

Abstract
Complete loss of platelet-derived growth factor (PDGF) receptor signaling results in embryonic lethality around embryonic day 9.5, but the cause of this lethality has not been identified. Because cardiovascular failure often results in embryonic lethality at this time point, we hypothesized that a failure in cardiovascular development could be the cause. To assess the combined role of PDGF receptor α (PDGFRα) and PDGFRβ, we generated embryos that lacked these receptors in cardiomyocytes and vascular smooth muscle cells (VSMC) using conditional gene ablation. Deletion of either PDGFRα or PDGFRβ caused no overt vascular defects, but loss of both receptors using an SM22α-Cre transgenic mouse line led to a disruption in yolk sac blood vessel development. The cell population responsible for this vascular defect was the yolk sac mesothelial cells, not the cardiomyocytes or the VSMC. Coincident with loss of PDGF receptor signaling, we found a reduction in collagen deposition and an increase in MMP-2 activity. Finally, in vitro allantois cultures demonstrated a requirement for PDGF signaling in vessel growth. Together, these data demonstrate that PDGF receptors cooperate in the yolk sac mesothelium to direct blood vessel maturation and suggest that these effects are independent of their role in VSMC development.
Vascular remodeling and maturation are complex processes that transform an endothelial plexus into vessels of various calibers and stabilities. Although angiogenesis has been studied extensively, the mesodermal signals directing these cellular processes are not well understood. One of the first tissues to undergo remodeling during development is the yolk sac, and the proper formation of yolk sac blood vessels is essential for embryonic development and hematopoiesis. Disruption of yolk sac vascular development, either directly or indirectly by aberrant cardiac function, often results in embryonic lethality between embryonic day 9.5 (E9.5) and E11.5 (9). In a majority of cases, the primary cell type responsible for yolk sac vessel abnormalities is the endothelial cell (1). While endothelial cells are commonly implicated in yolk sac phenotypes, the contribution of other yolk sac cell populations should not be discounted. For example, BMP-4 and retinoic acid secretion by the visceral endoderm is required in a paracrine manner for hematopoietic and endothelial development (3, 4, 11, 73), while fibronectin and laminin deposition by the yolk sac mesothelium is required for endothelial remodeling (18).
Due to their close proximity to endothelial cells, vascular smooth muscle cells (VSMC) are also believed to influence blood vessel integrity. In the absence of these support cells, some endothelial vessels are hyperplastic, tortuous, dilated, and leaky (20, 38). In the yolk sac vasculature, it has been difficult to ascertain the function of VSMC because many relevant regulatory molecules are expressed by both endothelial cells and VSMC. Mice that have mutations in transforming growth factor β (TGF-β) signaling exhibit defects in VSMC formation and recruitment, but they also possess cardiovascular and endothelial cell defects (11, 26, 35, 41, 72). Therefore, the lack of VSMC in these mutants may be secondary to aberrant circulation and not the cause of yolk sac vascular demise.
Platelet-derived growth factor (PDGF) receptors have been implicated in cardiovascular development by their functions in cardiac neural crest cells (53, 64), retinal astrocytes (17), mesoderm precursors to endothelial cells (55), VSMC (60), and tumor stroma (50), but few investigations have looked at a role for these receptors in cardiac and yolk sac development. To address this topic, we used Cre/loxP technology to remove PDGF receptors from cardiomyocytes and VSMC. We learned that PDGF receptor expression in the yolk sac mesothelium is essential for yolk sac blood vessel development and that one function of these receptors may be to direct extracellular matrix (ECM) deposition to promote vascular remodeling. These data demonstrate that PDGF receptor function in vascular development may be broader than once thought, and potentially, these receptors may play similar roles in vascular development in other tissues.
MATERIALS AND METHODS
Mouse lines.
