

Abstract
Germline mutations in PTEN (MMAC1/TEP1) are found in patients with Cowden syndrome, a familial cancer syndrome which is characterized by a high risk of breast and thyroid neoplasia. Although somatic intragenic PTEN mutations have rarely been found in benign and malignant sporadic thyroid tumors, loss of heterozygosity (LOH) has been reported in up to one fourth of follicular thyroid adenomas (FAs) and carcinomas. In this study, we examined PTEN expression in 139 sporadic nonmedullary thyroid tumors (55 FA, 27 follicular thyroid carcinomas, 35 papillary thyroid carcinomas, and 22 undifferentiated thyroid carcinomas) using immunohistochemistry and correlated this to the results of LOH studies. Normal follicular thyroid cells showed a strong to moderate nuclear or nuclear membrane signal although the cytoplasmic staining was less strong. In FAs the neoplastic nuclei had less intense PTEN staining, although the cytoplasmic PTEN-staining intensity did not differ significantly from that observed in normal follicular cells. In thyroid carcinomas as a group, nuclear PTEN immunostaining was mostly weak in comparison with normal thyroid follicular cells and FAs. The cytoplasmic staining was more intense than the nuclear staining in 35 to 49% of carcinomas, depending on the histological type. Among 81 informative tumors assessed for LOH, there seemed to be an associative trend between decreased nuclear and cytoplasmic staining and 10q23 LOH (P = 0.003, P = 0.008, respectively). These data support a role for PTEN in the pathogenesis of follicular thyroid tumors.
The tumor suppressor PTEN, also known as MMAC1 and TEP1,1-3 has recently been shown to play an important role in the pathogenesis of a variety of human cancers.1,2,4 PTEN is located on chromosome subband 10q23.32,5 and encodes a dual-specificity phosphatase with lipid and protein phosphatase activity. The major substrate for PTEN is phosphatidylinositol-3,4,5-trisphosphate, a direct product of phosphoinositol-3-kinase activity.6-8 Phosphatidylinositol-3,4,5-trisphosphate mediates growth factor-induced activation of intracellular signaling, in particular through the serine-threonine kinase Akt (also referred to as Akt1, RAC1, or PKB), which is known to promote cell survival and cell proliferation. High levels of PTEN are associated with low levels of phosphorylated Akt which leads to the induction of apoptosis; hence, loss of PTEN function leads to increased activity of Akt and subsequently cell survival.7-10 PTEN may also affect other pathways, such as the focal adhesion kinase and mitogen activated protein kinase pathways.11,12 In other words, PTEN has been shown to mediate G1 cell-cycle arrest and/or apoptosis via the phosphoinositol-3-kinase-Akt pathway in several cell lines such as glioma, breast, and prostate cell lines.13-15 PTEN therefore seems to play an important role in cell cycle growth, migration, and death. Germline PTEN mutations have been identified in the autosomal dominant hamartoma Cowden syndrome (CS) and Bannayan-Riley-Ruvalcaba syndrome.16,17 Benign thyroid disease is characteristic of both CS and Bannayan-Riley-Ruvalcaba syndrome.18 The risk of nonmedullary thyroid carcinomas is increased in CS.19-21 In humans, these thyroid cancers are most often follicular and very rarely papillary thyroid carcinoma (PTC). In contrast, histopathological analyses of Pten ± chimeric and heterozygous mice showed lesions consistent with well-differentiated PTC22 as well as follicular or papillary thyroid neoplasia.23 Although LOH (loss of heterozygosity) on chromosome band 10q23 has been identified in ∼26% of follicular thyroid adenomas (FAs)24-26 and up to 27% of FTCs,27 somatic intragenic mutations of PTEN are rare.25,27 The human syndromic and murine data for PTEN involvement in thyroid neoplasia is strong. In accordance with the Knudson “two hit” hypothesis of carcinogenesis, if a somatic mutation is found on one allele of PTEN and LOH or a deletion is found on the opposite allele, then biallelic inactivation of PTEN at the structural level is said to occur. However, to date, all genetic studies of PTEN in human primary thyroid tumors have only demonstrated monoallelic structural mutation (either a heterozygous deletion or a single-hit somatic intragenic mutation). Whether PTEN inactivation at the protein level or via other mechanisms apart from structural alteration applies to thyroid tumorigenesis is unknown. Thus, we sought to determine whether functional biallelic inactivation of PTEN occurs in sporadic nonmedullary thyroid adenomas and carcinomas by examining them for PTEN expression using immunohistochemistry in conjunction with LOH analysis.
Materials and Methods
Thyroid Samples
Paraffin blocks from 139 unselected benign and malignant nonmedullary thyroid tumors were ascertained from Germany, Australia, and Switzerland. Histological classification of the thyroid tumors was in accordance with the World Health Organization.28 Of note, five papillary tumors were classified as follicular type (Lindsay tumor), and 13 tumors (seven FAs, five FTCs, one PTC) had a prominent granular eosinophilic-appearing cytoplasm (also known as oxyphilic or Hürthle cell).
