mean intensity of j observations from a reflection hkl and its symmetry equivalents | |||
| cR-work=∑hkl∣∣Fobs∣–k∣Fcalc∣∣/∑hkl∣Fobs∣; R-free=R-work for 4–5% of reflections that were omitted from refinement | |||
In the present PDK3 structure, the N-terminal domain comprises residues 12–178 and 366–381, and the C-terminal domain encompasses residues 182–365 (Figure 2A). Structures of these domains are essentially identical to the corresponding domains in PDK2, except that certain disordered regions in PDK2 are ordered in PDK3–L2. One of the ordered regions is the loop between helices α2 and α3 of the N-terminal domain in PDK3. The lipoyl-lysine residue of L2 binds to this region, which is part of the lipoyl-binding pocket, to stabilize the loop conformation. The C-terminal region, which is not visible in PDK2, is ordered in PDK3–L2 (Figure 2A). This region joins the N-terminal domain with the formation of helix α13, and projects out from that domain in an extended conformation. The extended segment (residue 382–401), which is designated as the C-terminal tail, is critical to L2 binding (see below).
Figure 2.
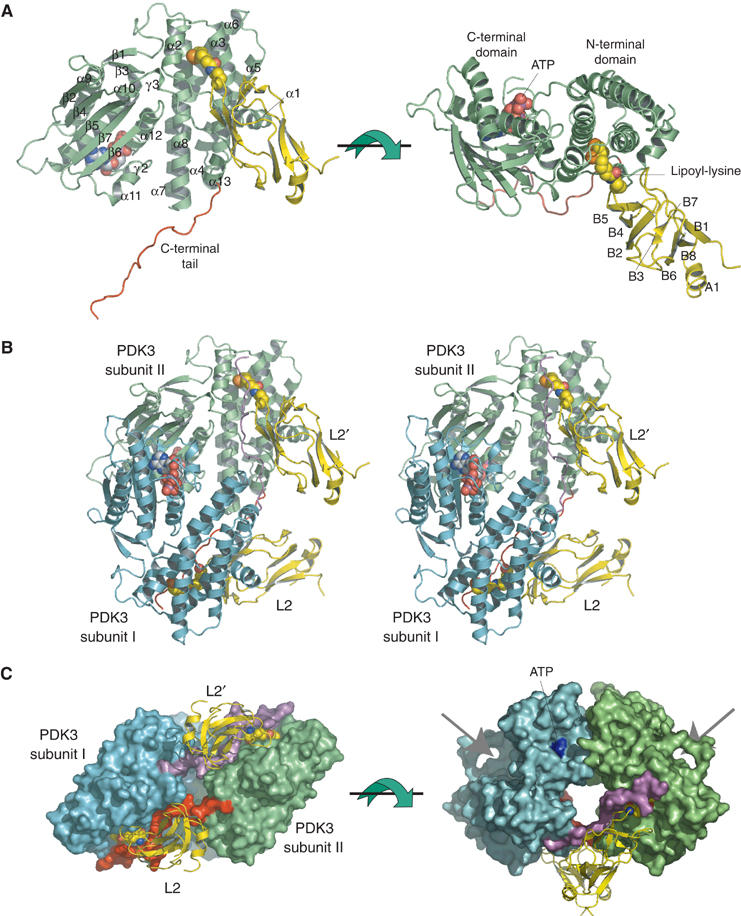
Monomeric and dimeric structures of the PDK3–L2 complex. (A) The monomeric structure of the PDK3–L2 complex is shown as ribbon models (PDK3 in green, L2 in yellow). The lipoyl-lysine residue of L2 (residues 126–233) bound to the N-terminal domain and ATP bound to the C-terminal domain are shown as space-filling models. The extended C-terminal tail is colored in red. The secondary structures of PDK3 and L2 are labeled as follows: α and A—alpha helix, β and B—beta strand, γ—310 helix. The first 310-helix of PDK3 (γ1), which is not labeled, is located between helices α6 and α7. (B) Stereo view of the dimeric structure of the PDK3–L2 complex with the same view angle as that of the left figure in (A). The C-terminal tails from subunits I (cyan) and II (green) of the PDK3 dimer are highlighted in purple and red, respectively. (C) Each subunit of the PDK3 dimer is shown in molecular-surface representation with the same color configuration as in (B). The two bound L2 (yellow) are shown as ribbon models with L2 bound to subunit I and L2′ to subunit II. The lipoyl-lysine residue of L2 is shown as a space-filling model. In the left panel, the extended C-terminal tail (red) from subunit II crosses over with the equivalent (purple) from subunit I to interact with both its N-terminal domain and L2 bound to subunit I. In the right panel, the arrows point to the active-site clefts between the N- and C-terminal domains of each subunit. The bound ATP is colored in blue.
The L2 domain is a member of the biotinyl/lipoyl-carrier protein superfamily (http://scop.mrc-lmb.cam.ac.uk/scop/data/scop.b.c.baf.b.b.html). In the complex, L2 adopts a β-barrel structure comprising two antiparallel β-sheets (Figure 2A). This fold is similar, but not identical, to the NMR structure previously reported (RMSD: 2.1 Å for 65 Cα atoms) (Supplementary Figure S1B) (Howard et al, 1998). The crystal structure of L2 shows a helix (A1) in the C-terminal region, whereas in the NMR structure this region is in an extended conformation. The lipoyl-lysine residue at position 173 is located in a short loop connecting strands β4 and β5. The long side chain of the lipoyl-lysine protrudes into its binding pocket in the N-terminal domain of PDK3 (Figure 2).
Each asymmetric unit contains one PDK3 monomer bound to one L2 (Figure 2A), with two symmetry-related monomers forming a functional dimer (Figure 2B and C) (Bowker-Kinley et al, 1998; Baker et al, 2000; Korotchkina and Patel, 2001). This result is consistent with the presence of PDK3 dimers in solution, as indicated by analytical ultracentrifugation (Hiromasa and Roche, 2003) and gel filtration (JL Chuang, unpublished data, 2004). The dimer formation of PDK3 is achieved by extensive interactions in a head-to-tail fashion between two β-sheets each from the C-terminal domains of the two subunits. This dimerization mode is similar to that described for PDK2 (Steussy et al, 2001) and BCK dimers (Machius et al, 2001). The PDK3 dimer binds two L2 with no interactions observed between these L2 domains (Figure 2C, left). Remarkably, the extended C-terminal tail from PDK3 subunit II crosses over to subunit I and interacts with L2 bound to subunit I, becoming an integral part of the lipoyl-binding pocket in the latter subunit.