The mouse lines used in these studies were PDGFRαfl/fl (64), PDGFRβfl/fl (53), Tg(Tagln-cre)1Her/J (SM22α-CreTg) (23), Meox2Cre/+ (65), myocardinCre/+ (39), Tie2CreTg (30), ROSA26 reporter LacZ (R26R) (61), Tie2GFPTg (59), and PDGFRαGFP (19) lines. SM22α-CreTg mice were purchased from Jackson Laboratories. Transgene levels of SM22α-CreTg animals were detected by Southern blot analysis using a probe for the Cre gene. These mice were maintained by inbreeding lines that were homozygous for SM22α-CreTg. Control embryos and yolk sacs were either SM22α-CreTg littermate embryos bearing heterozygous floxed alleles or wild-type embryos (somite stage matched). Detection of a vaginal plug was defined as E0.5. PDGFRMKO embryos were recovered up to E18.5, but we recovered fewer than expected after birth. Often, we found postnatal day 1 animals with spina bifida and a cleft palate. Previously, we had determined that the PDGFRα floxed allele is hypomorphic and is lethal in combination with a null allele. These data, combined with the fact that myocardinCre can lead to germ line deletion of floxed alleles, suggested that the lethality was not caused by the conditional deletion of the PDGF receptors but by loss of PDGFRα signaling regardless of the myocardincre status of the mice.
Histology and immunohistochemistry.
Samples stained for β-galactosidase were fixed in 2% formaldehyde/0.2% glutaraldehyde for 10 min, stained in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside overnight at room temperature, and postfixed in 10% buffered formalin for 20 min. Whole embryos were stained for PECAM and αSMA according to standard procedures and cleared using benzylalcohol-benzyl benzoate (1:2) for imaging (22). Yolk sacs were fixed for 1 h in 4% paraformaldehyde (PFA) at 4°C and blocked for 30 min in 1.5% normal serum. For section analysis, samples were fixed for 1 to 3 h in 4% PFA, embedded, and sectioned at 7 to 10 μm. Immunohistochemistry was performed by incubation in primary antibody for 2 h to overnight at 4°C and visualized using Alexafluor secondary antibodies or a Vectastain Elite ABC kit and a DAB peroxidase substrate kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Antigen retrieval for PECAM and αSMA paraffin sections was performed as previously described (64). Hematoxylin and eosin (HE) staining and picrosirius red staining were performed according to standard methods.
Western blotting and immunoprecipitation.
For immunoprecipitation, whole yolk sac lysates were incubated overnight at 4°C with 1 μg of antibody and then for 1 h with protein A-Sepharose beads. After being washed, the precipitated proteins were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For Western blotting, yolk sac lysate was quantified using Bradford reagent, and equal amounts of protein were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes following standard protocols. The membranes were incubated with 1% bovine serum albumin/0.05% Tween 20 in Tris-buffered saline for 30 min, with primary antibody overnight at 4°C, and with secondary antibody for 1 h. The results were visualized using enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ).
Antibodies.
The primary antibodies used for immunohistochemistry and whole mount included PDGFRβ (eBioscience, San Diego, CA), 1:250; PECAM (clone MEC13.3; BD Bioscience, San Jose, CA), 1:250; anti-SMA-fluorescein isothiocyanate (clone 1A4; Sigma, St. Louis, MO), 1:500; green fluorescent protein (GFP) (abcam, Cambridge, MA), 1:250; SM22α (abcam, Cambridge, MA), 1:250; collagen 1 (abcam, Cambridge, MA), 1:250; collagen 4 (Chemicon, Temecula, CA), 1:250; fibronectin (Sigma), 1:500; and laminin (Sigma), 1:250. The secondary antibodies used included anti-rabbit, anti-rat, and anti-mouse Alexafluor 488, 543, 594, and 633 (Molecular Probes, Eugene, OR), 1:500. Antibodies used for Western blots included integrin β1 (abcam), 1:1,000; integrin β1 phospho-S785 (abcam), 1:1,000; PDGFRβ (Upstate, Charlottesville, VA), 1:1,000; PDGFRα (Upstate), 1:1,000; and cytoskeletal actin (Novus Biologicals, Littleton, CO), 1:1,000.