Anti-PTEN Antibody Specificity
The monoclonal antibody 6H2.1 raised against the last 100 C-terminal amino acids of PTEN29 was used in all immunohistochemical analyses. As biochemical proof of antibody specificity for PTEN, total protein lysates were obtained10,15 from a series of thyroid cell lines for which PTEN status is known: NPA-87, K-1, FTC-133, and WRO-82–1 (gifts from D. V. Canlapan and D. Wynford-Thomas). Further, as an additional positive control, the wild-type full-length human PTEN cDNA sequence was cloned into the expression vector pcDNA3 and transfected into the PTEN null line FTC-133. Western blot analysis was performed as previously described10 except that 6H2.1 was used at a 1:250 dilution. Thyroid lines with endogenously expressing or exogenously introduced PTEN all demonstrated a single band at 55 kd, the molecular weight predicted for PTEN, although the PTEN null lines did not cross-react with 6H2.1. No other nonspecific bands were noted, thus proving antibody-specificity (Figure 1)▶ . Control antibody against α-tubulin (Sigma, St. Louis, MO), used at 1:10,000 dilution, immunoreacted evenly across protein lysates from all cell lines (Figure 1)▶ . The specificity of the antibody 6H2.1 and its suitability for immunohistochemistry in paraffin-embedded tissue has been demonstrated previously.29 In brief, we used the antibody against embedded PTEN-transfected U2OS cells, BALBc/3T3, Nalm6, and DU145 as positive controls; MDA-MB-468 with hemizygous deletion of PTEN and a truncation of the remaining allele; A172 which has loss of one PTEN allele, and a truncating mutation in exon 2 of the remaining allele; and PC3, which is null for PTEN.29 Further, commercially available peptide corresponding to PTEN has been used to successfully compete away 6H2.1 immunostaining in paraffin-embedded tissue (GL M, unpublished data).
Figure 1.

Western analysis of whole-cell protein lysates from thyroid cancer cell lines using the anti-PTEN monoclonal antibody 6H2.1 (top panel) and using the anti-α-tubulin antibody as a control (bottom panel). NPA-87 and K-1, which are two PTC lines, and WRO-82–1, a FTC line, have endogenous PTEN. FTC-133 is a FTC line that is PTEN null. Lane 1, NPA-87; lane 2, K-1; lane 3, FTC-133; lane 4, FTC-133 transfected with empty vector; lane 5, FTC-133 transfected with vector containing PTEN; lane 6, WRO-82–1. The same membrane was used for Western blot with 6H2.1 as well as anti-tubulin antibody.
Recently, it has been shown that a processed PTEN pseudogene (psiPTEN) can be transcribed in a number of cell lines and tissue types.30 For this reason, RNA in situ hybridization is not reliable. PsiPTEN, however, does not seem to be translated, at least in thyroid tumors, and therefore it would not be expected to complicate the analysis in this study.
Immunohistochemistry
The tissue samples were fixed by immersion in 10% buffered formalin and subsequently embedded in paraffin according to standard protocols. Four-mm sections were cut, mounted on Superfrost plus slides, and baked for 2 hours at 60°C. Subsequently, the sections were deparaffinized and rehydrated by passing through xylene and a graded series of ethanol solutions. Antigen retrieval was performed by boiling at 98°C in 0.01 mol/L sodium citrate buffer, pH 6.4, in a microwave oven for 20 minutes. To block endogenous peroxidase activity, the sections were incubated with 0.3% hydrogen peroxide in methanol for 30 minutes after cooling to room temperature. After blocking for 30 minutes in 0.75% horse serum, the sections were incubated with a PTEN monoclonal antibody 6H2.1 (dilution 1:100) for 1 hour at room temperature. Primary antibody binding was localized by using an avidin-biotin-peroxidase kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instruction. The chromogenic reaction was carried out with 0.05% 3,3′diaminobenzidine (Sigma, St. Louis, MO) using nickel cobalt amplification which gives a black product.31 After counterstaining with Nuclear Fast Red (Rowley Biochemical Institute, Danvers, MA) and mounting, the slides were independently evaluated under a light microscope by two investigators (OG and AP) and randomly spot evaluated by a third investigator (CE). Intensity of staining was classified separately for the nucleus/nuclear membrane and the cytoplasm and graded strong (+++), moderate (++), weak (+), or absent (−). These independent assessments did not differ by more than one grading level.