The present structure is consistent with earlier biochemical studies that indicated the subunit stoichiometry of PDK3:L2=1:1 (Tuganova et al, 2002; Hiromasa and Roche, 2003). Isothermal titration calorimetry (ITC) was performed to dissect interactions between MBP–PDK3 and lipoylated L2 (Figure 3). The binding isotherm gives rise to a dissociation constant (Kd) of 1.17±0.23 μM for L2 binding to PDK3. This constant is 10-fold lower than those for L2 binding to PDK1 and PDK2 previously determined by gel filtration (Tuganova et al, 2002). The close to unity (0.74) subunit stoichiometry (PDK3 monomer: L2 monomer) and the iterative-best-fit monophasic binding isotherm indicate that both L2-binding sites within the PDK3 dimer are equivalent.
Figure 3.
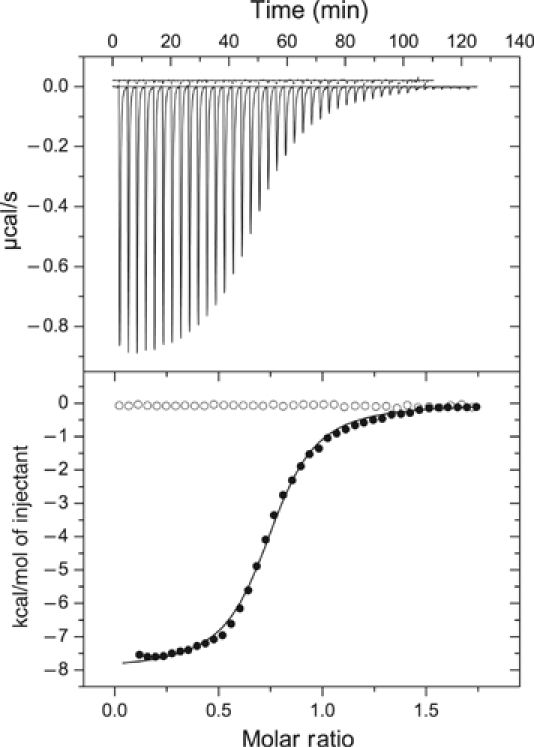
Binding of L2 domain to wild-type and C-terminally truncated PDK3. The binding of lipoylated L2 to wild-type and C-terminally truncated (Δ392–406) MBP–PDK3 was measured by ITC as described in ‘Materials and methods'. The upper panel shows the raw isothermal data for the titration of L2 against the truncated and wild-type MBP–PDK3 (upper and lower data, respectively). The lower panel represents the binding isotherms versus the molar ratio of L2 monomer: MBP–PDK3 monomer (•, wild-type MBP–PDK3; ○, truncated MBP-PDK3). The solid line in the lower panel depicts the fitting of the binding isotherm, based on a single type of the L2-binding site using ORIGIN software package. The wild-type MBP–PDK3 shows a dissociation constant (Kd) of 1.17±0.23 μM (n=3) for L2; for the truncated MBP–PDK3 (Δ392–406), there is no detectable L2 binding.
The C-terminal domain of PDK3 binds ATP or ADP through conserved residues in the common motifs of the GHKL superfamily (Dutta and Inouye, 2000). The ATP-binding site is located on the sidewall of the C-terminal domain facing the cleft between the N- and C-terminal domains (Figure 2A). The γ-phosphate of bound ATP is accessible to the E1p substrate from within the cleft, which is likely to be the active site for the phosphoryl-transfer reaction. Based on previous studies with PDK2 (Tuganova et al, 2001), the invariant D247 in PDK3 located in the active-site cleft presumably serves as the general base for the activation of phosphorylatable serine residues in E1p. The bound L2 in the N-terminal domain of subunit I is located more than 30 Å away from the active-site cleft of this subunit, and is at more than 23 Å distance from the opposing active-site cleft in subunit II (Figure 2B and C). Thus, there appears no direct interaction between the bound L2 and either of the active-site clefts, suggesting that L2 exerts its effects on PDK3 through an allosteric mechanism.
Extensive interactions exist between PDK3 and L2
Figure 4A shows the 619 Å2 surface area in the N-terminal domain of PDK3 subunit I that constitutes the binding interface with L2. Superimposition of electrostatic potential surfaces on PDK3 subunit I and L2 (Figure 4C) with interacting surfaces on the corresponding subunit/domain (Figure 4A) reveals two types of interactions: (1) hydrophobic interactions in the lipoyl-binding pocket and its surrounding regions, and (2) electrostatic interactions in the lower part of the N-terminal domain including helix α13. In the PDK2 structure without L2, the C-terminal region comprising α13 is largely disordered (Steussy et al, 2001); therefore, the formation of helix α13 in the complex structure is apparently induced by L2 binding.
Figure 4.
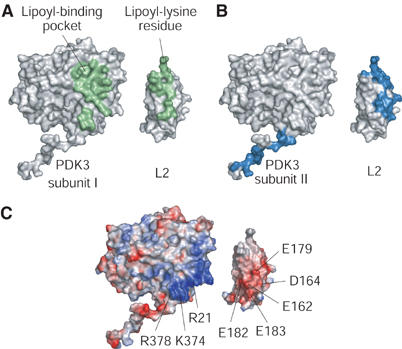
Interaction surfaces between PDK3 and L2. Interfaces between PDK3 and L2 are shown in molecular surface representations. L2 is separated from PDK3 and rotated by 180° to show the binding interfaces on both proteins. (A) A single L2 binds to the N-terminal domain of PDK3 subunit I. The corresponding interfaces are in green. (B) The C-terminal tail of PDK3 subunit I binds to L2′ in complex with subunit II (cf. Figure 2A). Binding surfaces between the tail and L2′ are in navy blue. (C) The electrostatic potential surfaces of PDK3 and L2 were calculated with APBS based on the Poisson–Boltzmann equation (Baker et al, 2001). Positive-charged regions are shown in blue, and negative-charged regions in red. The interfaces between the N-terminal domain of PDK3 and L2 (cf. A) consist of hydrophobic interactions in the lipoyl-binding pocket and its surrounding regions as well as electrostatic interactions on the lower portion of the N-terminal domain. Conserved residues in human PDKs and the lipoyl domains of human PDC are indicated.
