

Abstract
Activation of Toll-like receptor (TLR) signaling by microbial signatures is critical to the induction of immune responses. Such responses demand tight regulation. RP105 is a TLR homolog, thought to be largely B cell-specific, which lacks a signaling domain. We report that RP105 expression is wide, directly mirroring that of TLR4 on antigen presenting cells. We further show that RP105 is a specific inhibitor of TLR4 signaling in HEK293 cells, a function conferred by its extracellular domain. Notably, RP105 and its helper molecule, MD-1, interacted directly with the TLR4 signaling complex, inhibiting its ability to bind microbial ligand. Finally, we demonstrate that RP105 regulates TLR4 signaling in dendritic cells, as well as endotoxin responses in vivo. Thus, these results identify RP105 as a physiological negative regulator of TLR4 responses.
The field of innate immunity has undergone a recent renaissance, fueled largely by the molecular identification of critical receptors and signaling pathways involved in pathogen recognition. Study of the TLR family has led the way. Activation of TLR signaling by conserved microbial molecular signatures promotes the induction of both innate and adaptive immune responses1,2. It has long been clear that such immune responses need to be kept under tight control. Responses that are delayed or of insufficient vigor can lead to a failure to control infection. On the other hand, excessive or inappropriate inflammation can be harmful or even fatal. The hyper-inflammatory responses that characterize sepsis provide a paradigmatic example, as do the more localized inappropriate inflammatory processes leading to inflammatory bowel disease and arthritis3–5
Mammalian TLRs are characterized structurally by an extracellular leucine-rich repeat (LRR) domain, a conserved pattern of juxtamembrane cysteine residues, and an intracytoplasmic signaling domain (Toll/IL-1 resistance [TIR]) that is highly conserved across the TLRs as well as the receptors for interleukin 1 (IL-1) and IL-182. The TLR-like molecule RP105 was originally cloned as a B cell-specific molecule able to drive B cell proliferation6,7. Like TLRs, RP105 has a conserved extracellular LRR domain and a TLR-like pattern of juxtamembrane cysteines6–10. Unlike the TLRs, however, RP105 lacks a TIR domain, containing a mere 6–11 intracytoplasmic amino acids (depending upon the prediction algorithm). In parallel with TLR4, whose surface expression and signaling depend upon co-expression of the secreted extracellular protein MD-2, surface expression of RP105 is dependent upon the co-expression of the MD-2 homolog, MD-111–14.
Here we show that RP105 is a specific homolog of TLR4. We further show that RP105 is not B cell-specific as originally proposed: RP105 protein expression directly mirrors that of TLR4 on antigen-presenting cells. In Toll and TLR4, mutation of the conserved juxtamembrane cysteine residues, or significant deletion of the extracellular portion, results in a constitutively active molecule15–17. This suggests that Toll/TLR activation is normally restrained through extracellular protein-protein interactions, likely through the LRR domain. On the other hand, deletions or mutations in the TIR domain of Toll/TLRs can yield inactive or dominant negative molecules15,18,19. Thus, RP105 has the apparent structure of an inhibitory TLR4. These considerations led us to the hypothesis that RP105 is a physiological regulator of TLR4 signaling. We show here that RP105 is a specific inhibitor of TLR4 signaling in HEK293 cells, a function conferred by its extracellular domain. Mechanistically, the RP105–MD-1 complex interacts directly with TLR4–MD-2, inhibiting the ability of this LPS signaling complex to bind LPS. Finally, we demonstrate that RP105 is a physiological regulator of TLR4 signaling in primary dendritic cells, and of responses to endotoxin in vivo.
RESULTS
RP105 expression mirrors that of TLR4
Phylogenetic analysis of the TLR family has revealed 5 TLR subfamilies: TLR4; TLR3; TLR5; TLR7, TLR8, TLR9; and TLR2, TLR1, TLR6, TLR10 2. Similar analytic techniques unequivocally place RP105 in the TLR4 subfamily (Supplementary Fig. 1 online). The domain structure of the 641 amino acid mature RP105 protein has previously been reported7,9,10,13. The extracellular portion of this type I transmembrane protein contains 22 leucine-rich repeats (repeats 7–10 being atypical), along with a pattern of juxtamembrane cysteines that is conserved among the Toll and TLR families. Prediction algorithms disagree on the C-terminal boundary of the transmembrane domain. Although the 6–11 amino acid intracellular domain of RP105 contains 1–2 tyrosine residues, it is devoid of conserved motifs that would suggest potential sites for phosphorylation.
RP105 is reported as a B cell-specific molecule in mice7. Further, despite the fact that RP105 is expressed at the mRNA level in primary human myeloid cells8–10, the only published examination of RP105 protein expression in such cells (by immune blot analysis) was reported to be negative10. We examined RP105 expression by FACS in human and murine monocytic cells. In neither species was RP105 expression limited to B cells. In humans, RP105 was also expressed by human monocytes, as well as myeloid dendritic cells (DC) (Fig. 1a–c), cells that express considerably more TLR4 than do B cells 20 (and data not shown). RP105 was not expressed by human plasmacytoid DC (Fig. 1d), cells that also fail to express TLR420–22 (and data not shown).
Figure 1.
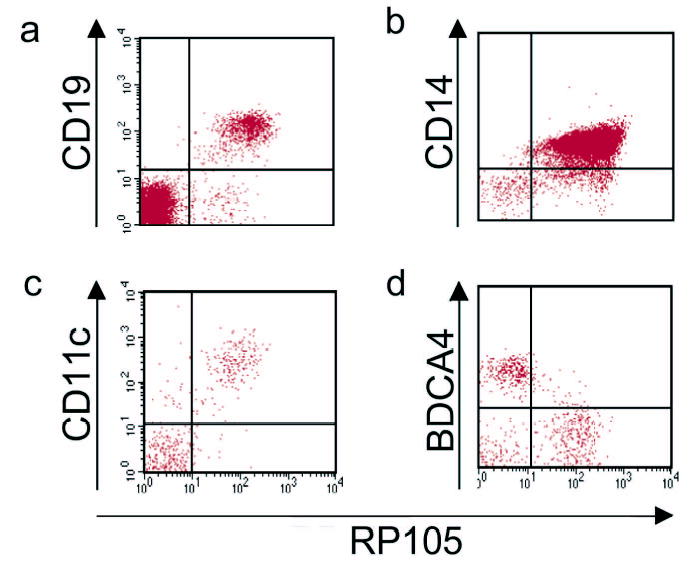
RP105 expression by human peripheral blood leukocytes. Flow cytometric analysis of PBMC from healthy human donors. (a) B cells; (b) monocytes; (c) myeloid DC; (d) plasmacytoid DC. Myeloid DC were identified as lineage negative (CD3−,CD14−,CD19−,CD20−,CD56−), HLA-DR+, CD11c+. Plasmacytoid DC were identified as lineage negative, HLA-DR+, CD11c−, BDCA-4+. Data are representative of an experimental n > 15 for monocytes; n = 3 for the other cell types.
Similarly, FACS analysis revealed that mice express RP105 on resident peritoneal macrophages, splenic DC subsets, as well as bone marrow-derived DC (Fig. 2). To confirm the specificity of the mAb used in this work, RP105-deficient mice were examined alongside wild-type mice in these studies; no RP105 staining was found in B cells, macrophages or DCs from RP105 knockout mice (data not shown). Further, to rule out any potential artifacts (from, e.g., a soluble isoform of RP105; yet to be described), RT-PCR analysis of bone marrow-derived DC was performed to confirm endogenous RP105 expression by such cells (data not shown). Unlike those found in human peripheral blood, essentially all of the splenic plasmacytoid DC from flt3L-treated mice expressed both RP105 and TLR4 (Fig 2e; and data not shown). Thus, RP105 is not B cell-specific, and its expression directly mirrors that of TLR4 on human and murine macrophages and DC.
Figure 2.
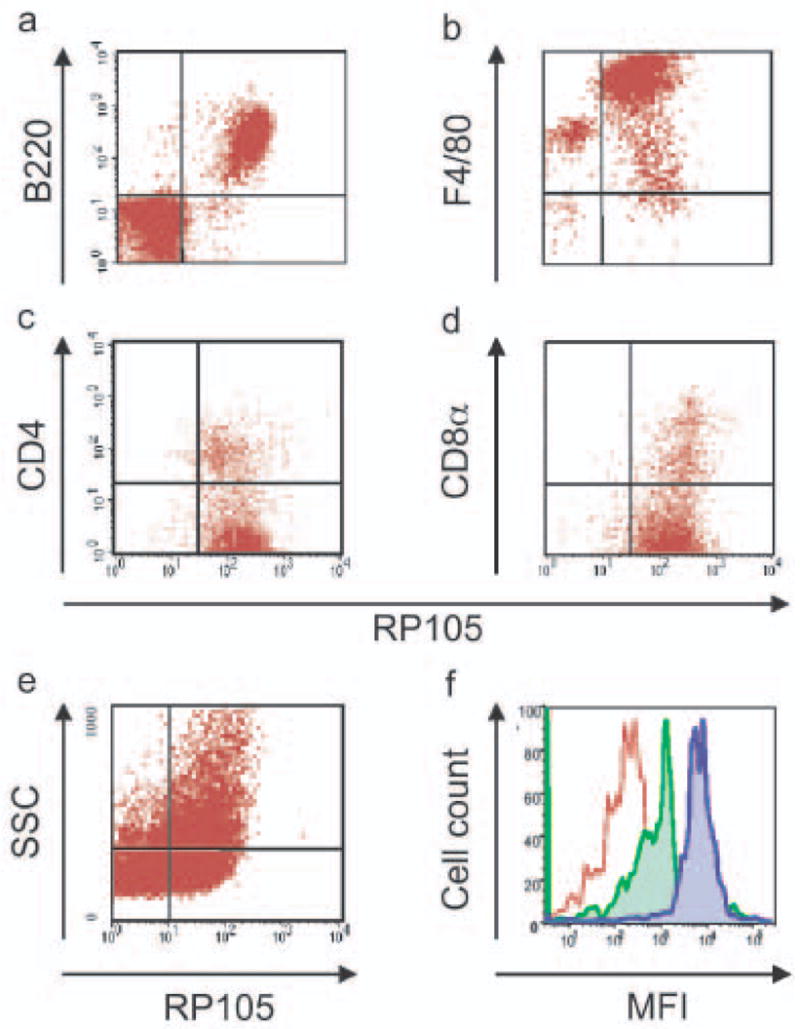
RP105 expression by murine leukocytes. Flow cytometric analysis of cell populations. (a) splenic B cells (representative of an experimental n > 50). (b) resident peritoneal macrophages (n = 5). (c) splenic CD11c+CD11b+ CD4− and CD11c+CD11b+CD4+ DC (n = 6). (d) splenic CD11c+CD11b−CD8α+DC (n = 6). (e) bone marrow-derived DC (n = 6). (f) splenic plasmacytoid DC (CD19+B220+CD11c+GR-1+) [n = 5]; red: isotype control, green: TLR4, blue: RP105. Analysis of splenic DC subsets was performed after 10 d of in vivo treatment with flt3L.