LOH Analysis
In 95 samples, tumor tissue and blood or corresponding normal tissue (either normal thyroid tissue or adjacent muscle tissue) were available for extraction of paired somatic and germline DNA to study LOH. DNA extraction after microdissection was performed using standard protocols.32 All subsequent polymerase chain reactions were carried out using 0.6 μM each of forward and reverse primer in 1× polymerase chain reaction buffer (Qiagen, Valencia, CA), 4.5 mmol/L MgCl2 (Qiagen), 1× Q-buffer (Qiagen), 2.5 U HotStarTaq polymerase (Qiagen), and 200 μmol/L dNTP (Gibco, Gaithersburg, MD) in a final volume of 50 μl. Reactions were subjected to 35 cycles of 94°C for 1 minute, 55°C to 60°C for 1 minute, and 72°C for 1 minute followed by 10 minutes at 72°C. Potential hemizygosity at the PTEN locus was assessed by screening for a T/G polymorphism within PTEN intron 8 (IVS8 + 32G/T) detected by differential digestion with the restriction endonuclease HincII as previously described25 except for using the primers PTEN-E8-F (5′-GCGTGCAGATAATGACAAGG-3′) and PTEN-I8-R (5′-TGTCAAGCAAGTTCTTCATCG-3′). If the result of the digestion was not informative, LOH analysis was performed using markers flanking PTEN, D10S541 (telomeric) and D10S579 (centromeric)1,16 as well as the marker D10S2491 that lies within PTEN.33 All forward primers were 5′-labeled with either HEX or 6-FAM fluorescent dye (Research Genetics, Huntsville, AL). Polymerase chain reactions were carried out as described above and separated by electrophoresis through 6% denaturing polyacrylamide gels using an Applied Biosystems model 377 automated DNA sequencer (Applied Biosystems, Perkin-Elmer Corp., Norwalk, CT). The results were analyzed by automated fluorescence detection using the GeneScan collection and analysis software (GeneScan, Applied Biosystems). Scoring of LOH was performed by inspection of the GeneScan analysis output. A double peak, observed in the microsatellite marker that was amplified from DNA extracted from the germline sample, indicated heterozygosity. A single peak in DNA extracted and amplified from the corresponding tumor sample indicated a loss of one allele. If normal cells were admixed with tumor cells, a ratio of 1.5:1 or greater of germline DNA peak to tumor DNA peak was also considered LOH.24
To examine the correlative trend between PTEN staining intensity and LOH, we performed a Mantel-Haenszel test34 for trend in the association between the row and column variables. A P < 0.05 was considered statistically significant.
Results
PTEN Immunohistochemistry in Normal Thyroid and Primary Thyroid Tumors
Of the total 139 thyroid tumor samples examined for PTEN expression using the monoclonal antibody 6H2.1, 50 had accompanying normal thyroid tissue. Normal follicular thyroid cells showed a uniform strong (+++) to moderate (++) nuclear or nuclear membrane (hereafter referred to as nuclear) signal whereas the cytoplasmic staining was less strong, + to ++ (Figure 2A)▶ . Endothelial cells showed strong (+++) to moderate (++) PTEN expression with a nuclear predominance and were useful as internal positive controls (Figure 2▶ , B and E). In contrast, nuclear and cytoplasmic staining intensity of fibrocytes was very heterogeneous and varied from weak (Figure 2A)▶ to strong (Figure 2▶ F).
Figure 2.
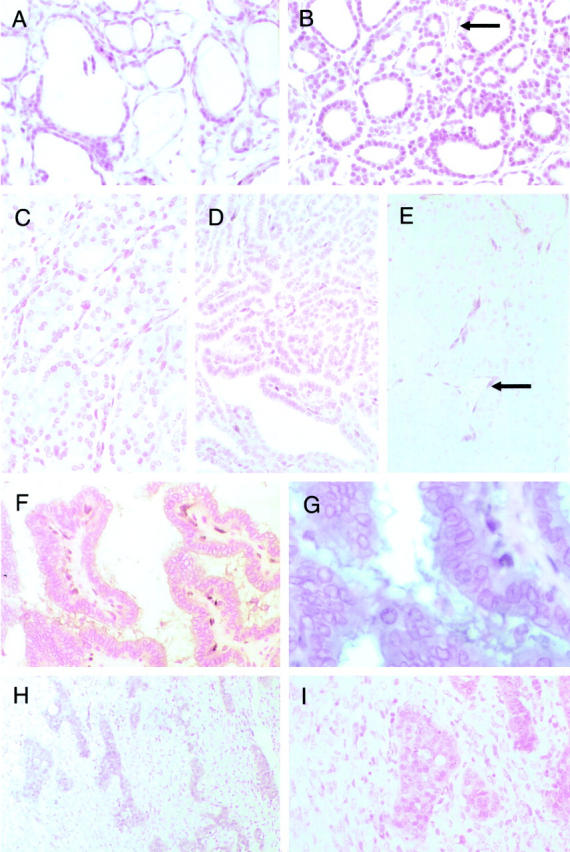
Immunohistochemical analysis of normal thyroid, FA, FTC, PTC, and UTCs using the anti-PTEN monoclonal antibody 6H2.1. A: Normal thyroid tissue (magnification, ×20). Note strong (+++) to moderate (++) nuclear and weak (+) cytoplasmic staining in the follicular epithelial cells. B: Follicular thyroid adenoma (magnification, ×20). In this particular sample, the intensity of nuclear PTEN staining in the majority of follicular cells is strong (graded +++ to ++); the intensity of cytoplasmic staining is weak (+), not greatly different from that of normal epithelial cells. Note the strong staining intensity of the endothelial cells that serve as positive controls (←). C: FTC (magnification, ×20) with mainly weak (+) nuclear staining. D: PTC (magnification, ×20) with weak (+) nuclear staining. E: UTC (magnification, ×20) with absent (−) nuclear and cytoplasmic staining. Note the intense immunostaining in the endothelial cells, which serve as an internal positive control (←). F: PTC (magnification, ×20) with absent (−) nuclear immunostaining but moderate (++) cytoplasmic staining. G: Same PTC (magnification, ×40). Note the completely absent (−) staining in the nucleus compared to the cytoplasm. H: UTC (magnification, ×10) with heterogeneous PTEN immunostaining pattern. Islands of immunopositivity interspersed among large areas of immunonegativity. I: Same UTC (magnification, ×20). Note that the immunopositive cells are small and round whereas the immunonegative cells are large with large pleomorphic nuclei.