The hydrophobic interactions occur mainly in the lipoyl-binding pocket in PDK3. The pocket is formed by the loop between helices α1 and α2 as well as helices α2 and α3, with helix α8 serving as the backwall (Figure 5). These structural components confer a 9-Å deep cylindrical pocket. The long aliphatic side chain of the lipoyl-lysine residue in L2 bends approximately 60° at the C4 atom, allowing the lipoamide group to protrude into the lipoyl-binding pocket. Hydrophobic residues L27, F32, F35, F48, L49, L164, L171, and F172, which are highly conserved among human PDKs (Supplementary Figure S1), constitute the inner lining of the pocket and appear to be essential for hydrophobic interactions with the aliphatic side chain of lipoamide. In particular, the reduced dithiolane moiety of lipoamide (Figure 5, inset) is sandwiched between the invariant F35 and F48. Crystallization of the PDK3–L2 complex was carried out in the presence of 10 mM DTT; at this DTT concentration the oxidized form of lipoamide is readily reduced (Bao et al, 2004a; Hiromasa et al, 2004); this apparently facilitated the crystallization of the PDK3–L2 complex. The reduced form of lipoamide in L2 has higher affinity for PDKs than its oxidized counterpart, as a feedback inhibitory mechanism in the PDC (Baker et al, 2000; Roche et al, 2003; Bao et al, 2004b). Hydrophobic residues in the lipoyl-binding pocket are also conserved in the related BCK (Supplementary Figure S1). The conservation of these residues strongly suggests that the recognition of the lipoyl-lysine residue by PDKs and BCK is similar. Surrounding the lipoyl-binding pocket in PDK3, highly conserved residues F22, P26, and V55 as well as methylene groups of the K51 side chain contribute to interactions with highly conserved hydrophobic residues in L2 (L140, P142, A174, and I176) (Supplementary Figures S1 and S2). The structural data explain the preliminary findings that substitutions of L140 and A174 of L2 both remove 80% of L2-stimulated PDK3 activity (Roche et al, 2003).
Figure 5.
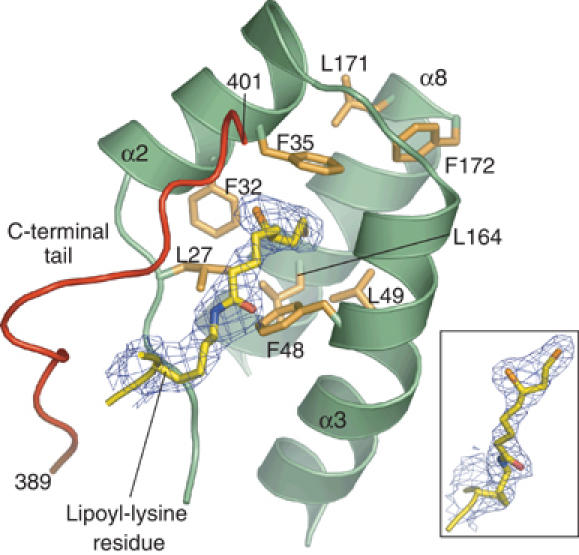
Structure of the hydrophobic lipoyl-binding pocket in PDK3. The lipoyl-binding pocket is formed by a loop region between helices α1 (not shown) and α2, as well as helices α2, α3, and α8. These regions form a cylindrical pocket containing conserved hydrophobic residues (brown). The lipoyl-lysine residue (yellow) projects into the hydrophobic pocket. The final 2Fo−Fc electron density (blue, contoured at 1σ) is superimposed on the lipoyl-lysine residue. The inset shows a different view of the bound reduced dihydrolipoamide. The C-terminal tail (red) from the other PDK3 subunit is integrated into the lipoyl-binding pocket. The first and last residue numbers of the C-terminal tail are indicated.
The second interface between the N-terminal domain of PDK3 and L2 consists of electrostatic interactions. Basic residues R21 (α1), K374, and R378 (α13), which are highly conserved in human PDKs (Supplementary Figure S1), are located underneath the lipoyl-binding pocket (Figure 4C). This basic region interacts with an acidic cluster on L2 formed by conserved residues E162, D164, E179, E182, and E183 (Supplementary Figures S1 and S2). E162 and E179 have also previously been implicated in interactions of the L2 domain with PDK3 in site-directed mutagenesis studies (Roche et al, 2003).
As described above, it will be of particular significance for diabetes therapy to design drugs that specifically inhibit elevated PDK activities. To date, two types of specific PDK inhibitors have been developed (Aicher et al, 2000; Morrell et al, 2003). One of these compounds (AZD7545) shows ATP-independent inhibition of PDK2 activity, and it is speculated that this inhibitor acts as a competitive inhibitor to prevent L2 from binding to PDK2. Thus, our structural data depicting the interactions between PDK3 and L2 may constitute a framework for designing a new generation of specific PDK inhibitors to eradicate impaired glucose utilization in type II diabetes.
Interactions of the PDK3 C-terminal tail with the L2 domain
As described above, a significant area (603 Å2) of the extended C-terminal tail from subunit II provides additional interface with L2 bound to subunit I (Figure 4B). The additional surface accounts for 49% of the total interface of L2 for PDK3. To facilitate the illustration of these interactions, the C-terminal tail is divided into three consecutive regions in the N- to C-terminus direction (Figure 6).
Figure 6.