RP105 suppresses TLR4 signaling in HEK293 cells
HEK293 cells lack expression of endogenous TLR2, TLR4, TLR9, MD-2 and CD1423, as well as RP105 and MD-1 (data not shown). Their TLR signaling machinery is fully functional, however23. As a result, HEK293 cells are used extensively for the in vitro analysis of TLR function23,24. Given the homology of RP105 to TLR4, we first examined whether RP105–MD-1 could act as a signaling receptor for LPS in HEK293 cells. HEK293 cells that stably express CD14 were transiently transfected with cDNA encoding MD-1, MD-2, RP105, and/or TLR4. Although TLR4–MD-2 expression conferred LPS-sensitivity with resultant LPS-driven IL-8 production, RP105–MD-1 expression did not (Fig. 3a). It should be noted that these data are consistent with data generated previously using Ba/F3 cells: even in B cell lines, no direct role for RP105 as a signaling receptor for LPS was shown25. We confirmed that in vitro overexpression of TLR4–MD-2 in HEK293 cells led to an increase in baseline IL-8 production in the absence of stimulation (Fig. 3a).
Figure 3.
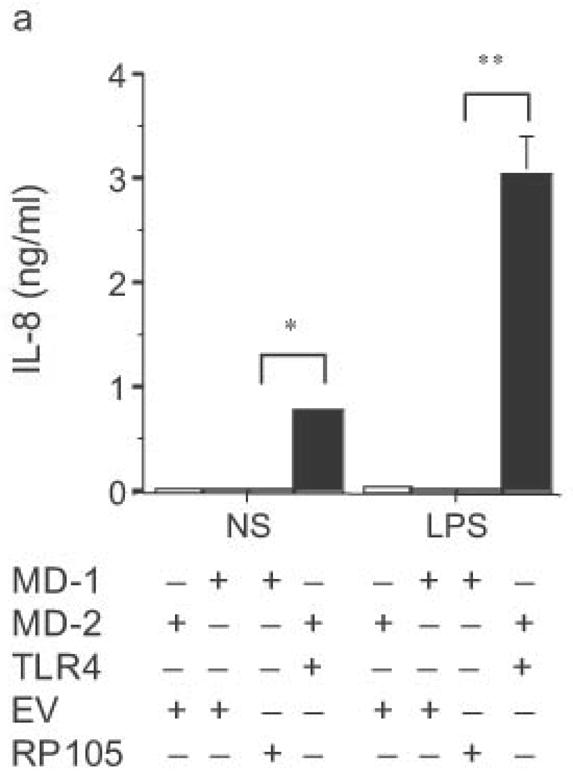
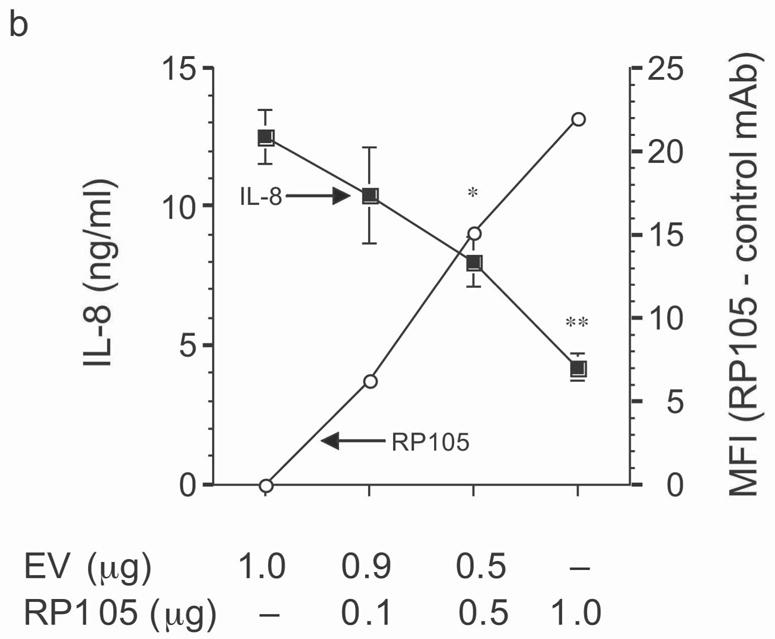
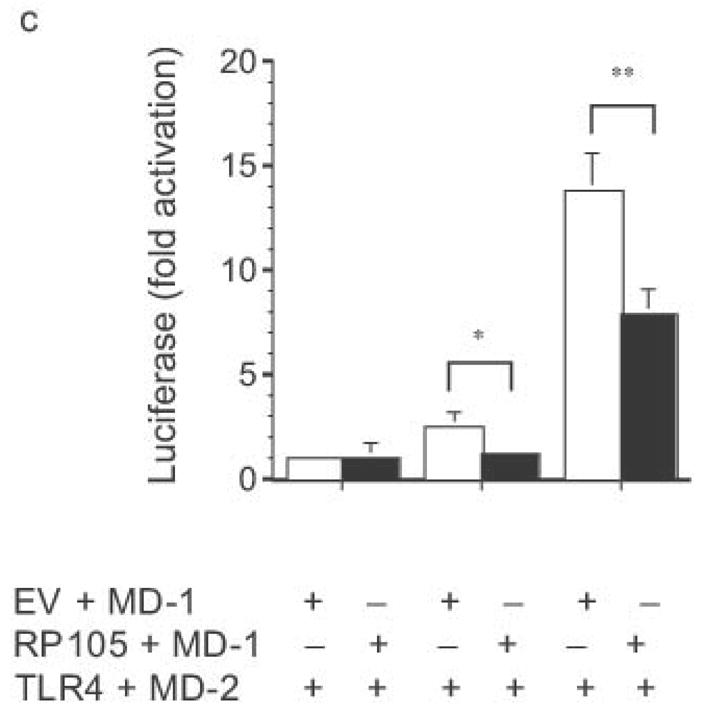
Dose-dependent suppression of TLR4 signaling in HEK293 cells by RP105 expression. (a) HEK293 cells stably expressing CD14 were transiently transfected with cDNA encoding MD-1, MD-2, TLR4, empty vector control cDNA (EV) and/or RP105. Cells were subsequently stimulated with purified E. coli K235 LPS (10 ng/ml). (b) HEK293 cells stably expressing CD14 and TLR4 were transiently transfected with MD-1 and MD-2 along with the indicated concentrations of RP105 and/or EV cDNA, and secondarily stimulated with LPS (10 ng/ml). (c) HEK293 cells stably expressing MD-2 and TLR4 were transiently co-transfected with an NF-κB-firefly luciferase reporter plasmid, a TK-renilla luciferase reporter plasmid and MD-1, along with EV (open bars) or RP105 (filled bars). Cells were stimulated with the indicated concentrations of LPS. * p < 0.03, **p < 0.004, compared with RP105-deficient cells. Means +/− SE of triplicate cultures in a single experiment are depicted, representative of an experimental n = 4 (a and b); n = 2 (c). NS: no stimulation.
The effects of RP105–MD-1 expression on LPS-driven TLR4 signaling were also examined. Notably, RP105 expression inhibited TLR4-driven IL-8 production by HEK293 cells in a dose-dependent manner (Fig. 3b). Further, RP105-mediated inhibition of LPS-driven IL-8 production was associated with inhibition of LPS-driven NF-κB transactivation (Fig. 3c), suggesting that modulation of IL-8 production by RP105 was upstream of NF-κB activation.
The specificity of RP105-mediated suppression of proinflammatory signaling was subsequently examined. RP105 did not inhibit IL-1- or TLR2-driven IL-8 production (Fig. 4). Indeed, RP105 overexpression led to variable augmentation of TLR2-mediated IL-8 production in some experiments (shown in Fig. 4b). Thus, RP105-mediated suppression exhibits specificity among the TLR/IL-1R family of receptors.
Figure 4.