The quality and intensity of PTEN immunostaining in the nucleus and cytoplasm in 132 of 139 thyroid tumors was relatively uniform throughout each specimen. However, in seven carcinomas, PTEN expression differed significantly within different regions of each tumor (see below). Because PTEN expression in each of these seven tumors could not be classified into a single category, these different regions were classified separately as if they were two separate tumors, ie, PTEN expression was classified in 139 + 7 = 146 thyroid tumors. Hence, the intensity of PTEN staining in the nucleus and cytoplasm were assessed for 146 thyroid tumors (Figure 3)▶ for purposes of this study.
Figure 3.
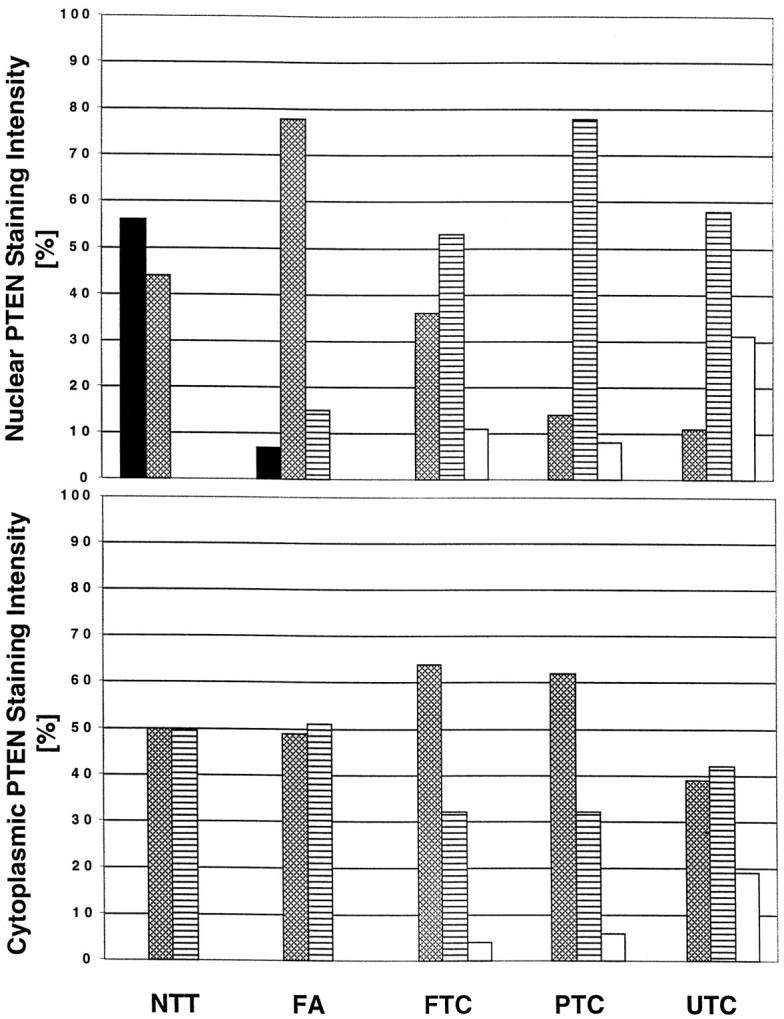
Distribution of PTEN immunostaining intensity in and cytoplasm in thyroid samples (50 normal thyroid tissues, 55 thyroid adenomas, 28 FTC, 37 PTC, and 26 UTC). Strong ▪, moderate ▩, weak ▤, and absent staining □. NTT, normal thyroid tissue. The y-axis represents percentage of samples with various intensities of nuclear (top) or cytoplasmic (bottom) staining.