Real-time PCR.
RNA was isolated from yolk sacs using Trizol (Invitrogen, Carlsbad, CA) and an RNeasy kit (Qiagen, Valencia, CA). Samples were isolated and homogenized in Trizol; 20% choloroform was added, mixed well, and centrifuged for layer separation. The top layer was mixed with an equal volume of 70% ethanol, added to an RNeasy kit minispin column, and centrifuged. Washes were followed according to the RNeasy protocol and eluted with diethyl pyrocarbonate-H2O. RNA was quantified and DNase treated. RNA (1 μg) was used to generate cDNA using PowerScript reverse transcriptase (Clontech, Mountain View, CA) and random hexamers. Gene expression was quantified using standard real-time PCR methods using Sybr green master mixture on an ABI7000 instrument (Applied Biosystems, Foster City, CA). Samples were analyzed in triplicate on a minimum of three samples per genotype. Primer sequences will be provided upon request.
MMP assays.
DQ gelatin assay (Molecular Probes) was performed on fresh frozen tissue sections according to manufacturer's instructions. Briefly, samples were sectioned and incubated for 30 min in substrate buffer and then transferred to DQ gelatin (3 μg/ml) at 37°C and imaged after incubation for 0, 15, and 30 min. Zymography was performed according to standard protocols (32) with human matrix metalloprotease 2 (MMP-2) and MMP-9 standards (Chemicon). Yolk sac lysates were quantified using Bradford reagent, and equal amounts of protein were analyzed.
Allantois assay.
E8.5 allantoides were isolated from wild-type and PDGFRαfl/fl; PDGFRβfl/fl embryos and plated on gelatin-coated coverslips. The medium was supplemented with 1% fetal bovine serum (FBS), 10% FBS, or 20 ng PDGFBB (R&D Systems, Minneapolis, MN). Cultures were grown for 24 to 26 h at 37°C in 5% CO2. Samples were fixed with 4% PFA for 10 min and stained for PECAM as described above. Adenovirus transduction was performed using 1.4 × 107 PFU per 1 ml of medium at the time of plating. PDGF receptor inhibitor (AG1296 [2 μM]; Calbiochem, Gibbstown, NJ) was added to appropriate stimulation medium at the time of plating. For collagen 4 assays, wells were coated for 1 h with 0.5 μg/ml, 5 μg/ml, or 50 μg/ml collagen 4 (R&D Systems), and then medium was added to the collagen 4 at the time of allantois plating.
Image acquisition and manipulation.
Whole-mount and section samples were analyzed using Nikon SMZ1000 and Zeiss Axiovert200 (Carl Zeiss, Thornwood, NY) microscopes. Images were captured using Hamamatsu Orca-ER (Hamamatsu, Bridgewater, NJ) and Olympus DP71 (Olympus, Center Valley, PA) cameras with OpenLab 3.5.1 and DP Controller software, respectively. Fluorescent images were colored using OpenLab and then processed in Adobe Photoshop. Confocal images were captured using an LSM510Meta confocal microscope (Carl Zeiss) and processed in ImageJ and Adobe Photoshop. Color images and Western blots were processed in Adobe Photoshop for a white background. Quantification and threshold measurements were calculated through ImageJ. Graphs and statistics were generated using Prism (Graphpad). The final figures were compiled using Canvas 9.
RESULTS
Generation and survival of PDGF receptor smooth muscle cell knockout embryos.
Previous data from PDGFRβ and PDGFB null embryos indicated that PDGFRβ is required for formation of VSMC and that myocardial development is also affected (21, 34, 37, 60, 66). Because we wanted to investigate the cell lineages dependent on PDGFRβ signal transduction, we used loxP/Cre technology to generate animals that lacked PDGFRβ in both cardiomyocytes and VSMC. We used two Cre-expressing mouse lines to accomplish the deletion, SM22α-CreTg (23) and myocardinCre (39) lines. SM22α is expressed as early as E8.5 in cardiomyocyte development and during VSMC terminal differentiation (36), while myocardin is expressed by E8.5 in cardiomyocytes and is one of the earliest genes expressed in VSMC precursors (68). Surprisingly, deletion of PDGFRβ using either SM22αCreTg (referred to as PDGFRβSKO) or myocardinCre/+ (referred to as PDGFRβMKO) mice did not phenocopy PDGFRβ−/− mice. Both PDGFRβSKO and PDGFRβMKO embryos and mice were viable and fertile.