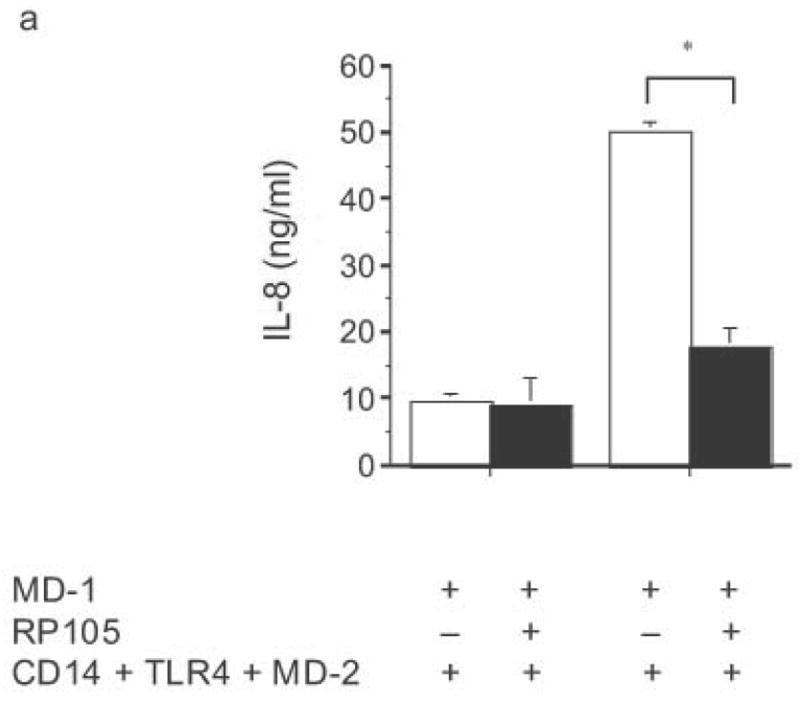
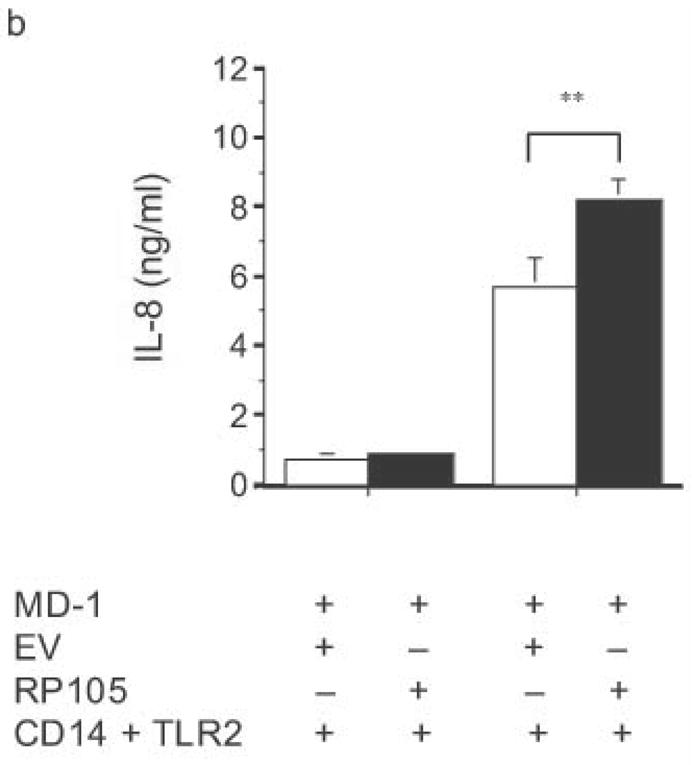
Specificity of RP105-mediated suppression. (a) HEK293 cells stably expressing CD14 and TLR4 (open bars) or CD14, TLR4 and RP105 (filled bars) were transiently transfected with MD-1 and MD-2, and subsequently stimulated with purified E. coli K235 LPS (10 ng/ml) or IL-1β (100 ng/ml). (b) HEK293 cells stably expressing CD14 and TLR2 were transiently transfected with MD-1 and EV (open bars) or MD-1 and RP105 (filled bars) and subsequently stimulated with Zymosan A (10 μg/ml) or IL- 1β (100 ng/ml). * p < 0.0001, ** p = 0.05, compared with RP105-deficient cells. Means +/− SE of triplicate cultures in a single experiment are depicted, representative of an experimental n = 2–4.
The necessity for MD-1 expression in this system was formally examined. In the absence of MD-1 co-expression, RP105 did not inhibit TLR4 signaling (Supplementary Fig. 5a online). Consistent with the previous observation that MD-1 expression is required for surface expression of RP10513, RP105 failed to be detected on cells in the absence of MD-1 co-expression (Supplementary Fig. 5b online). Thus, RP105–MD-1 is a specific inhibitor of TLR4 signaling in HEK293 cells.
Suppression by the RP105 extracellular domain
Antibodies to RP105 can induce signaling events and proliferation in B cells, although there is no evidence that RP105 signals directly6,10,25–27. Use of such antibodies in human primary monocyte/macrophages did not induce a measurable signal (cytokine production; data not shown), an observation that suggested that the mechanism of suppressive action of RP105 might be independent of any putative signaling through its intracellular tail. Consistent with this, RP105 did not inhibit NF-κB transactivation induced through the overexpression of TLR4 signaling molecules (Supplementary Fig. 3 online)28. A soluble mutant of RP105, lacking the transmembrane and intracellular domains, was thus constructed. Initial analysis by immunoprecipitation revealed that the RP105 extracellular domain protein was secreted into the medium of transfected cells (data not shown). Mechanistic analysis revealed that co-expression of MD-1 and the extracellular portion of RP105 is sufficient to effect RP105-mediated suppression of TLR4 signaling (Fig. 5a). Identical to findings with the full-length construct, suppression of signaling by the extracellular domain of RP105 had considerable specificity, failing to inhibit TLR2 or IL-1R signaling in HEK293 cells (Fig. 5b and 5c). Thus, inhibition of TLR4 signaling is mediated by the extracellular domain of RP105.
Figure 5.
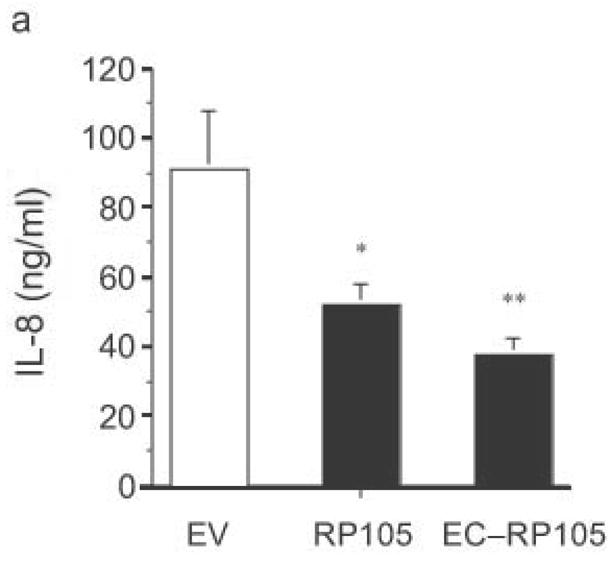
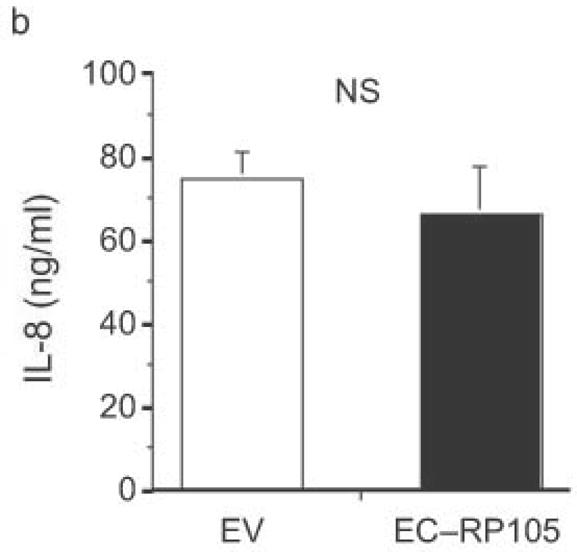
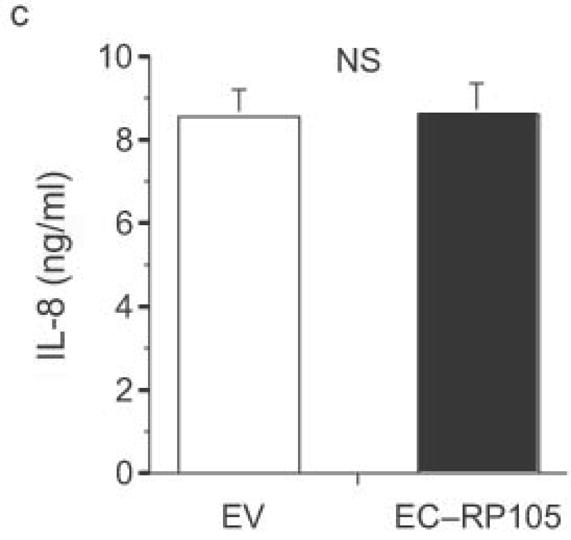
The extracellular domain of RP105 is sufficient to effect suppression of TLR4 signaling. (a) HEK293 cells stably expressing CD14 and TLR4 were transiently transfected with MD-2 and MD-1, along with EV, RP105 or the extracellular domain of RP105 (EC-RP105), as indicated. Cells were subsequently stimulated with purified E. coli K235 LPS (10 ng/ml). Means +/− SE of triplicate cultures in a single experiment are depicted, representative of an experimental n = 4 *p < 0.02, **p < 0.002, compared with RP105-deficient cells. (b, c) HEK293 cells stably expressing CD14 and TLR2 were transiently transfected with MD-2 and MD-1, along with EV or EC-RP105, as indicated. Cells were subsequently stimulated with (b) Zymosan A (10 μg/ml) or (c) IL-1β (100 ng/ml) as noted. Means +/− SE of replicate cultures (n =9) are depicted. NS, not significant.
Direct Interaction of RP105–MD-1 withTLR4–MD-2
The extracellular localization of the inhibitory effects of the RP105 suggested a mechanistic model involving direct interactions between the RP105–MD-1 and TLR4–MD-2 complexes. Co-immunoprecipitation techniques were used to probe the association of these complexes. These complexes co-immunoprecipitate bi-directionally, demonstrating physical association between TLR4–MD-2 and RP105–MD-1 (Fig. 6). As MD-1 failed to associate with TLR4 in the absence of MD-2 (data not shown), the ability of MD-1 to associate with TLR4–MD-2 (Fig. 6, lanes 2 and 3) suggested the possibility that MD-1 and MD-2 interact directly. Indeed, immunoprecipitation analysis revealed the presence of such interactions (Supplementary Fig. 4 online).
Figure 6.