In FA, the neoplastic nuclei had less intense PTEN immunostaining (+ to ++) compared to normal follicular epithelium whereas the cytoplasmic PTEN staining intensity did not differ significantly from that observed in normal follicular cells (Figures 2B and 3)▶ ▶ . In thyroid carcinomas (FTC, PTC, and undifferentiated thyroid carcinoma [UTC]) as a group, nuclear PTEN immunostaining was mostly weak in comparison with normal thyroid follicular cells and FAs. Among the three classes of carcinomas, FTCs had the strongest immunostaining in both the nucleus and cytoplasm and UTCs the weakest (Figures 2, C–E, and 3)▶ ▶ . A few carcinomas, in particular UTCs, showed no PTEN staining in the nucleus (FTC, 3 of 28; PTC, 3 of 37; UTC, 8 of 26) or in the cytoplasm (one FTC, two PTCs, five UTCs; Figure 2▶ E); eight carcinomas (one FTC, two PTCs, five UTCs) had no immunoreactivity in the nucleus or in the cytoplasm. In almost half of all thyroid carcinomas (FTC, 46%; PTC, 49%; UTC, 35%), the cytoplasmic staining was more intense than the nuclear staining (Figures 2, F and G, and 3)▶ ▶ . In contrast, the more intense cytoplasmic over nuclear staining was observed in only 7% of FAs. In other words, the stepwise decrease in PTEN immunoreactivity in the nucleus seemed to precede that in the cytoplasm from normal thyroid tissue to FA to carcinomas and finally to UTC (Figure 3)▶ . There was no obvious difference in PTEN staining pattern and intensity in Hürthle cell tumors compared to non-Hürthle cell tumors. Fibrocytes seemed to stain a little more intensely in tumor stroma (Figure 2▶ F) compared to normal thyroid stroma (Figure 2A)▶ .
Seven carcinomas (four UTCs, two PTCs, one FTC), but no adenomas, showed dichotomous regional PTEN staining within each sample. These were characterized either by islands of strongly immunopositive cells among sheets of cells staining more weakly (Figure 2▶ , H and I) or by single cells staining weakly randomly distributed among cells staining strongly. In one UTC, the positive cells (graded +) were small and more regular whereas the PTEN-negative cells were larger, pleiomorphic, and had a more undifferentiated appearance. This correlation, however, was not seen in the other three UTCs. This pattern of immunostaining in the former UTC was replicated several times, thus indicating that this was not an artifact.
Comparison of Immunohistochemical and LOH Data
In 95 tumors, normal and tumor tissue samples were available for LOH analysis. Similar to the immunohistochemical analysis, the four available carcinomas with dichotomous staining intensity within each sample, which had paired normal and tumor tissue, were considered as eight samples. Therefore, 99 total samples were considered. For purposes of this study, which was to compare the immunohistochemical data to the LOH data, one copy of PTEN was considered to be physically deleted only when one or more of the markers, which lie within or closely flanking the gene, showed LOH. Using this definition for monoallelic PTEN deletion, LOH data from 92 tumors were informative. Of the 92 informative tumors, 27 tumors (29%) were shown to have loss of one allele of PTEN and 65 tumors were classified as having no LOH (Table 1)▶ .
Table 1.
LOH Analysis of Markers on Chromosome Band 10q23 in 92 Informative Thyroid Tumors
| Tumor type | LOH 10q23 | % | % of LOH literature* |
|---|---|---|---|
| Follicular thyroid adenoma | 8 /41 | 20 | 6–26 (10q23) |
| Follicular thyroid carcinoma | 6 /20 | 30 | 7–27 (10q23) |
| Papillary thyroid carcinoma | 3 /14 | 21 | 5 (10q) |
| Undifferentiated thyroid carcinoma | 10 /17 | 59 | 35 (10q) |
*Data based on References 24, 25, 27, 44, and 45 .
LOH analysis was performed separately for regions with different staining intensity in those four carcinomas with dichotomous staining intensity and available paired normal and thyroid tissue. One of these four tumors showed LOH whereas the other three tumors did not. Interestingly, in this tumor, LOH was identified in both immunoreactive-positive and -negative regions.
Among the 92 informative tumors assessed for LOH and PTEN immunostaining, there seemed to be an associative trend between decreased or absent staining and 10q23 LOH (Table 2▶ , Figure 3▶ ). The proportion of tumors that had LOH steadily increased from tumors with +++ nuclear staining (0% with LOH), ++ staining (21% LOH), + staining (32% LOH) to no (−) staining (75% LOH) (P = 0.003). This trend was also mirrored if we considered only cytoplasmic staining and LOH status (++ cytoplasmic staining [18% with LOH], + staining [36% LOH], to no [−] staining [67% LOH]) (Table 2▶ ; Figure 3▶ ; P = 0.008). Two samples showed no PTEN staining without evidence for LOH. One of the samples was heterozygous for IVS8 + 32G/T whereas the other markers were not informative. The other sample showed regions of heterozygosity at D10S579 and was not informative for the other markers.
Table 2.