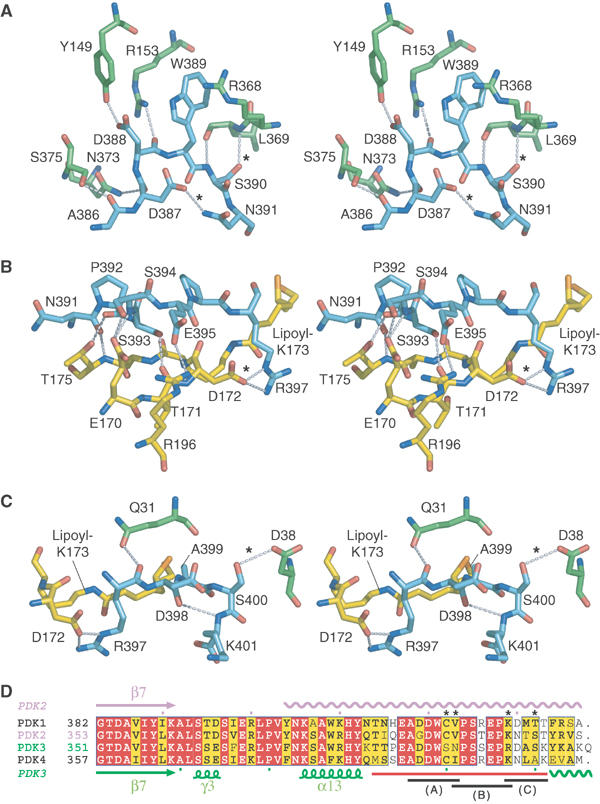
Stereo views of interactions between residues from L2 and the C-terminal tail of PDK3 and sequence alignments of human PDK C-terminal regions. (A) The N-terminal region of the C-terminal tail from subunit I (cyan) is stabilized by formation of hydrogen bonds with conserved residues from subunit II (green). (B) The following region of the C-terminal tail forms many hydrogen bonds with residues on the side of L2′ bound to subunit II. (C) The terminal region of the C-terminal tail is stabilized by hydrogen bonds formed with residues from subunit II above the lipoyl-binding pocket. (D) Sequence alignments of C-terminal regions of human PDKs suggest that hydrogen bonds indicated with ‘*' in (A)–(C) are uniquely present in PDK3. The C-terminal tail is depicted with a red line at the bottom of the alignments. The three regions shown in (A), (B), and (C) are indicated as black lines at the bottom. Conserved residues are colored in red and similar residues in yellow. The secondary structures of PDK2 and PDK3 are shown at the top and bottom of the alignments, respectively. Wavy lines indicate disordered regions in the crystal structures of PDK2 and PDK3. Sequences were aligned with ClustalW (Thompson et al, 1994) and drawn with ESPript (Gouet et al, 1999). For complete alignments including the cognate BCK, see Supplementary Figure S1.
In the PDK3 dimer, the region of residues 386–391 in the C-terminal tail from subunit I forms an extensive hydrogen-bond network with conserved residues in subunit II (Figure 6A). Specifically, S390 and N391 from subunit I form hydrogen bonds with L369 of subunit II and D387 of subunit I, respectively. In other PDKs, S390 and N391 of PDK3 are substituted by cysteine and valine/isoleucine, respectively (Figure 6D); the side chain of cysteine is a poor hydrogen donor and that of valine or isoleucine is unable to form hydrogen bonds.
Residues 392–397 of subunit I interact with the L2 domain bound to subunit II (Figure 6B). This region largely recognizes residues from L2 flanking the lipoyl-lysine residue. R196, which is located on the back of L2 opposite to the PDK3–L2 binding interface, forms a hydrogen bond with the conserved E395 from PDK3. Another notable residue is R397 in subunit I, whose side chain makes two hydrogen bonds to D172 of L2. In other human PDKs, R397 in PDK3 is replaced by a lysine residue whose side chain is capable of forming a single hydrogen bond (Supplementary Figure S1). The present results provide the structural basis for the earlier findings that substitutions of R196 and D172 of L2 significantly diminish L2-stimulated PDK3 activity (Roche et al, 2003).
The end of the C-terminal tail from residues 397 to 401 is integrated into the lipoyl-binding pocket (Figure 6C). Side chains of R397 and A399 in the C-terminal tail make van der Waals contacts with the aliphatic lipoamide backbone. This region is further stabilized by three hydrogen bonds between: (1) R397 in subunit I and Q31 in subunit II, (2) S400 in subunit I and D38 in subunit II, and (3) an internal hydrogen bond between D398 and K401 of the C-terminal tail in subunit I. The hydrogen bond between S400 and D38 appears to occur only in PDK3 and is not present in other PDKs (Supplementary Figure S1). A C-terminal truncation (Δ392–406) of PDK3 abolishes L2 binding to PDK3 as determined by ITC (Figure 3). The data support the notion that the interactions between the C-terminal tail and L2 are essential for L2 binding to PDK3. The C-terminal regions of PDKs from Ascaris suum and Caenorhabditis elegans were previously shown to be required for the binding of these kinases to the respective nematode E2p (Chen et al, 1999).
Previous biochemical studies have shown that PDK isoforms exhibit different affinities for L2, with PDK3 possessing the highest affinity (Tuganova et al, 2002). As shown above, the PDK3 dimer binds L2 through two regions, namely the N-terminal domain and the C-terminal tail. The interface between the N-terminal domain and L2 consists of hydrophobic and electrostatic interactions (Figure 4) that occur similarly in all four PDKs based on sequence conservation (Supplementary Figure S1). These results suggest that the hydrogen-bond formation in the C-terminal tail of PDKs contains specific determinants that confer differential affinity for L2. In the PDK3–L2 complex, but not in other PDKs, the C-terminal tail is further stabilized by additional interactions with residues from the other subunit (Figure 6A and C). Moreover, the two hydrogen bonds described above occur between R397 in PDK3 and D172 in L2 (Figure 6B), as opposed to only one potential hydrogen bond in other PDKs. These structural determinants in combination likely impart the higher affinity of PDK3 for L2 than other PDKs.
L2 binding-induced conformational changes in PDK3
To decipher L2-induced conformational changes in PDK3, it is necessary to determine the structure of PDK3 free of L2. However, PDK3 alone is sparingly soluble, which prevented the crystallization of free PDK3. Since amino-acid sequences are highly conserved between PDK3 and PDK2 (65% identity), the available PDK2 structure free of L2 (Steussy et al, 2001) was compared with the PDK3–L2 complex. In the two β-sheets of the C-terminal domains, many residues that participate in the dimer formation are conserved in both the PDK2 and PDK3 structures. Therefore, the structure at the dimer interface is expected to be similar between these two PDKs.