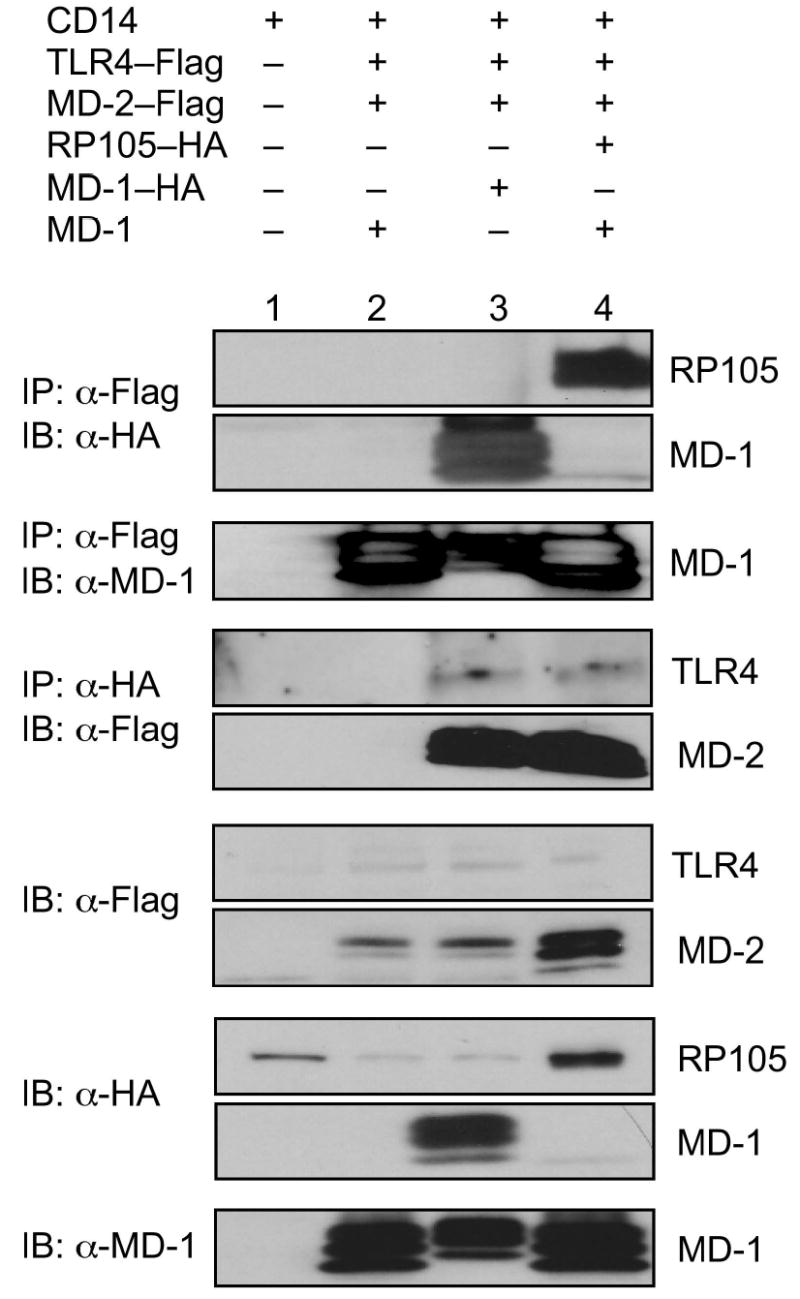
RP105–MD-1 interacts directly with TLR4–MD-2. HEK293 cells stably expressing CD14 (lane 1) or CD14 and FLAG-tagged TLR4 (lanes 2–4) were transiently transfected with MD-2, RP105 and/or MD-1 constructs, as indicated. Lysates were immunoprecipitated with antibodies to FLAG or HA, and the association between RP105–MD-1 and TLR4–MD-2 was examined by immunoblotting using antibodies to FLAG, HA and MD-1, as indicated. The expression of RP105, MD-1, TLR4, and MD-2 in cell lysates was characterized by immunoblotting using anti-FLAG, anti-HA and anti-MD-1 antibodies, as indicated. Data are from a single experiment, representative of an n = 3.
Biotin-labeled LPS has been used to demonstrate that LPS binds directly to MD-2, leading to the association of LPS/MD-2 complexes with TLR4, and to TLR4 signaling29. In replication of these data, incubation of biotinylated LPS with TLR4-expressing HEK293 cells allowed for precipitation of TLR4 (and MD-2) only in the presence of MD-2 expression (Fig. 7a). Of note, co-expression of RP105–MD-1 in this system inhibited LPS–TLR4–MD-2 complex formation (Fig. 7a), providing direct evidence that RP105–MD-1 inhibits LPS signaling complex formation. The fact that, consistent with previously reported data30, no direct interactions between LPS and RP105–MD-1 were shown in this system (Fig. 7b) indicates that RP105–MD-1-mediated interference with LPS signaling complex formation is not due to RP105–MD-1 acting as a molecular sink for LPS. Thus, RP105–MD-1 interacts directly with the TLR4 signaling complex, inhibiting its ability to bind microbial ligand.
Figure 7.
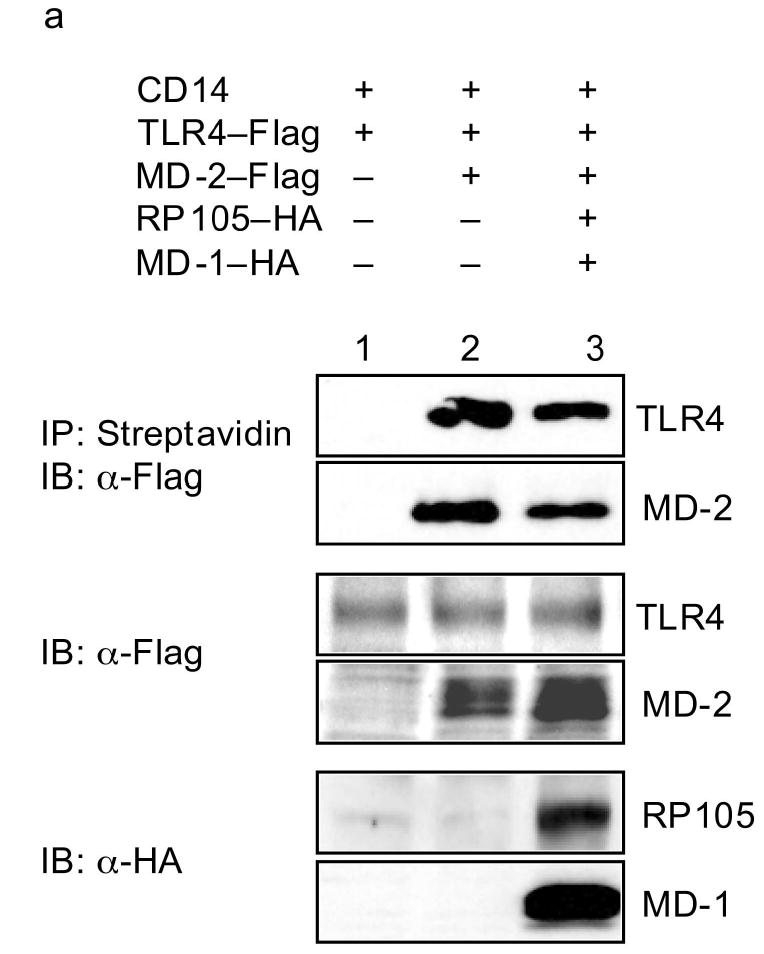
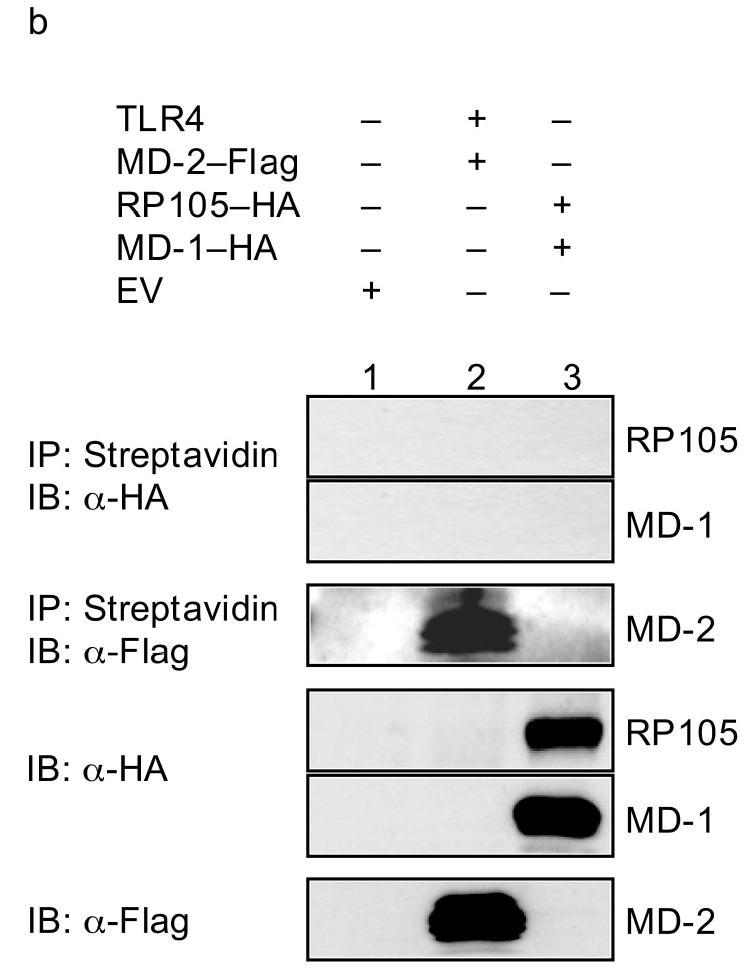
RP105–MD-1 inhibits LPS binding to TLR4–MD-2. HEK293 cells stably expressing CD14 and FLAG-tagged TLR4 (a), or HEK293FT cells (b), were transiently transfected with empty vector (EV), MD-2, RP105, MD-1 and/or TLR4 constructs, as indicated. After incubation with biotinylated LPS, cells were lysed, lysates were immunoprecipitated with streptavidin-conjugated Sepharose A beads, and the association of LPS with (a) TLR4–MD-2 or (b) RP105–MD-1 was examined by immunoblotting using anti-FLAG or anti-HA antibodies, respectively. The expression of RP105, MD-1, TLR4, and MD-2 in cell lysates was characterized by immunoblotting using anti-FLAG and anti-HA antibodies as indicated. Data are from a single experiment, representative of an n = 2 (a), or 1 (b).
RP105 is a physiological regulator of cytokine production
Although expression studies in cell lines have considerable utility, they also have obvious drawbacks; principally, that overexpression may drive non-physiological interactions and processes. To confirm and extend our findings in transfected cell lines, we thus examined RP105-deficient mice, generated by _targeted disruption of exon 3 of RP10525 and backcrossed for more than 10 generations onto the C57BL/6 background. Bone marrow-derived DC were generated by standard techniques31 from age-matched RP105-deficient mice and wild-type littermate controls. DC generated from RP105-deficient mice appeared to be phenotypically normal. In particular, FACS analysis revealed no differences between DC generated from RP105-sufficient and -deficient mice in baseline expression of CD14, TLR4, CD83, CD11c, CD80, CD86 or major histocompatibility complex (MHC) class II (data not shown). Further, RT-PCR analysis revealed no differences in expression of any of the TLRs, MD-1, MD-2, or the TLR pathway inhibitors IRAK-M, single immunoglobulin IL-1R-related protein (SIGIRR), ST2, Tollip or SOCS1 in such DC (Supplementary Fig. 5 online; and data not shown).