Correlation between Intensity of Nuclear PTEN Staining and LOH Analysis
| Intensity of PTEN staining | Nucleus | Cytoplasm | ||
|---|---|---|---|---|
| LOH (%) | No LOH (%) | LOH (%) | No LOH (%) | |
| +++ | 0 (0) | 3 (100) | 0 (0) | 0 (0) |
| ++ | 9 (21) | 34 (79) | 8 (18) | 36 (82) |
| + | 12 (32) | 26 (68) | 15 (36) | 27 (64) |
| − | 6 (75) | 2 (25) | 4 (67) | 2 (33) |
Statistical correlation of PTEN intensity versus LOH performed using a Mantel-Haenszel test yielded results of χ21 = 8.70, P = 0.003 for nuclear and χ21 = 7.11, P = 0.008 for cytoplasmic PTEN staining, indicating a significant increase in LOH at 10q23 with decreasing PTEN stain intensity.
Discussion
In this study, we have demonstrated that PTEN is strongly expressed in normal thyroid epithelium, in particular in the nucleus and to a lesser extent in the cytoplasm. Endothelial cells also show strong PTEN expression with a nuclear predominance. In benign thyroid tumors, the nuclear expression is weaker compared to normal thyroid follicular cells but still remains at relatively high levels whereas the cytoplasmic staining remains unchanged. In contrast, in thyroid carcinomas, nuclear and cytoplasmic PTEN expression, as judged by immunohistochemistry, is reduced. The reduced intensity of the nuclear staining often predominated the reduced intensity in the cytoplasm, in particular in FTC and PTC. In some advanced or aggressive malignant tumors, eg, UTCs, no PTEN staining was detectable at all. These data support a role for PTEN in the pathogenesis of nonmedullary thyroid adenomas and carcinomas, although the marked reduction to no PTEN immunostaining as well as the relatively higher percentage of 10q23 deletion in UTCs does argue that PTEN′s role in thyroid tumorigenesis may be in tumor progression rather than tumor initiation.
Because PTEN has not been shown to have a nuclear localization signal,1,2 cytoplasmic expression of PTEN is expected. In normal thyroid cells, as expected, PTEN is strongly expressed. The decrease in PTEN immunoreactivity in the cytoplasm from normal cells to differentiated carcinomas and to UTC supports our hypothesis that PTEN is inactivated and plays a role in thyroid tumorigenesis. Decreasing PTEN expression in this progression from adenoma to UTC would result in poor control of G1 arrest, apoptosis, and/or cell-cell adhesion.13,15,35,36
Given that PTEN does not have a nuclear localization signal,1 our observation of prominent and differential nuclear PTEN staining is puzzling. Our initial postulate that this represented nuclear membrane staining would appear to be more plausible, and such staining could reflect PTEN′s role in regard to the cytoskeleton. Other unrelated studies have noted this nuclear staining with different PTEN antibodies, without explanation. A weak or absent intensity of nuclear PTEN staining along with a strong cytoplasmic PTEN staining was also observed in prostate cancer xenografts37 and fibroblasts.11 Together with these previous incidental observations, our immunohistochemical evidence of nuclear staining is likely real, given that the decreasing nuclear signal with less-differentiated carcinomas predominates the decreased cytoplasmic staining. The precise mechanism for our observations is yet unknown.
The equally puzzling observation that decreased nuclear staining often precedes the decreased cytoplasmic staining in thyroid cancers is intriguing. Because PTEN per se does not have a nuclear localization signal, it is possible that PTEN is shuttled into the nucleus by another molecule. Such a mechanism has been described for the tumor-suppressor TP53 which is shuttled between the nucleus and the cytoplasm via the oncoprotein MDM2.38 Other examples exist in the literature. Phosphorylated forkhead-related transcription factor is excluded from the nucleus by interaction with 14-3-3.39 When not bound to 14-3-3, forkhead-related transcription factor enters the nucleus and acts as a transcription factor for various genes, including, presumably, FAS ligand.40 VHL and β-catenin have also been documented to play different roles depending on their subcellular localization.41,42 Based on the current state of knowledge, we would speculate that PTEN could be shuttled into the nucleus related to the cell cycle and/or as a response to cell division and cell growth. It is very well known that intracellular substrates can show a distinct distribution within different compartments (eg, cytoplasm, nucleus, microsomes), ie, differential intracellular compartmentalization. Hence, differential compartmentalization of PTEN might play some as yet undefined role in the tumorigenetic process.