As described above, PDK3, but not PDK2, is activated by the isolated L2 domain (Baker et al, 2000; Tuganova et al, 2002). However, recent studies by the Popov group showed that the dissociation constant (Kd) of PDK2 for Mg-ATP is increased by 10-fold in the presence of L2 (Tuganova and Popov, 2005). These authors suggest that L2 binding promotes a conformational change in PDK2, which accounts for the reduced affinity of PDK2 for ATP. The L2-induced conformational change in PDK2 was also suggested previously by the Roche group, based on the modest activation of PDK2 activity by a dimeric L2 construct and an E2p construct lacking the E1p-binding domain (Baker et al, 2000; Bao et al, 2004b). These findings provide additional rationale for using the PDK2 structure to deduce possible L2-mediated conformational changes in PDK3.
When the right subunit of PDK3–L2-ADP is superimposed onto that of the PDK2–ADP dimer, the left subunit of PDK2 swings outward by 15° relative to the corresponding subunit of PDK3 (Figure 7A). If one assumes that the PDK3–ADP structure free of L2 is similar to the PDK2–ADP structure, the crossover of the two C-terminal tails is likely to restrict subunit movements in the PDK3–L2 complex, resulting in a clamping of the space between two subunits in the structure. This clamping has long-range effects on the conformation of the active-site cleft in each monomer. This becomes apparent when the monomeric structure of PDK2–ADP is superimposed onto that of PDK3–L2–ADP. As shown in Figure 7B, helix α7 of the N-terminal domain and helix α9 of the C-terminal domain in PDK3 move outward in opposite directions by ∼2 Å each, relative to the corresponding helices in PDK2. This domain shift results in the widening of the active-site cleft in PDK3–L2–ADP compared to PDK2–ADP. In addition, the ATP lid of PDK2–ADP is partially ordered, whereas the equivalent in PDK3–L2–ADP is largely invisible (Figure 7B) (for stereo-view, see Supplementary Figure S3A). The order-to-disorder transition of the ATP lid is caused by loss of interactions between the lid and the phosphate group of the bound ADP. The disordered conformation is not due to the loss of crystal lattice interactions, because this region is not involved in crystal packing. Significant movements occur in Y326 and M304 in the ATP lid, which result in the repositioning of their side chains in the space vacated by reciprocal shifts of conserved residues V130 (α7) and I147 (α8) in the N-terminal domain of PDK3. These concerted side-chain movements are likely to contribute to the disordering of the ATP-lid conformation.
Figure 7.
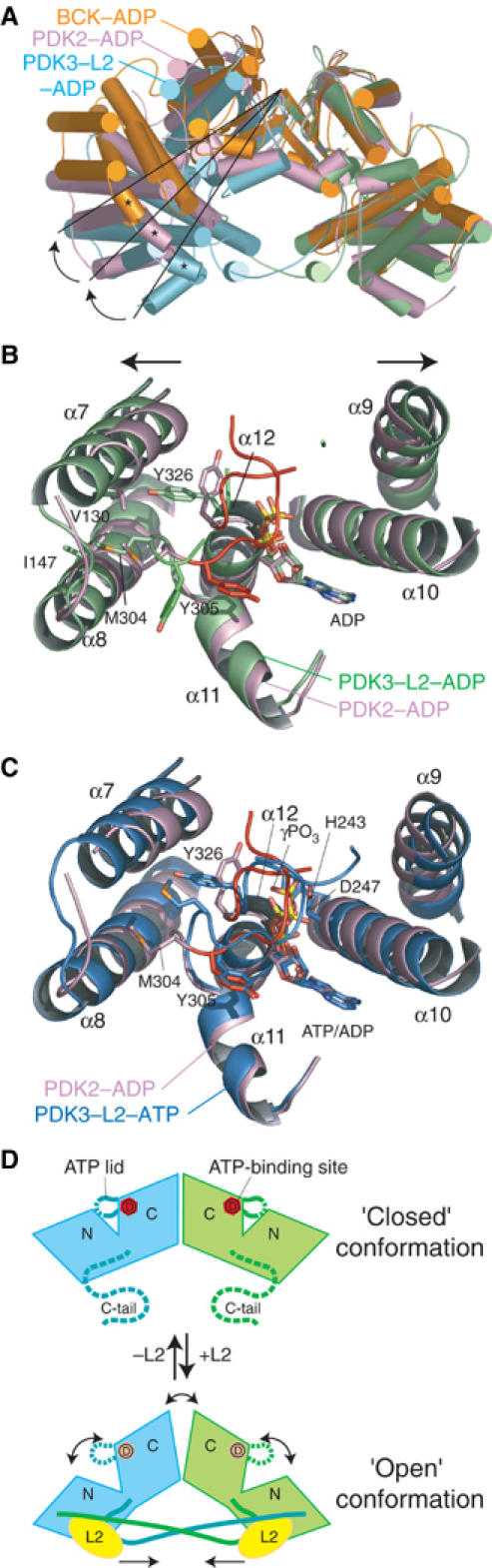
L2-induced conformational changes in PDK3 destabilize the ATP lid. (A) Superimposition of the dimer structure of the PDK3–L2–ADP (cyan and green) onto the structures of PDK2-ADP (pink, PDB code 1JM6) and BCK–ADP (orange, 1GKZ). Only the right subunits were superimposed, resulting in RMSD values of 1.07 Å (321 Cα atoms) between PDK3 and PDK2, and 1.40 Å (277 Cα atoms) between PDK3 and BCK. Black lines are drawn from the dimer interfaces to the edges of helix α4 of PDK3 and the corresponding helices of PDK2 and BCK (marked with ‘*'), which show outward shifts of the left subunits from the right subunits in PDK2 and BCK. (B) Superimposition of ATP-binding sites from the PDK3–L2-ADP (green) and PDK2–ADP (pink) structures. ADP and conserved residues are shown as stick models. The arrows show domain shifts associated with α7 of the N-terminal domain and α9 of the C-terminal domain in PDK3–L2–ADP compared to PDK2-ADP. The ATP lid (red) is ordered in PDK2–ADP, but largely disordered in PDK3–L2-ADP. (C) Superimposition of PDK3–L2–ATP (blue) onto PDK2–ADP (pink). The ATP lid, which is disordered in PDK3–L2–ADP (cf. B), is ordered in PDK3–L2–ATP to the extent similar to that of PDK2-ADP (red) through the formation of hydrogen bonds between the lid and the γ-phosphate of the bound ATP (cf. Supplementary Table SI). Conserved residues essential for the phosphoryl transfer reaction (H243 and D247) are shown as stick models. (D) The proposed R↔T equilibrium for L2-stimulated PDK3 activity. The ‘closed' (tight) and ‘open' (relaxed) conformations correspond to PDK3 in the absence and presence of bound L2, respectively. In the closed conformation, the C-terminal tails in the PDK3 dimer are disordered; the ATP lid in the active-site cleft is partially ordered, which traps ADP with high affinity (D in red circles). In the L2-binding-induced open conformation, both C-terminal tails are ordered and exist in a crossover configuration. This configuration promotes the opening of the active-site cleft with a completely disordered ATP lid in the ADP-bound form. The accelerated release of ADP caused by low-affinity binding (D in pink circles) removes product inhibition by ADP, resulting in L2-stimulated PDK3 activity.