DC from RP105-deficient mice produced significantly higher concentrations of proinflammatory cytokines after stimulation with purified E. coli LPS than did DC from wild-type controls (Fig. 8). Thus, RP105-mediated suppression of TLR4 signaling is not merely an overexpression artifact in cell lines. Increased TLR4-driven cytokine production occurred for tumor necrosis factor (TNF), IL-12p70 and IL-6 (Fig. 8a–c), cytokines whose TLR4-driven production occurs through the signaling adaptor proteins Mal–MyD88, as well as for IP-10 (Fig. 8d), a chemokine whose TLR4-driven production occurs through a complementary signaling pathway mediated by the TRIF–TRAM adaptor proteins. RP105-mediated modulation of cytokine production by DC also exhibited specificity; for example, CpG (TLR9)-driven cytokine production was unaltered in DC generated from RP105-deficient mice (Fig. 8e). RP105-mediated modulation of proinflammatory cytokine production was also observed in macrophages; resident peritoneal macrophages from RP105-deficient mice produced significantly higher concentrations of TNF after stimulation with purified E. coli LPS than did macrophages from wild-type controls (Supplementary Fig. 6 online)
Figure 8.
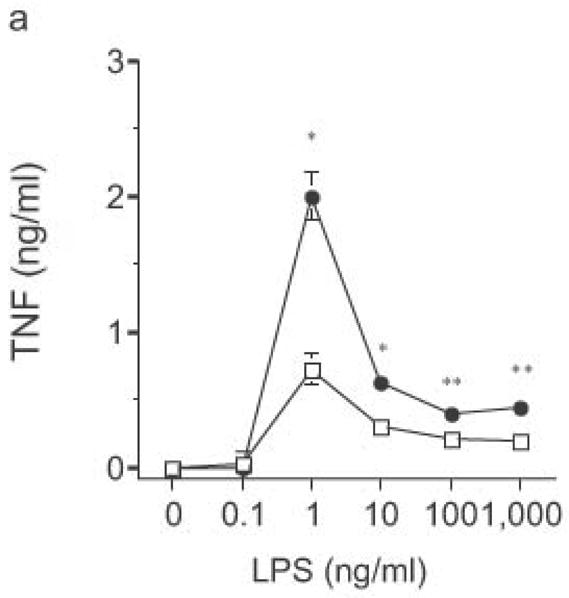
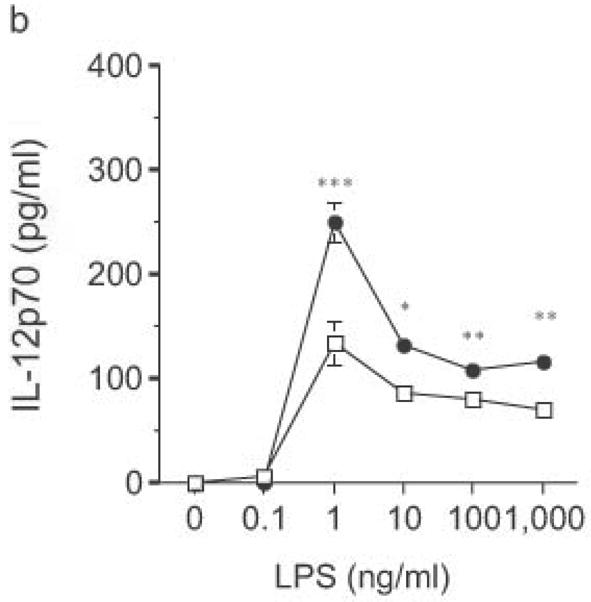
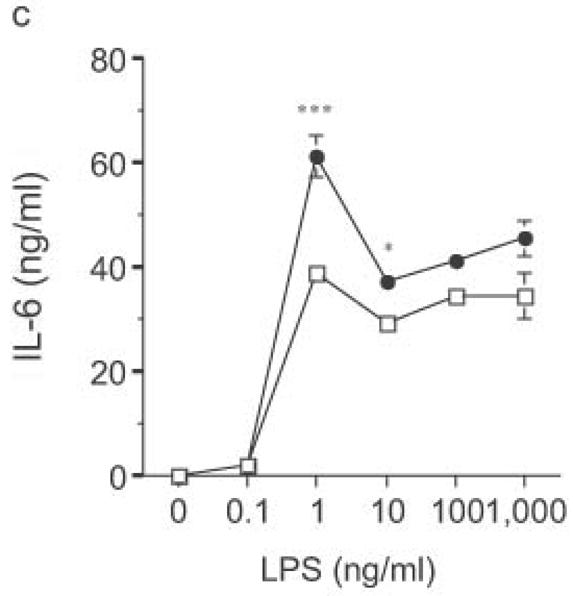
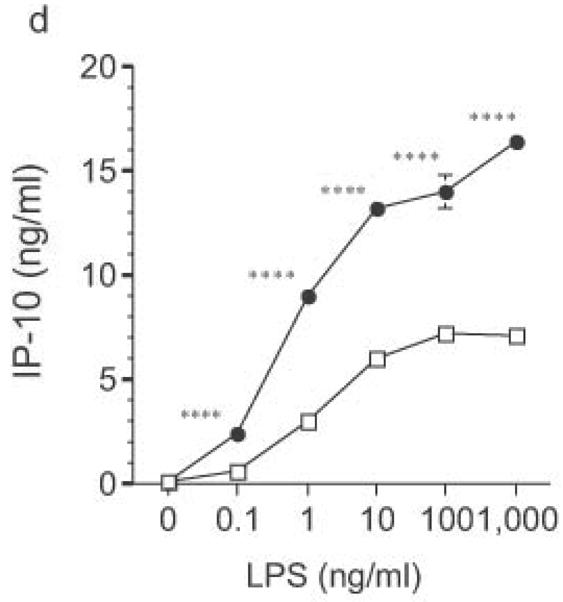
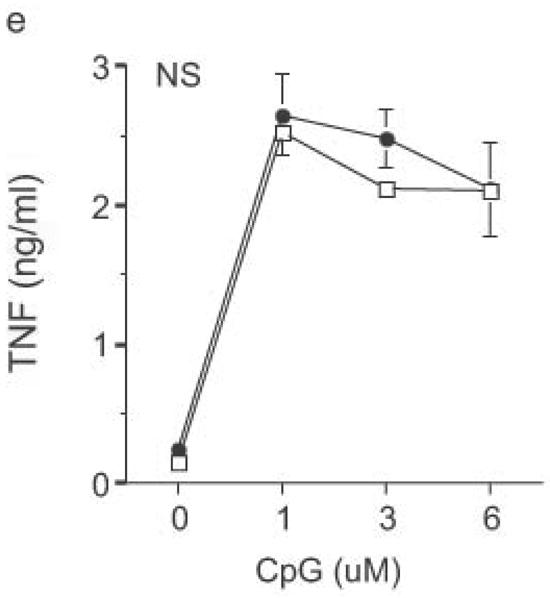
Altered TLR4-induced cytokine production by dendritic cells from RP105-deficient mice. Bone marrow-derived DC from wild-type (open symbols) or RP105-deficient (filled symbols) mice were stimulated with purified E. coli K235 LPS (a–d) or CpG DNA (e). Supernatants were harvested after 24 h. (a) TNF. (b) IL-12p70. (c) IL-6. (d) IP-10. (e) TNF. * p < 0.05, ** p < 0.001, *** p < 0.01, **** p < 0.0001. Means + SE of triplicate cultures in a single experiment, representative of an n = 8 (a); 4 (b, c); 7 (d); or 3 (e).
The biphasic dose-response for TNF, IL-12p70 and IL-6 production observed in DC from both wild-type and knockout mice (Fig. 8) was examined further. Flow cytometric analysis revealed that the reduced cytokine production at higher doses of LPS was not associated with greater DC apoptosis (data not shown). Commercial LPS preparations are typically contaminated with lipopeptide ligands for TLR232. Such preparations of LPS continue to drive increasing TNF production at higher doses (Fig. 9a), the very doses at which such preparations drive IL-8 production by HEK293 cells transfected with TLR2 (data not shown). Notably, stimulation of DC with these higher doses of commercial LPS preparations led to similar amounts of TNF production by DC from RP105-deficient and wild-type mice (Fig. 9a). Indeed, stimulation with combinations of purified TLR4 and TLR2 agonists revealed that TLR2 agonists are able to effect functional reversal of RP105-mediated inhibition of TLR4 signaling (Fig. 9b). These data indicate that, as with many biological agonistic responses, the response to a pure TLR4 ligand is biphasic, something modifiable by secondary agonists; and that signaling through other TLRs can overcome RP105-mediated modulation of TLR4 signaling.
Figure 9.
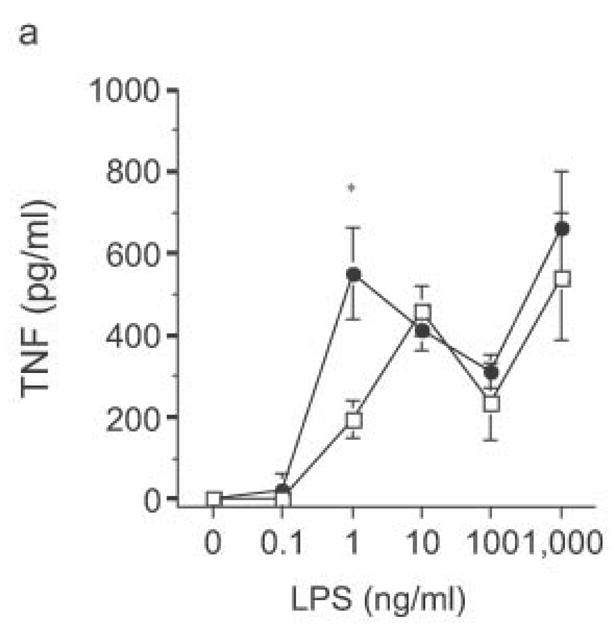
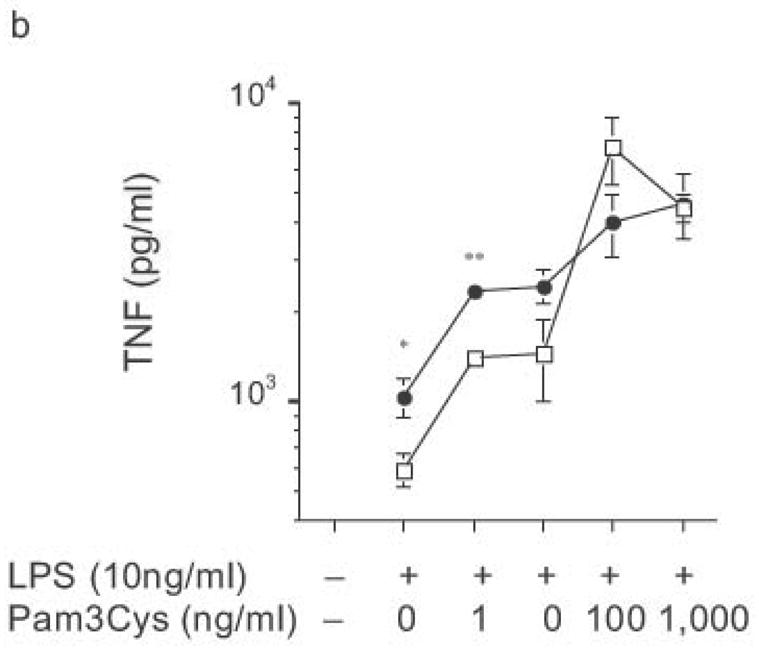
Ability of heterologous TLR signaling to overcome RP105-mediated inhibition of TLR4 signaling. Bone marrow-derived DC from wild-type (open symbols) or RP105-deficient (filled symbols) mice were stimulated with: (a) commercial E. coli K235 LPS; or (b) purified E. coli K235 LPS plus the TLR2 agonist, Pam 3Cys. * p < 0.05, ** p < 0.001. Means + SE of triplicate cultures in a single experiment; representative of an experimental n = 4.