As more studies are performed, it is becoming apparent that inactivation of PTEN relies on multiple diverse mechanisms and not merely structural abnormalities such as somatic mutations.10,43 In the present study, approximately one fourth of FAs and one fourth of the differentiated carcinomas had LOH. Both FAs and FTCs have a similar frequency of LOH as previously noted. The relatively high frequency of LOH in PTCs (21%) and UTCs (59%) is higher than that reported in the literature possibly because we used PTEN-specific markers. The markers in other studies were not specifically chosen for 10q23 but rather broadly covered the whole long arm of chromosome 10. Somatic intragenic mutations of PTEN are also not likely to play a major role in PTEN silencing because they have been rarely found in thyroid carcinomas.25,27 Another mechanism which has been implicated in prostate cancer and non-Hodgkin’s lymphoma is gene silencing by hypermethylation of CpG islands, presumably in the putative promoter,10,37 although the situation in prostate cancer is still controversial.33 Finally, at least in hematological malignancies, reduced or absent PTEN protein levels seem to involve either the postranscriptional, translational, or protein degradation pathways.10 Given our observations, all of these mechanisms likely come into play in the situation of epithelial thyroid tumorigenesis. We also add another possible mechanism: differential compartmentalization of PTEN. These issues need to be addressed and characterized at the functional level in the future.
Acknowledgments
We thank Albert de la Chapelle for helpful discussions, Patricia L. M. Dahia for critical reading of the manuscript, Fred Wright and Xin Gao for providing excellent statistical assistance, and Terry Bradley for outstanding technical assistance. We are grateful to Danilo V. Canlapan and David Wynford-Thomas for providing the thyroid cancer cell lines WRO-82–1 (DVC), NPA-87 (DVC), K-1 (DWT), and FTC-133 (DWT).
Footnotes
Address reprint requests to Dr. Charis Eng, Human Cancer Genetics Program, Ohio State University Comprehensive Cancer Center, 420 W. 12th Avenue, Room 690C Medical Research Facility, Columbus, OH 43210. E-mail: eng-1@medctr.osu.edu.
Supported in part by grants RPG-98–211-01-CCE from the American Cancer Society (to C. E.) and P30CA16058 from the National Cancer Institute (Ohio State University Comprehensive Cancer Center). O. G. is a Fellow of the Deutsche Forschungsgemeinschaft, Germany, and A. P. is a Fellow of the Lydia Hochstrasser-Stiftung, Zürich, Switzerland (to P. K.)
O. G. and A. P. contributed equally to this study.
References
- 1.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 1943, 275:277. [DOI] [PubMed] [Google Scholar]
- 2.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997, 15:356-362 [DOI] [PubMed] [Google Scholar]
- 3.Li DM, Sun H: TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res 1997, 57:2124-2129 [PubMed] [Google Scholar]
- 4.Teng DH, Hu R, Lin H, Davis T, Iliev D, Frye C, Swedlund B, Hansen KL, Vinson VL, Gumpper KL, Ellis L, El-Naggar A, Frazier M, Jasser S, Langford LA, Lee J, Mills GB, Pershouse MA, Pollack RE, Tornos C, Troncoso P, Yung WK, Fujii G, Berson A, Bookstein R, Bolen JB, Tavtigian SV, Steck PA: MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res 1997, 57:5221-5225 [PubMed] [Google Scholar]
- 5.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK: P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA 1997, 94:9052-9057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maehama T, Dixon JE: The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998, 273:13375-13378 [DOI] [PubMed] [Google Scholar]
- 7.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK: The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc Natl Acad Sci USA 1998, 95:13513-13518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW: Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95:29-39 [DOI] [PubMed] [Google Scholar]
- 9.Haas-Kogan D, Shalev N, Wong M, Mills G, Yount G, Stokoe D: Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol 1998, 8:1195-1198 [DOI] [PubMed] [Google Scholar]
- 10.Dahia PL, Aguiar RC, Alberta J, Kum JB, Caron S, Sill H, Marsh DJ, Ritz J, Freedman A, Stiles C, Eng C: PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in haematological malignancies. Hum Mol Genet 1999, 8:185-193 [DOI] [PubMed] [Google Scholar]
- 11.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM: Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280:1614-1617 [DOI] [PubMed] [Google Scholar]
- 12.Gu J, Tamura M, Yamada KM: Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol 1998, 143:1375-1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furnari FB, Huang HJ, Cavenee WK: The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res 1998, 58:5002-5008 [PubMed] [Google Scholar]
- 14.Li DM, Sun H: PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci USA 1998, 95:15406-15411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weng LP, Smith WM, Dahia PL, Ziebold U, Gil E, Lees JA, Eng C: PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res 1999, 59:5808-5814 [PubMed] [Google Scholar]
- 16.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R: Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997, 16:64-67 [DOI] [PubMed] [Google Scholar]