Recently, we have shown that the two PDGF receptors (PDGFRα and -β) act cooperatively in the neural-crest-derived smooth muscle of the aortic arch (53). To determine if PDGFRα was compensating for loss of PDGFRβ signaling in other VSMC populations, we generated mice that contained SM22α-CreTg and conditional alleles of both PDGF receptors. Genotyping of offspring from these crosses revealed that few PDGFRαfl/fl; PDGFRβfl/fl; SM22α-CreTg (PDGFRSKO) pups were recovered at birth, indicating that PDGF receptor signaling through both receptors was required for viability in an SM22α-expressing cell population. Using timed matings, we recovered the expected Mendelian ratios of embryos up to E10.5 but few PDGFRSKO embryos past that time point. The few viable mice that were doubly homozygous for the PDGF receptor floxed alleles were SM22α-CreTg/0 (hemizygous for the transgene) (data not shown). We surmised that the expression level of Cre in mice hemizygous for the transgene was not sufficient to recombine all four floxed alleles efficiently. Indeed, no PDGFRSKO pups resulted from breeders that were SM22α-CreTg/Tg. All subsequent analyses were performed using mice homozygous for SM22α-CreTg. Commonly, transgenic Cre lines are not maintained as homozygotes due to potential transgene insertion effects. We have ruled out these effects, because lethality occurs only in double-homozygous PDGF receptor SM22α-CreTg/Tg embryos, not single PDGF receptor SM22α-CreTg/Tg embryos. Because SM22α-CreTg was likely to _target deletion in VSMC similarly to myocardinCre, we predicted that excision of the two PDGF receptor genes in this mouse line (PDGFRMKO) would phenocopy the embryonic lethality observed in PDGFRSKO embryos. However, PDGFRMKO embryos survived until birth.
Expression of SM22-CreTg before VSMC differentiation in the yolk sac.
The dramatic difference in survival between PDGFRSKO and PDGFRMKO embryos led us to investigate the profiles of Cre activity in the two Cre mouse lines. Using ROSA26 reporter mice, we determined SM22α-CreTg and myocardinCre expression between E8.5 and E10.5. Cre activity was not detected in any tissue at E7.5 using either of the Cre lines. By E8.5, SM22α-Cre activity was observed in many cells of the yolk sac, as well as a small number of cells in the primitive heart (Fig. 1A). In contrast, myocardin-Cre activity was detected in the cardiac crescent, and only a few β-galactosidase-positive cells were observed in myocardinCre yolk sacs (Fig. 1E). Because Cre expression in the yolk sac was the most obvious difference between these lines, we examined histological sections of this tissue. At E8.5, SM22α-Cre activity was present throughout the yolk sac mesothelium, but no β-galactosidase-positive cells were detected in the myocardinCre yolk sacs (Fig. 1A and E). The yolk sac mesothelium is a mesoderm-derived epithelium-like component of the yolk sac that rests on a thin basement membrane and is believed to be important for transport and movement of fluid from the yolk sac. At E9.5, β-galactosidase-positive cells were present both in the mesothelial layer and surrounding some vessels in the SM22α-CreTg mice, while myocardinCre activity was present in only a few cells associated with blood vessels, presumably VSMC progenitors (Fig. 1B and F). At E10.5, SM22α-CreTg and myocardinCre yolk sacs both possessed β-galactosidase expression in cells surrounding blood vessels, but β-galactosidase expression in myocardinCre was lacking in yolk sac mesoderm populations that were not vessel associated (Fig. 1C and G). These data pointed to the possibility that deletion of the PDGF receptors in the yolk sac mesothelium caused the embryonic lethality.