Other modulators of TLR signaling have, themselves, been found to be regulated by TLR signaling33. Quantitative RT-PCR was used to examine RP105 expression after LPS stimulation of murine DC. TLR4 signaling induced transient upregulation of RP105 mRNA expression, along with the previously-described downregulation of TLR4 mRNA expression34, something unaltered by a lack of RP105 expression in such cells (Supplementary Fig. 7a online). Despite these findings, preliminary experiments have not revealed a role for RP105 in endotoxin tolerance (data not shown). Of interest, LPS stimulation of human DC induced coordinate down-regulation of both TLR4 and RP105 (Supplementary Fig. 7b online), adding to growing findings of divergent regulation of TLRs in humans and mice35.
Finally, LPS-driven in vivo responses were compared in RP105-deficient and wild- type mice. Notably, RP105-deficient mice produced significantly more TNF in response to low dose intraperitoneal challenge with E. coli LPS (Fig. 10a). Further, high dose LPS challenge led to significant acceleration and amplification of endotoxicity in RP105-deficient mice (Fig. 10b). Thus, RP105 is a physiological regulator of responses to LPS, in primary myeloid cells in vitro as well as in vivo.
Figure 10.
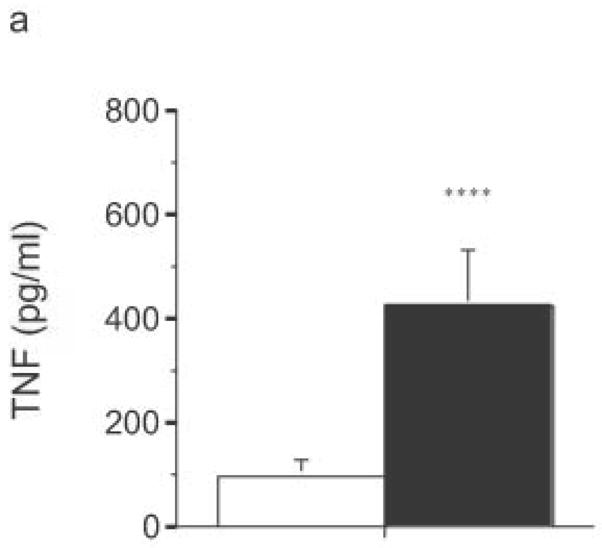
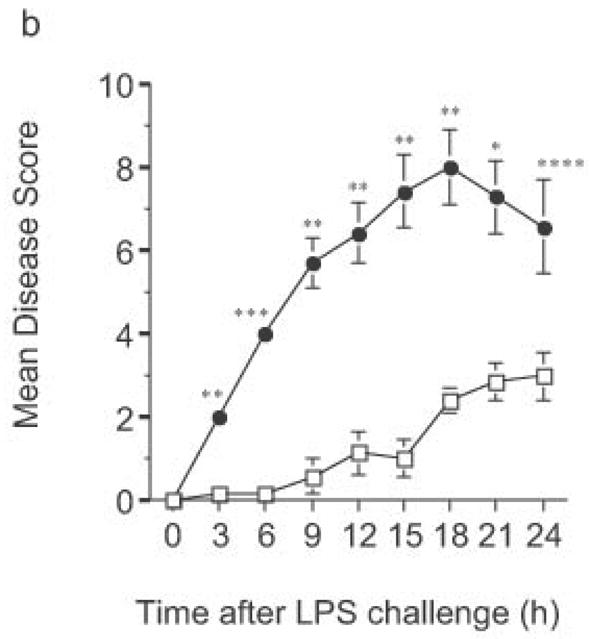
Exaggerated in vivo responses to LPS in RP105−/− mice. (a) Wild-type mice (n = 17) (open bars) or RP105-deficient mice (n = 18) (filled bars) were challenged intraperitoneally with 25 μg of purified E. coli K235 LPS. Serum was harvested 60 min later. (b) Wild type (open symbols) or RP105−/− mice (filled symbols) were challenged with 8 mg/kg of purified E. coli K235 LPS (n = 7/group). Data represent means + SE. *p < 0.0005, ** p < 0.00005, *** p < 10−8, **** p < 0.01.
DISCUSSION
The studies reported here show that RP105, a TLR4 homolog whose expression mirrors that of TLR4 on antigen presenting cells, is a negative regulator of TLR4 signaling. RP105 specifically inhibited TLR4 signaling when co-expressed in HEK293 cells. Furthermore, RP105 was a physiological regulator of endotoxin-driven TLR4 signaling in DC, as well as of endotoxicity in vivo.
Although the activation of proinflammatory responses through TLRs is critical for host defense, excessive or inappropriate inflammation can itself be maladaptive. RP105 joins a growing list of molecules that can inhibit TLR signaling. Regulation of TLR expression provides one point of control2, as does the complex phenomenon of endotoxin tolerance36. Several direct, negative regulators of TLR signaling have also been found, including SIGIRR33, IRAK-M37, MyD88s38, Tollip39, ST228, NOD240, and Triad3A41. Among this list, RP105 stands out for its apparent specificity for inhibition of TLR4 signaling.
This specificity, together with the structural homologies between RP105 and TLR4 suggested an attractive hypothesis for the mechanism of negative regulation of TLR4 signaling by RP105: interference with TLR4 signaling via direct interactions of RP105–MD-1 with the TLR4–MD-2 cell surface signaling complex. Such a model received theoretical support from the apparent lack of anti-RP105-mediated signaling in myeloid cells, the fact that RP105-mediated inhibition of TLR4 signaling suppressed both signaling pathways (Mal–MyD88 and TRIF–TRAM) known to be downstream of TLR4, and the fact that RP105–MD-1 was not able to inhibit NF-κB transactivation driven by overexpression of TLR4 signaling intermediates. Such findings suggested an upstream, likely extracellular, locus for RP105-mediated inhibition. In fact, structure and function analysis of RP105 was consistent with this: the extracellular domain of RP105 was sufficient to effect specific, negative regulation of TLR4 signaling. Finally, co-immunoprecipitation experiments revealed direct physical association between RP105–MD-1 and TLR4–MD-2, an association that inhibits LPS binding to this signaling complex. The exact stoichiometry of RP105–MD-1–MD-2–TLR4 interactions remains to be defined.
The current data suggest that RP105 regulates LPS responses differently in myeloid cells and B cells6,13,25–27. The fact that antibodies to RP105 drive B cell proliferation was integral to the discovery of this molecule6. Anti-RP105-mediated B cell proliferation appears to involve signaling through the Lyn–CD19–Vav complex, along with protein kinase CßI/II, and the Erk2-specific MAP kinase kinase, MEK26,27. However, RP105 does not appear to signal directly in B cells. As noted, treatment with antibodies to RP105 did not activate human monocyte/macrophages, failing, for example, to drive proinflammatory cytokine production. As for LPS-driven B cell responses, while LPS-induced murine B cell proliferation is strictly dependent upon TLR4, B cells from RP105 knockout mice have reduced LPS-driven proliferative responses as well as diminished humoral immune responses to LPS25. It is reasonable to suspect that the dichotomous effects of RP105 on TLR4 signaling in B cells and myeloid cells are due to differential interactions with cell surface molecular partners in these different cell types. Widely differing expression of TLR4 (greater on myeloid cells; barely detectable on B cells25,42) may provide the key. TLR4 multimerization appears to be necessary for signaling29. Although TLR4–MD-2 would be expected to have a higher affinity for homodimerization than for heterodimerization with RP105–MD-1, data presented here suggest the likelihood that both homo- and heterodimers can multimerize with further TLR4–MD-2 complexes. This suggests that when TLR4–MD-2 is highly expressed (e.g., on myeloid cells), lower affinity heterodimeric interactions inhibit TLR4 multimerization and signaling. In contrast, when TLR4 is limiting (e.g., on B cells), such heterodimeric interactions serve to facilitate further TLR4 recruitment and signaling. The fact that RP105 appears to promote B cell activation, while inhibiting DC activation, suggests the possibility that, overall, immunoregulation by RP105 leads to augmentation of humoral immunity (through effects on B cells) along with concomitant inhibition of cellular immune responses (through effects on DCs and macrophages).
The apparent TLR4-specificity of RP105 raises another issue. Concurrent stimulation of TLR4 and other TLRs is able to reverse RP105-mediated inhibition of TLR4 signaling. Clearly, most microbes that express ligands that stimulate TLR4 signaling also express ligands that stimulate signaling through other TLRs. TLR4 is notable for leading to a more robust and complex response, both in terms of signaling and in terms of subsequent gene expression, than the other TLRs43. TLR4 may also stand out among the TLRs for its ability to signal in response to a variety of endogenous “danger signals”2. The ability of TLR4 to recognize endogenous heat shock proteins and extracellular matrix components unmasked by tissue injury suggests the possibility that RP105 may well be of special importance in down-modulation of the injurious inflammatory responses observed in the systemic inflammatory response syndrome and in autoimmune diseases. In this regard, it is notable that RP105-deficient mice spontaneously develop splenomegaly with age (data not shown).
Recent renewed mechanistic interest in the innate immune system should lead to the development of novel therapeutic approaches to a variety of infectious and autoimmune diseases. The identification here of RP105 as a physiological, endogenous inhibitor of TLR4 signaling would seem to hold similar translational promise.