In the PDK3–L2–ATP structure, the C-terminal ordered region (residues 325–327) of the ATP lid is stabilized by forming hydrogen bonds with the γ-phosphate of bound ATP (Figure 7C) (see also Supplementary Figure S3B and Table SI). G325 in this region in turn forms a hydrogen bond with M304 in the N-terminal segment of the same ATP lid, further stabilizing the lid conformation. However, the L2-induced widening of the active-site cleft in PDK3–L2–ADP occurs similarly in PDK3–L2–ATP. The subsequent weakened interactions between the ATP lid and the N-terminal domain sidewall could conceivably render the lid in PDK3–L2–ATP more flexible than that in PDK2–ADP.
L2 binding markedly decreases affinities of PDK3 for both ADP and ATP
We have previously shown that the ATP lid is significantly ordered in the ADP-bound form of rat BCK free of its cognate lipoyl domain (Machius et al, 2001). The ordering of the ATP lid, which effectively traps ADP, accounts for product inhibition during the phosphoryl-transfer reaction catalyzed by BCK. We proposed that E2 or the lipoyl domain bound to BCK functions as a nucleotide exchange factor to promote the substitution of inhibitor ADP for substrate ATP in the nucleotide-binding site. The complete disordering of the ATP lid in PDK3–L2–ADP (Figure 7B) may be related to a reduced affinity of PDK3 for ADP induced by L2. To support this notion, ITC was employed to measure dissociation constants (Kd's) for ADP binding to MBP–PDK3 in the absence or presence of the L2 construct (Table II). PDK3 without L2 exhibits a Kd of 0.16±0.04 μM for ADP, whereas when a two-fold molar excess of lipoylated L2 is present, PDK3 shows a Kd of 1.30±0.14 μM for the same nucleotide. The data indicate a significantly (>8-fold) decreased affinity of PDK3–L2 for ADP compared to free PDK3. The good fits of monophasic binding isotherms in the presence of lipoylated L2 (data not shown) indicate that the two nucleotide-binding sites in the PDK3 dimer are equivalent and not interacting with each other, in accordance with that recently reported for PDK2 (Tuganova and Popov, 2005). Thus, the present study validates and extends the above hypothesis, in that L2 binding promotes the release of ADP from PDK3 to remove product inhibition by this nucleotide. This model provides the structural basis for recent findings that show the rate of ADP dissociation is a rate-limiting step in E2p-stimulated PDK2 activity (Bao et al, 2004a, 2004b).
Table 2. Dissociation constants (Kd) for nucleotide binding to PDK3 with and without L2.
| Kinase | Nucleotide | Kd (μM)a |
|---|---|---|
| PDK3 | ADP | 0.16±0.04 (n=3) |
| PDK3-L2 | ADP | 1.30±0.14 (n=3) |
| PDK3 | ATP | 0.16±0.005 (n=3) |
| PDK3-L2 | ATP | 0.84±0.10 (n=3) |
| PDK3 | ATPγS | 0.17 |
| PDK3-L2 | ATPγS | 0.96 |
| Dissociation constants for the binding of Mg-ADP, Mg-ATP, and Mg-ATPγS to MBP–PDK3 were measured by isothermal titration calorimetry in the absence and presence of lipoylated L2, as described in ‘Materials and methods'. | ||
| aFor Mg-ADP and Mg-ATP in the absence or presence of L2, Kd values are expressed as mean±standard deviation. n=the number of experiments. | ||
As described above, the ATP lid in PDK3–L2–ATP is apparently destabilized by weakened interactions with the N-terminal domain even though the lid is still partially visible in the crystal structure (Figure 7C). The flexible ATP lid accounts for the more than five-fold decrease in affinity for ATP, when one compares the dissociation constants for ATP between free PDK3 (0.16±0.005 μM) and PDK3–L2 (0.84±0.10 μM) (Table II). Similar dissociation constants were obtained for ATPγS, with Kd=0.16 μM for PDK3 and Kd=0.96 μM for PDK3–L2 (Table II). The significantly lower affinity of PDK3 for ADP than ATP is likely to facilitate ATP/ADP exchange to remove the product inhibition by ADP. The flexible ATP lid may also explain the recent biochemical findings that Kd for ATP binding to PDK2 is elevated by 10-fold in the presence of L2 (Tuganova and Popov, 2005). Similarly, Km for ATP of PDK2 kinase activity is increased by eight-fold by E2p (Bao et al, 2004a).