FIG. 1.
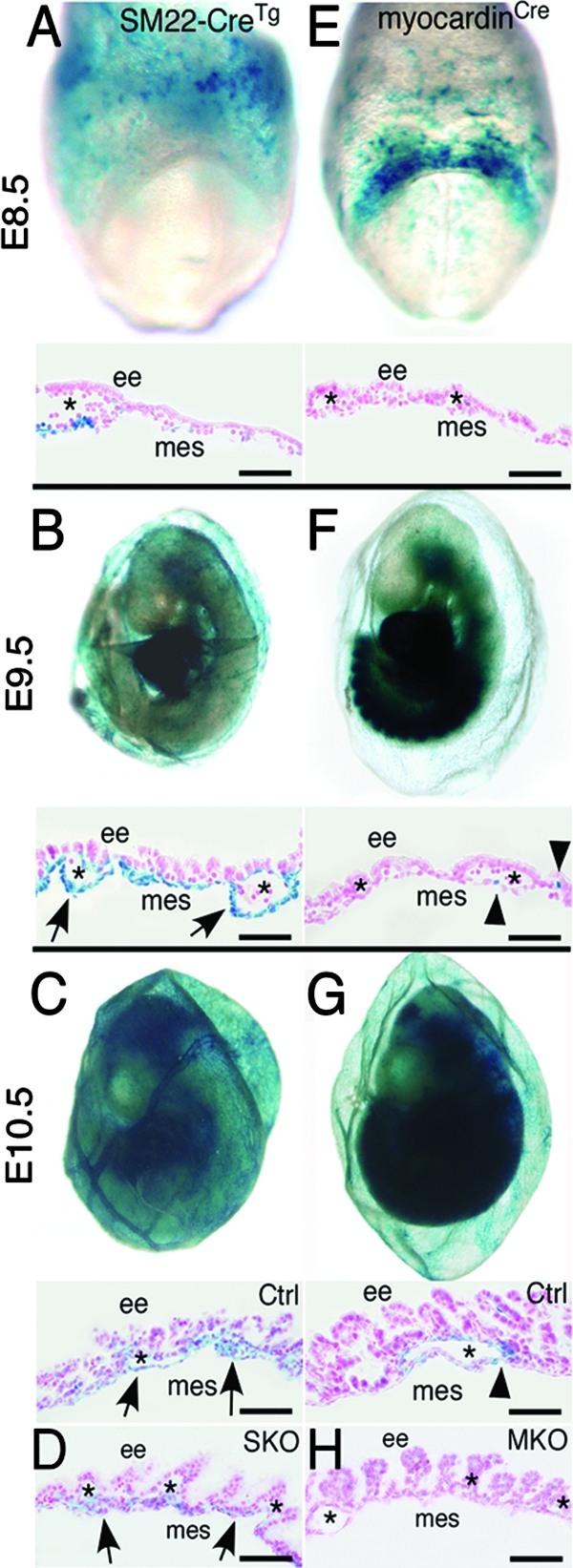
SM22α-CreTg is expressed in yolk sac mesothelium and VSMC, while myocardinCre is specific to VSMC. (A to C) Whole-mount LacZ staining of the yolk sac demonstrating SM22α-CreTg expression at the indicated stages. (A) Yolk sac expression of SM22α-CreTg can be detected as early as E8.5. (C) At E10.5, the yolk sac vasculature is undergoing vascular remodeling, and Cre expression can be detected along the remodeled vasculature and mesothelium. (D) β-Galactosidase expression in PDGFRSKO yolk sacs is restricted to mesothelial cells. (E) myocardinCre expression is not detected in the yolk sac at E8.5 by whole-mount or section analysis. (F and G) β-Galactosidase expression in VSMC begins at E9.5 and is maintained at E10.5. (H) Loss of β-galactosidase-positive cells in PDGFRMKO yolk sacs. The arrows indicate the mesothelium. The arrowheads point to myocardin-Cre-expressing cells. The asterisks indicate blood vessels. ee, extraembryonic endoderm; mes, mesothelium. Scale bars = 20 μm.