METHODS
Reagents
Zymosan A and commercial LPS were from Sigma. Highly purified E. coli LPS was generated as described44. Pam3Cys was from EMC Microcollections. CpG (5′ TCCATGACGTTCCTGATGCT 3′) was from TriLink Biotechnologies. Recombinant cytokines were from Peprotech. All reagents contacting cultured cells were endotoxin-free to the limits of detection of the Limulus amebocyte lysate assay (Bio-Whittaker) at the concentrations employed, unless otherwise stated.
Cloning and Expression Constructs
Human RP105 and MD-1 were cloned from primary human monocytes by RT-PCR. RP105 deletion mutants and epitope-tagged RP105 and MD-1 constructs, were generated by PCR. The HSV thymidine kinase promoter-driven Renilla luciferase reporter plasmid (pRL-TK) was from Promega; the NF-κB-dependent ELAM-1 promoter-driven Firefly Luciferase plasmid (P-ELAM) has been described23. cDNA for TLR4 was from R. Medzhitov (Yale Univ.); that for MD-2 was from K. Miyake (Univ. of Tokyo). Plasmids encoding Mal, MyD88, IRAK-1 and TRIF were from K. Fitzgerald (Univ. of Massachusetts). The IκB super-repressor expression plasmid was from R. Hay (Univ. of St. Andrews).
Cell lines
HEK293 cell lines stably expressing CD14, CD14–TLR4, CD14–TLR2, and TLR4–MD-2 have been described23,45. HEK293 cells stably expressing RP105–CD14–TLR4 were generated from CD14–TLR4 HEK293 cells. Transient transfections were performed using PolyFect (Qiagen). Construct expression was quantified by flow cytometry. All cell lines were Mycoplasma-free.
In vitro stimulation
24 h after transient transfection, HEK293 cells were washed and stimulated for an additional 24 h. Cell-free supernatants were collected and assayed for IL-8 production (ELISA; Pharmingen). To assay NF-κB-driven luciferase expression23, cells were co-transfected with pELAM (0.5 μg) and pRL-TK (0.1 μg) plasmids, stimulated for 5 h, lysed, and luciferase activity was quantified on a Monolight 3010 luminometer (Pharmingen).
Immunoprecipitation and Western Blotting
48 h after transfection, HEK293 cells were washed and lysed. For immunoprecipitation (IP) with HA antibody (Y-11; Santa Cruz), cell lysates were incubated with antibody, followed by incubation with protein-G Sepharose beads (Zymed). For IP with FLAG mAb, cell lysates were incubated with anti-FLAG (M2) affinity gel (Sigma). Immunoprecipitates and lysates were resolved by SDS-PAGE, and electrotransferred onto Immobilon-P PVDF membranes (Millipore). After blocking, proteins were revealed by probing with unconjugated M2 mAb (FLAG; Sigma); Y-11 rabbit polycolonal Ab (HA; Santa Cruz); Ab-1 rabbit polyclonal Ab (MD-1;Oncogene); followed by horseradish peroxidase-conjugated secondary mAb (Santa Cruz). LPS labeling, and IP with biotinylated LPS, was performed as described29.
Mice
RP105-deficient mice, on a C57BL/6 background (>10 generations) have been described25. Mice were genotyped by PCR, as well as phenotyped for RP105 surface expression on peripheral blood B cells 25 (Supplementary Fig. 8 online). In vivo DC amplification was performed through daily i.p. administration of 10 μg of flt3 ligand (provided by Immunex/Amgen) for 10 d. Mice were housed in a specific pathogen-free facility in high-efficiency particulate-filtered laminar flow hoods with free access to food and water. Animal care was provided in accordance with National Institutes of Health guidelines. These studies were approved by the CCHMC IACUC.
Ex vivo stimulation
Bone marrow-derived DC (BMDDC), generated using standard protocols31, were stimulated for 24 h. Cell-free supernatants were collected and assayed by ELISA for TNF (Becton Dickinson), IL-12p70, IL-6 and IP-10 (R&D Systems). RP105, TLR4, IRAK-M, SIGIRR, Tollip and ST2 mRNA expression was analyzed by quantitative RT-PCR in BMDDC prior to and following stimulation with 10 ng of purified E. coli K235 LPS. PCR (LightCycler; Roche) used the following primers: (1) RP105: AGTCTCCTCCCCATCTTGTCC, GATAGCGTCACATCGGAGAGC; (2) TLR4: CATCCAGGAAGGCTTCCACA, GGCGATACAATTCCACCTGC; (3) IRAK-M: GAGAATTGCTCTGGTCCTGGG, CACCTCAAGTGGGAAGCTGG; (4) SIGIRR: GGCCCCTAATTTCCTTTCCC, CATGGAGGCTGAAGTGGCTT; (5) Tollip: TGGACCCACATCACCATCC, GTTGGCATCAGGACCACAGG; (6) ST2: GGCTCTCACTTCTTGGCTGATG, GCCAGACAGTCATATTCCAGGG; (7) β-actin: GGCCCAGAGCAAGAGAGGTA, GGTTGGCCTTAGGGTTCAGG.
In vivo stimulation
Six-8 week old mice were challenged i.p. with 25 μg of purified E. coli K235 LPS. One h later, serum was collected. TNF concentrations were assayed by ELISA. For endotoxicity studies, mice were challenged i.p. with 8 mg/kg of purified E. coli K235 LPS. Clinical endotoxicity was scored using a quantitative scale integrating 4 cardinal signs of systemic toxicity46 (piloerection, ocular discharge, lethargy, diarrhea; each scored from 0–3) by an investigator blinded to mouse genotype.
Human leukocytes
PBMC were isolated from healthy volunteers by Ficoll/Hypaque sedimentation. Monocytes were purified by countercurrent elutriation from PBMC isolated by leukapheresis47. Purified monocytes were differentiated into DC using standard techniques using IL-4 and GM-CSF. RP105 and TLR4 mRNA expression was analyzed by qRT-PCR in human monocyte-derived DC using the following primers: (1) RP105: TCAGTGCTGCCAATTTCCC, CTGCAGCAGTCAGAAGCCTCT; (2) TLR4: AGTTTCCTGCAATGGATCAAGG, GGACCGACACACCAATGATG; (3) Ubiquitin: CACTTGGTCCTGCGCTTGA, CAATTGGGAATGCAACAACTTTAT. These studies were approved by the CCHMC and University of Cincinnati College of Medicine IRBs.
Flow Cytometry
Surface and intracellular protein expression by HEK293 cells was quantified by FACS techniques described previously48. Flow cytometric analysis was performed using a FACSCalibur along with CellQuest Software (Becton Dickinson). At least 20,000 events were acquired for each data point.
Phylogenetic and domain analysis
Sequence alignments were performed using the ClustalW49 and Pileup50 programs. MEMSAT, SOSUI and SABLE software were used to predict transmembrane boundaries.
Statistical analysis
Data analysis was performed using the unpaired Student t test, or ANOVA with post-hoc analysis by Student t test or Fisher’s PLSD, as appropriate.
Supplementary Material
Supplementary Figure 1 RP105 phylogeny. Phylogenetic relationships among the human TLRs as inferred by sequence alignment using ClustalW software. Similar results were obtained with PileUp software. Horizontal branch lengths are proportional to the degree of inferred evolutionary change.
Supplementary Figure 2 MD-1 expression is necessary for RP105-mediated suppression of TLR4 signaling. (a) HEK293 cells stably expressing CD14 and TLR4 were transiently transfected with MD-2; along with EV, MD-1 and/or RP105 as indicated. Cells were subsequently stimulated with purified E. coli K235 LPS (10 ng/ml). Means +/- SE of triplicate cultures in a single experiment are depicted, representative of an experimental n = 4. *p < 0.008. (b) HEK293 cells were analyzed for surface and intracellular RP105 expression by FACS (representative of 2 separate experiments).
Supplementary Figure 3 RP105 fails to inhibit NF-κB transactivation driven by overexpression of TLR4 signaling molecules. HEK293FT cells were co-transfected with an NF-κB-firefly luciferase reporter plasmid (pELAM; 0.5 μg), along with plasmids encoding an I-κB super-repressor, RP105 (100 ng) plus MD-1 (50 ng), Mal (100ng), IRAK-1 (100 ng), Myd88 (100 ng), TRIF (100 ng) and/or empty vector (EV; 100 ng). After 48 h, cells were lysed and luciferase activity was quantified. Means +/- SE of triplicate cultures in a single experiment are depicted, representative of an experimental n=3.
Supplementary Figure 4 MD-1 and MD-2 can interact directly with each other. HEK293T cells were transiently transfected with MD-2 and/or MD-1 constructs, as indicated. Lysates were immunoprecipated with antibodies to FLAG or HA, and the association between MD-1 and MD-2 was examined by immunoblotting using the antibodies indicated.
Supplementary Figure 5 Similar expression of molecules regulating TLR signaling in bone marrow-derived dendritic cells from RP105-deficient and wild-type mice. mRNA expression was analyzed by quantitative RT-PCR in bone marrow-derived DC from wild type (open symbols) or RP105−/− (filled symbols) mice prior to and following stimulation with 10 ng of purified E. coli K235 LPS. (a) IRAK-M, (b) Tollip, (c) ST2, (d) SIGIRR.
Supplementary Figure 6 Altered TLR4-induced cytokine production by resident peritoneal macrophages from RP105-deficient mice. Resident peritoneal macrophages from wild type (open symbols) or RP105-deficient (filled symbols) mice were stimulated with purified E. coli K235 LPS. Supernatants were harvested after 24 h. * p < 0.005, ** p < 0.05.
Supplementary Figure 7 Regulation of RP105 expression by LPS. DC were stimulated with purified E. coli K235 LPS. Kinetic analysis of RP105 and TLR4 mRNA expression was performed by quantitative RT-PCR. (a) Regulation of TLR4 and RP105 expression in mouse bone marrow-derived DC. Means + SE of duplicate PCR reactions in a single experiment, representative of 3 different experiments. (b) Regulation of TLR4 and P105 expression in human monocyte-derived DC. Means + SE of duplicate PCR reactions from 3 different donors. RP105 expression: square symbols; TLR4 expresssion: round symbols. RP105-deficient mice: filled symbols.
Supplementary Figure 8 Genotyping and phenotyping of RP105-deficient mice. (a) Genotyping via standard techniques. (b) FACS analysis of peripheral blood cells.