Allosteric R↔T equilibrium as the mechanism for L2-stimulated PDK3 activity
Based on the present structural and biochemical data, we hypothesize that the PDK3 dimer in the absence of L2 adopts a ‘closed' conformation similar to that in the published PDK2–ADP structure (Figure 7D, top). In this conformation, the active-site cleft is closed because the C-terminal tail of each subunit is disordered and not interacting with the lipoyl-binding pocket in the other subunit. In the closed active-site cleft, ADP binds to PDK3 with the same affinity as ATP (Table II) because the ATP lid is stabilized by interactions with the sidewall of the N-terminal domain. The high-affinity binding of ADP prevents an efficient ADP/ATP exchange, resulting in product inhibition by ADP with low kinase activity. Upon the binding of L2 to PDK3, the C-terminal tails are ordered in the crossover configuration, which promotes the widening of the active-site cleft as well as the dimer interface, which in turn leads to an ‘open' conformation in the PDK3 dimer (Figure 7D, bottom). In this open conformation, the interactions of the ATP lid with the sidewall from the N-terminal domain are significantly weakened, which renders the ATP lid largely disordered. The subsequent lower-affinity binding of ADP than ATP in the PDK3–L2 complex accelerates the release of trapped ADP to mitigate product inhibition by this nucleotide. The open conformation of PDK3 may also facilitate the access of the E1p substrate to the active-site cleft. Indeed, recent biochemical studies suggest that the Km for E1p of PDK2 decreases significantly when the kinase is bound to an E2p construct devoid of the E1p-binding domain (Bao et al, 2004a). This situation is analogous to cyclin-dependent kinase 2 (CDK2), where a pivotal T loop is utilized to regulate its kinase activity (Jeffrey et al, 1995). In the free form of CDK2, the T loop covers the active site to block the substrate access, whereas the binding of cyclin A to CKD2 induces a conformational change in the T loop, resulting in the opening of the active site. Thus, our data suggest that L2-induced stimulation of PDK3 activity may employ the classic allosteric mechanism of R (relaxed)↔T (tight) equilibrium (Monod et al, 1965) between the L2-bound and free PDK3, with the L2-bound form favoring the R or open conformation.
Materials and methods
This section is provided in Supplementary data.
Structural data deposition
The model coordinates and structural factor amplitudes have been deposited in the Protein Data Bank for structures of the apo- (1Y8N), ADP-bound (1Y8O) and ATP-bound PDK3–L2 complex (1Y8P).
Supplementary Material
Supplementary data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table SI
Supplementary data
Acknowledgments
The authors thank Dr Kirill Popov (University of Alabama at Birmingham) for providing the PDC E2p/E3BP plasmid. We are also indebted to Drs Mischa Machius, Diana Tomchick and Chad Brautigam in the Structural Biology Laboratory for the collection of synchrotron data. This work was supported by Grants DK62306 and DK26758 from the National Institutes of Health and Grant I-1286 from the Welch Foundation. Use of the Argonne National Laboratory Structural Biology Center beam lines at the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research under Contract No. W-31-109-ENG-38.
References
- Aicher TD, Anderson RC, Gao J, Shetty S, Coppola GM, Stanton JL, Knorr DC, Sperbeck DM, Brand LJ, Vinluan CC, Kaplan EL, Dragland C, Tomaselli HC, Islam A, Lozito RJ, Liu X, Maniara WM, Fillers WS, DelGrande D, Walter RE, Mann WR (2000) Secondary amides of (R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionic acid as inhibitors of pyruvate dehydrogenase kinase. J Med Chem 43: 236–249 [DOI] [PubMed] [Google Scholar]
- Alex LA, Simon MI (1994) Protein histidine kinases and signal transduction in prokaryotes and eukaryotes. Trends Genet 10: 133–138 [DOI] [PubMed] [Google Scholar]
- Baker JC, Yan X, Peng T, Kasten S, Roche TE (2000) Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem 275: 15773–15781 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban C, Junop M, Yang W (1999) Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell 97: 85–97 [DOI] [PubMed] [Google Scholar]
- Ban C, Yang W (1998) Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell 95: 541–552 [DOI] [PubMed] [Google Scholar]
- Bao H, Kasten SA, Yan X, Hiromasa Y, Roche TE (2004a) Pyruvate dehydrogenase kinase isoform 2 activity stimulated by speeding up the rate of dissociation of ADP. Biochemistry 43: 13442–13451 [DOI] [PubMed] [Google Scholar]
- Bao H, Kasten SA, Yan X, Roche TE (2004b) Pyruvate dehydrogenase kinase isoform 2 activity limited and further inhibited by slowing down the rate of dissociation of ADP. Biochemistry 43: 13432–13441 [DOI] [PubMed] [Google Scholar]
- Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P (1997) An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386: 414–417 [DOI] [PubMed] [Google Scholar]
- Bilwes AM, Alex LA, Crane BR, Simon MI (1999) Structure of CheA, a signal-transducing histidine kinase. Cell 96: 131–141 [DOI] [PubMed] [Google Scholar]
- Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329: 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Komuniecki PR, Komuniecki R (1999) Nematode pyruvate dehydrogenase kinases: role of the C-terminus in binding to the dihydrolipoyl transacetylase core of the pyruvate dehydrogenase complex. Biochem J 339 (Part 1): 103–109 [PMC free article] [PubMed] [Google Scholar]
- Chuang JL, Wynn RM, Chuang DT (2002) The C-terminal hinge region of lipoic acid-bearing domain of E2b is essential for domain interaction with branched-chain alpha-keto acid dehydrogenase kinase. J Biol Chem 277: 36905–36908 [DOI] [PubMed] [Google Scholar]
- Davie JR, Wynn RM, Meng M, Huang YS, Aalund G, Chuang DT, Lau KS (1995) Expression and characterization of branched-chain alpha-ketoacid dehydrogenase kinase from the rat. Is it a histidine-protein kinase? J Biol Chem 270: 19861–19867 [DOI] [PubMed] [Google Scholar]
- De Marcucci OG, Gibb GM, Dick J, Lindsay JG (1988) Biosynthesis, import and processing of precursor polypeptides of mammalian mitochondrial pyruvate dehydrogenase complex. Biochem J 251: 817–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 25: 24–28 [DOI] [PubMed] [Google Scholar]
- Gong X, Peng T, Yakhnin A, Zolkiewski M, Quinn J, Yeaman SJ, Roche TE (2000) Specificity determinants for the pyruvate dehydrogenase component reaction mapped with mutated and prosthetic group modified lipoyl domains. J Biol Chem 275: 13645–13653 [DOI] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15: 305–308 [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T (1988) The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241: 42–52 [DOI] [PubMed] [Google Scholar]