- 17.Marsh DJ, Dahia PL, Zheng Z, Liaw D, Parsons R, Gorlin RJ, Eng C: Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet 1997, 16:333-334 [DOI] [PubMed] [Google Scholar]
- 18.Eng C: Genetics of Cowden syndrome: through the looking glass of oncology. Int J Oncol 1998, 12:701-710 [DOI] [PubMed] [Google Scholar]
- 19.Wade TR, Kopf AW: Cowden’s disease: a case report and review of the literature. J Dermatol Surg Oncol 1978, 4:459-464 [DOI] [PubMed] [Google Scholar]
- 20.Thyresson HN, Doyle JA: Cowden’s disease (multiple hamartoma syndrome). Mayo Clin Proc 1981, 56:179-184 [PubMed] [Google Scholar]
- 21.Starink TM: Cowden’s disease: analysis of fourteen new cases. J Am Acad Dermatol 1984, 11:1127-1141 [DOI] [PubMed] [Google Scholar]
- 22.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP: Pten is essential for embryonic development and tumour suppression. Nat Genet 1998, 19:348-355 [DOI] [PubMed] [Google Scholar]
- 23.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R: Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A 1999, 96:1563-1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marsh DJ, Zheng Z, Zedenius J, Kremer H, Padberg GW, Larsson C, Longy M, Eng C: Differential loss of heterozygosity in the region of the Cowden locus within 10q22–23 in follicular thyroid adenomas and carcinomas. Cancer Res 1997, 57:500-503 [PubMed] [Google Scholar]
- 25.Dahia PL, Marsh DJ, Zheng Z, Zedenius J, Komminoth P, Frisk T, Wallin G, Parsons R, Longy M, Larsson C, Eng C: Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res 1997, 57:4710-4713 [PubMed] [Google Scholar]
- 26.Yeh JJ, Marsh DJ, Zedenius J, Dwight T, Robinson BG, Eng C: Fine structure deletion analysis of 10q22–24 demonstrates regions of loss and suggests that sporadic follicular adenomas and follicular thyroid adenomas develop along distinct parallel neoplastic pathways. Genes Chromosom Cancer 1999, 26:322-328 [DOI] [PubMed] [Google Scholar]
- 27.Halachmi N, Halachmi S, Evron E, Cairns P, Okami K, Saji M, Westra WH, Zeiger MA, Jen J, Sidransky D: Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosom Cancer 1998, 23:239-243 [DOI] [PubMed] [Google Scholar]
- 28.Hedinger C, Williams ED, Sobin LH: The WHO histological classification of thyroid tumors: a commentary on the ed 2. Cancer 1989, 63:908-911 [DOI] [PubMed] [Google Scholar]
- 29.Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PL, Komminoth P, Lees JA, Mulligan LM, Mutter GL, Eng C: Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 1999, 155:1253-1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujii GH, Morimoto AM, Berson AE, Bolen JB: Transcriptional analysis of the PTEN/MMAC1 pseudogene, psiPTEN. Oncogene 1999, 18:1765-1769 [DOI] [PubMed] [Google Scholar]
- 31.Werner M, Von Wasielewski R, Komminoth P: Antigen retrieval, signal amplification and intensification in immunohistochemistry. Histochem Cell Biol 1996, 105:253-260 [DOI] [PubMed] [Google Scholar]
- 32.Eng C, Thomas GA, Neuberg DS, Mulligan LM, Healey CS, Houghton C, Frilling A, Raue F, Williams ED, Ponder BA: Mutation of the RET proto-oncogene is correlated with RET immunostaining in subpopulations of cells in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1998, 83:4310-4313 [DOI] [PubMed] [Google Scholar]
- 33.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D: Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res 1997, 57:4997-5000 [PubMed] [Google Scholar]
- 34.Mantel N, Haenszel W: Statistical aspects for the analysis from retrospective studies of disease. J Natl Cancer Inst 1959, 22:719-748 [PubMed] [Google Scholar]
- 35.Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R: The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res 1998, 58:5667-5672 [PubMed] [Google Scholar]
- 36.Tamura M, Gu J, Takino T, Yamada KM: Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res 1999, 59:442-449 [PubMed] [Google Scholar]
- 37.Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL: Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci USA 1998, 95:5246-5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freedman DA, Wu L, Levine AJ: Functions of the MDM2 oncoprotein. Cell Mol Life Sci 1999, 55:96-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franke TF: A difficult Akt to follow. Neural Notes 1999, 5:3-7 [Google Scholar]
- 40.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME: Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96:857-868 [DOI] [PubMed] [Google Scholar]
- 41.Ye Y, Vasavada S, Kuzmin I, Stackhouse T, Zbar B, Williams BR: Subcellular localization of the von Hippel-Lindau disease gene product is cell cycle-dependent. Int J Cancer 1998, 78:62-69 [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL: Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res 1999, 59:1811-1815 [PubMed] [Google Scholar]
- 43.Wang SI, Parsons R, Ittmann M: Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res 1998, 4:811-815 [PubMed] [Google Scholar]
- 44.Ward LS, Brenta G, Medvedovic M, Fagin JA: Studies of allelic loss in thyroid tumors reveal major differences in chromosomal instability between papillary and follicular carcinomas. J Clin Endocrinol Metab 1998, 83:525-530 [DOI] [PubMed] [Google Scholar]
- 45.Zedenius J, Wallin G, Svensson A, Bovee J, Hoog A, Backdahl M, Larsson C: Deletions of the long arm of chromosome 10 in progression of follicular thyroid tumors. Hum Genet 1996, 97:299-303 [DOI] [PubMed] [Google Scholar]