Acknowledgments
The authors thank L. Flick, and H. Stanley for skillful technical assistance; and C. Chougnet, J. Bridges, K. Fitzgerald, B. Aronow, B. Sakthivel, and J. Meller for helpful discussions. This work was supported by National Institutes of Health grants R21 AI063183 (C.L.K.) and T32 AI 055406.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests in relation to this work.
Correspondence and requests for materials should be addressed to C.L.K. (email: chris.karp@chmcc.org).
References
- 1.Medzhitov R, Janeway C., Jr Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 3.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–76. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 4.Ardizzone S, Porro GB. Inflammatory bowel disease: new insights into pathogenesis and treatment. J Intern Med. 2002;252:475–96. doi: 10.1046/j.1365-2796.2002.01067.x. [DOI] [PubMed] [Google Scholar]
- 5.Bingham CO., 3rd The pathogenesis of rheumatoid arthritis: pivotal cytokines involved in bone degradation inflammation. J Rheumatol Suppl. 2002;65:3–9. [PubMed] [Google Scholar]
- 6.Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J Exp Med. 1994;180:1217–24. doi: 10.1084/jem.180.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyake K, Yamashita Y, Ogata M, Sudo T, Kimoto M. RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J Immunol. 1995;154:3333–40. [PubMed] [Google Scholar]
- 8.Miura Y, et al. Molecular cloning of a human RP105 homologue and chromosomal localization of the mouse and human RP105 genes (Ly64 and LY64) Genomics. 1996;38:299–304. doi: 10.1006/geno.1996.0632. [DOI] [PubMed] [Google Scholar]
- 9.Fugier-Vivier I, et al. Molecular cloning of human RP105. Eur J Immunol. 1997;27:1824–7. doi: 10.1002/eji.1830270734. [DOI] [PubMed] [Google Scholar]
- 10.Roshak AK, et al. Anti-human RP105 sera induces lymphocyte proliferation. J Leukoc Biol. 1999;65:43–9. doi: 10.1002/jlb.65.1.43. [DOI] [PubMed] [Google Scholar]
- 11.Shimazu R, et al. MD-2, a Molecule that Confers Lipopolysaccharide Responsiveness on Toll-like Receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyake K, et al. Mouse MD-1, a molecule that is physically associated with RP105 and positively regulates its expression. J Immunol. 1998;161:1348–53. [PubMed] [Google Scholar]
- 13.Miura Y, et al. RP105 is associated with MD-1 and transmits an activation signal in human B cells. Blood. 1998;92:2815–22. [PubMed] [Google Scholar]
- 14.Nagai Y, et al. Requirement for MD-1 in cell surface expression of RP105/CD180 and B-cell responsiveness to lipopolysaccharide. Blood. 2002;99:1699–705. doi: 10.1182/blood.v99.5.1699. [DOI] [PubMed] [Google Scholar]
- 15.Schneider DS, Hudson KL, Lin TY, Anderson KV. Dominant and recessive mutations define functional domains of Toll, a transmembrane protein required for dorsal-ventral polarity in the Drosophila embryo. Genes Dev. 1991;5:797–807. doi: 10.1101/gad.5.5.797. [DOI] [PubMed] [Google Scholar]
- 16.Winans KA, Hashimoto C. Ventralization of the Drosophila embryo by deletion of extracellular leucine-rich repeats in the Toll protein. Mol Biol Cell. 1995;6:587–96. doi: 10.1091/mbc.6.5.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 18.Vogel SN, et al. Cutting edge: functional characterization of the effect of the C3H/HeJ defect in mice that lack an lpsn gene: In vivo evidence for a dominant negative mutation. J Immunol. 1999;162:5666–70. [PubMed] [Google Scholar]
- 19.Ronni T, et al. Common interaction surfaces of the toll-like receptor 4 cytoplasmic domain stimulate multiple nuclear _targets. Mol Cell Biol. 2003;23:2543–55. doi: 10.1128/MCB.23.7.2543-2555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol. 2001;31:3388–93. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 21.Kadowaki N, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–9. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krug A, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol. 2001;31:3026–37. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 23.Latz E, et al. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem. 2002;277:47834–43. doi: 10.1074/jbc.M207873200. [DOI] [PubMed] [Google Scholar]
- 24.Kurt-Jones EA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 25.Ogata H, et al. The toll-like receptor protein RP105 regulates lipopolysaccharide signaling in B cells. J Exp Med. 2000;192:23–9. doi: 10.1084/jem.192.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan VW, et al. The molecular mechanism of B cell activation by toll-like receptor protein RP-105. J Exp Med. 1998;188:93–101. doi: 10.1084/jem.188.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yazawa N, et al. CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood. 2003;102:1374–80. doi: 10.1182/blood-2002-11-3573. [DOI] [PubMed] [Google Scholar]
- 28.Brint EK, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–9. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 29.Visintin A, Latz E, Monks BG, Espevik T, Golenbock DT. Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J Biol Chem. 2003;278:48313–20. doi: 10.1074/jbc.M306802200. [DOI] [PubMed] [Google Scholar]
- 30.Tsuneyoshi N, et al. The functional and structural properties of MD-2 required for lipopolysaccharide binding are absent in MD-1. J Immunol. 2005;174:340–4. doi: 10.4049/jimmunol.174.1.340. [DOI] [PubMed] [Google Scholar]
- 31.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 32.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–22. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 33.Wald D, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–7. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 34.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 35.Rehli M. Of mice and men: species variations of Toll-like receptor expression. Trends Immunol. 2002;23:375–8. doi: 10.1016/s1471-4906(02)02259-7. [DOI] [PubMed] [Google Scholar]
- 36.Cross AS. Endotoxin tolerance-current concepts in historical perspective. J Endotoxin Res. 2002;8:83–98. doi: 10.1179/096805102125000227. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi K, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 38.Burns K, et al. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–8. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–65. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–8. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 41.Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5:495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- 42.Akashi S, et al. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–5. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- 43.Vogel SN, Fitzgerald KA, Fenton MJ. TLRs: Differential adapter utilization by Toll-like receptors mediates TLR-specific patterns of gene expression. Mol Interventions. 2003;3:466–477. doi: 10.1124/mi.3.8.466. [DOI] [PubMed] [Google Scholar]
- 44.McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY. Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry. 1967;6:2363–72. doi: 10.1021/bi00860a011. [DOI] [PubMed] [Google Scholar]
- 45.Kurt-Jones EA, et al. Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood. 2002;100:1860–8. [PubMed] [Google Scholar]
- 46.Acred P, et al. Guidelines for the welfare of animals in rodent protection tests: A report from the rodent protection test working party. Lab Anim. 1994;28:13–18. doi: 10.1258/002367794781065870. [DOI] [PubMed] [Google Scholar]
- 47.Karp CL, et al. Mechanism of suppression of cell-mediated immunity by measles virus. Science. 1996;273:228–31. doi: 10.1126/science.273.5272.228. [DOI] [PubMed] [Google Scholar]
- 48.Atabani SF, et al. Natural measles causes prolonged suppression of interleukin-12 production. J Infect Dis. 2001;184:1–9. doi: 10.1086/321009. [DOI] [PubMed] [Google Scholar]
- 49.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998;23:403–5. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 50.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–95. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 RP105 phylogeny. Phylogenetic relationships among the human TLRs as inferred by sequence alignment using ClustalW software. Similar results were obtained with PileUp software. Horizontal branch lengths are proportional to the degree of inferred evolutionary change.
Supplementary Figure 2 MD-1 expression is necessary for RP105-mediated suppression of TLR4 signaling. (a) HEK293 cells stably expressing CD14 and TLR4 were transiently transfected with MD-2; along with EV, MD-1 and/or RP105 as indicated. Cells were subsequently stimulated with purified E. coli K235 LPS (10 ng/ml). Means +/- SE of triplicate cultures in a single experiment are depicted, representative of an experimental n = 4. *p < 0.008. (b) HEK293 cells were analyzed for surface and intracellular RP105 expression by FACS (representative of 2 separate experiments).
Supplementary Figure 3 RP105 fails to inhibit NF-κB transactivation driven by overexpression of TLR4 signaling molecules. HEK293FT cells were co-transfected with an NF-κB-firefly luciferase reporter plasmid (pELAM; 0.5 μg), along with plasmids encoding an I-κB super-repressor, RP105 (100 ng) plus MD-1 (50 ng), Mal (100ng), IRAK-1 (100 ng), Myd88 (100 ng), TRIF (100 ng) and/or empty vector (EV; 100 ng). After 48 h, cells were lysed and luciferase activity was quantified. Means +/- SE of triplicate cultures in a single experiment are depicted, representative of an experimental n=3.
Supplementary Figure 4 MD-1 and MD-2 can interact directly with each other. HEK293T cells were transiently transfected with MD-2 and/or MD-1 constructs, as indicated. Lysates were immunoprecipated with antibodies to FLAG or HA, and the association between MD-1 and MD-2 was examined by immunoblotting using the antibodies indicated.
Supplementary Figure 5 Similar expression of molecules regulating TLR signaling in bone marrow-derived dendritic cells from RP105-deficient and wild-type mice. mRNA expression was analyzed by quantitative RT-PCR in bone marrow-derived DC from wild type (open symbols) or RP105−/− (filled symbols) mice prior to and following stimulation with 10 ng of purified E. coli K235 LPS. (a) IRAK-M, (b) Tollip, (c) ST2, (d) SIGIRR.
Supplementary Figure 6 Altered TLR4-induced cytokine production by resident peritoneal macrophages from RP105-deficient mice. Resident peritoneal macrophages from wild type (open symbols) or RP105-deficient (filled symbols) mice were stimulated with purified E. coli K235 LPS. Supernatants were harvested after 24 h. * p < 0.005, ** p < 0.05.
Supplementary Figure 7 Regulation of RP105 expression by LPS. DC were stimulated with purified E. coli K235 LPS. Kinetic analysis of RP105 and TLR4 mRNA expression was performed by quantitative RT-PCR. (a) Regulation of TLR4 and RP105 expression in mouse bone marrow-derived DC. Means + SE of duplicate PCR reactions in a single experiment, representative of 3 different experiments. (b) Regulation of TLR4 and P105 expression in human monocyte-derived DC. Means + SE of duplicate PCR reactions from 3 different donors. RP105 expression: square symbols; TLR4 expresssion: round symbols. RP105-deficient mice: filled symbols.
Supplementary Figure 8 Genotyping and phenotyping of RP105-deficient mice. (a) Genotyping via standard techniques. (b) FACS analysis of peripheral blood cells.