- Harris RA, Huang B, Wu P (2001) Control of pyruvate dehydrogenase kinase gene expression. Adv Enzyme Regul 41: 269–288 [DOI] [PubMed] [Google Scholar]
- Hiromasa Y, Fujisawa T, Aso Y, Roche TE (2004) Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J Biol Chem 279: 6921–6933 [DOI] [PubMed] [Google Scholar]
- Hiromasa Y, Roche TE (2003) Facilitated interaction between the pyruvate dehydrogenase kinase isoform 2 and the dihydrolipoyl acetyltransferase. J Biol Chem 278: 33681–33693 [DOI] [PubMed] [Google Scholar]
- Holness MJ, Sugden MC (2003) Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans 31: 1143–1151 [DOI] [PubMed] [Google Scholar]
- Howard MJ, Fuller C, Broadhurst RW, Perham RN, Tang JG, Quinn J, Diamond AG, Yeaman SJ (1998) Three-dimensional structure of the major autoantigen in primary biliary cirrhosis. Gastroenterology 115: 139–146 [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP (1995) Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature 376: 313–320 [DOI] [PubMed] [Google Scholar]
- Kolobova E, Tuganova A, Boulatnikov I, Popov KM (2001) Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 358: 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotchkina LG, Patel MS (1995) Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem 270: 14297–14304 [DOI] [PubMed] [Google Scholar]
- Korotchkina LG, Patel MS (2001) Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem 276: 37223–37229 [DOI] [PubMed] [Google Scholar]
- Lindsay JG (1989) _targeting of 2-oxo acid dehydrogenase complexes to the mitochondrion. Ann N Y Acad Sci 573: 254–266 [DOI] [PubMed] [Google Scholar]
- Liu S, Baker JC, Roche TE (1995) Binding of the pyruvate dehydrogenase kinase to recombinant constructs containing the inner lipoyl domain of the dihydrolipoyl acetyltransferase component. J Biol Chem 270: 793–800 [DOI] [PubMed] [Google Scholar]
- Machius M, Chuang JL, Wynn RM, Tomchick DR, Chuang DT (2001) Structure of rat BCKD kinase: nucleotide-induced domain communication in a mitochondrial protein kinase. Proc Natl Acad Sci USA 98: 11218–11223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12: 88–118 [DOI] [PubMed] [Google Scholar]
- Morrell JA, Orme J, Butlin RJ, Roche TE, Mayers RM, Kilgour E (2003) AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem Soc Trans 31: 1168–1170 [DOI] [PubMed] [Google Scholar]
- Popov KM, Hawes JW, Harris RA (1997) Mitochondrial alpha-ketoacid dehydrogenase kinases: a new family of protein kinases. Adv Second Messenger Phosphoprotein Res 31: 105–111 [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH (1997) Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90: 65–75 [DOI] [PubMed] [Google Scholar]
- Radke GA, Ono K, Ravindran S, Roche TE (1993) Critical role of a lipoyl cofactor of the dihydrolipoyl acetyltransferase in the binding and enhanced function of the pyruvate dehydrogenase kinase. Biochem Biophys Res Commun 190: 982–991 [DOI] [PubMed] [Google Scholar]
- Reed LJ (2001) A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem 276: 28329–28336 [DOI] [PubMed] [Google Scholar]
- Reed LJ, Damuni Z, Merryfield ML (1985) Regulation of mammalian pyruvate and branched-chain alpha-keto acid dehydrogenase complexes by phosphorylation–dephosphorylation. Curr Top Cell Regul 27: 41–49 [DOI] [PubMed] [Google Scholar]
- Roche TE, Patel MS (1989) Alpha-keto acid dehydrogenase complexes: organization, regulation, and biomedical ramifications. Ann NY Acad Sci 573: 1–4622699393 [Google Scholar]
- Roche TE, Hiromasa Y, Turkan A, Gong X, Peng T, Yan X, Kasten SA, Bao H, Dong J (2003) Essential roles of lipoyl domains in the activated function and control of pyruvate dehydrogenase kinases and phosphatase isoform 1. Eur J Biochem 270: 1050–1056 [DOI] [PubMed] [Google Scholar]
- Sale GJ, Randle PJ (1981) Analysis of site occupancies in [32P]phosphorylated pyruvate dehydrogenase complexes by aspartyl–prolyl cleavage of tryptic phosphopeptides. Eur J Biochem 120: 535–540 [DOI] [PubMed] [Google Scholar]
- Smirnova IN, Kasho VN, Faller LD (1998) Inferences about the catalytic domain of P-type ATPases from the tertiary structures of enzymes that catalyze the same elementary reaction. FEBS Lett 431: 309–314 [DOI] [PubMed] [Google Scholar]
- Steussy CN, Popov KM, Bowker-Kinley MM, Sloan RB Jr, Harris RA, Hamilton JA (2001) Structure of pyruvate dehydrogenase kinase. Novel folding pattern for a serine protein kinase. J Biol Chem 276: 37443–37450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saha SK, Tomomori C, Ishima R, Liu D, Tong KI, Park H, Dutta R, Qin L, Swindells MB, Yamazaki T, Ono AM, Kainosho M, Inouye M, Ikura M (1998) NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature 396: 88–92 [DOI] [PubMed] [Google Scholar]
- Teague WM, Pettit FH, Yeaman SJ, Reed LJ (1979) Function of phosphorylation sites on pyruvate dehydrogenase. Biochem Biophys Res Commun 87: 244–252 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar-Mendez A, Miernyk JA, Randall DD (2002) Histidine mutagenesis of Arabidopsis thaliana pyruvate dehydrogenase kinase. Eur J Biochem 269: 2601–2606 [DOI] [PubMed] [Google Scholar]
- Tuganova A, Boulatnikov I, Popov KM (2002) Interaction between the individual isoenzymes of pyruvate dehydrogenase kinase and the inner lipoyl-bearing domain of transacetylase component of pyruvate dehydrogenase complex. Biochem J 366: 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuganova A, Popov KM (2005) Role of protein–protein interactions in the regulation of pyruvate dehydrogenase kinase activity. Biochem J 387: 147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuganova A, Yoder MD, Popov KM (2001) An essential role of Glu-243 and His-239 in the phosphotransfer reaction catalyzed by pyruvate dehydrogenase kinase. J Biol Chem 276: 17994–17999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley DB, Davies GJ, Dodson EJ, Maxwell A, Dodson G (1991) Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature 351: 624–629 [DOI] [PubMed] [Google Scholar]
- Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA (1998) Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem J 329: 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn RM, Chuang JL, Cote CD, Chuang DT (2000) Tetrameric assembly and conservation in the ATP-binding domain of rat branched-chain alpha-ketoacid dehydrogenase kinase. J Biol Chem 275: 30512–30519 [DOI] [PubMed] [Google Scholar]
- Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH (1978) Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 17: 2364–2370 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table SI
Supplementary data