Because Cre expression leads to indelible β-galactosidase expression, we could also use this marker to follow mesothelial and VSMC cell lineages in PDGFRSKO and PDGFRMKO embryos. In both PDGFRSKO and PDGFRMKO yolk sacs, few perivascular, β-galactosidase-positive cells were present at E10.5 (Fig. 1D and H). β-Galactosidase-positive cells were abundant in the mesothelial layer in the PDGFRSKO yolk sac, demonstrating that loss of PDGF receptors did not lead to failure in the formation of this cell population. In addition, examination of both PDGFRSKO and PDGFRMKO embryos revealed abundant β-galactosidase-positive cells in the hearts and trunk areas (data not shown), demonstrating that loss of the receptors did not result in a general reduction of mesoderm cells.
PDGFRα and PDGFRβ are coexpressed in the yolk sac mesothelium and perivascular cells.
While PDGFRα and PDGFRβ expression has been documented as early as E6.5 in the extraembryonic endoderm and ectoplacental cone, and the ligands are expressed in the chorionic ectoderm and parietal endoderm (42, 48, 58), analysis of coexpression of the receptors in the yolk sac has not been done. Therefore, we investigated the expression patterns of the receptors. Expression of PDGFRα was detected using mice that expressed a nuclear-localized GFP from the PDGFRα locus that faithfully traced PDGFRα expression (19) and a PDGFRβ-specific antibody. At E8.5, PDGFRα and PDGFRβ were expressed in the mesothelium and endoderm, as previously reported (Fig. 2A). At E9.5, PDGFRβ endoderm expression was reduced, but expression of both receptors was maintained in the mesothelium (Fig. 2B and data not shown). At E10.5, both receptors were expressed in the mesothelium, but only PDGFRβ was identified in cells surrounding endothelial vessels (Fig. 2C). Presumably, these cells were VSMC. The early and persistent coexpression of PDGF receptors in the mesothelial layer (Fig. 2D), along with lethality of PDGFRSKO but not PDGFRMKO, supports the possibility that PDGF receptor expression in the mesothelium is required for embryo viability.
FIG. 2.

PDGFRα- and PDGFRβ-positive cells in yolk sac mesothelium. (A to C) Section immunohistochemistry for PDGFRβ was performed on PDGFRαGFP/+ yolk sacs. Both receptors were expressed abundantly at E8.5, but their expression patterns became more restricted to mesothelial and perivascular cells at the later stages. (D) PDGFRα and PDGFRβ coexpression with the mesothelial marker SM22. DAPI (4′,6′-diamidino-2-phenylindole)-stained and SM22-negative cells represent the endoderm. (E) Immunohistochemistry for PDGFRβ and GFP on Tie2GFPTg frozen sections at E8.5 to E10.5 demonstrating PDGFRβ expression in close proximity to, but not overlapping with, Tie2GFP expression. The arrowhead indicates perivascular cells expressing only PDGFRβ. The arrows indicate PDGFRαGFP- and SM22-coexpressing cells. The asterisks indicate blood vessels. ee, extraembryonic endoderm; mes, mesothelium. Scale bar = 20 μm.
Because multiple reports have suggested that PDGFRβ is expressed by endothelial cells and to further refine our expression analysis in the yolk sac, we examined PDGFRβ expression in Tie2GFPTg mice. These mice express GFP in endothelial cells (59). We found no Tie2-positive cells that expressed detectable levels of PDGFRβ between E8.0 and E10.5 (Fig. 2E and data not shown). Tie2GFPTg cells were present in blood islands adjacent to PDGFRβ-expressing mesothelium at E8.5. This expression profile was consistent with a previous report that demonstrated PDGFRβ expression in mesoderm precursors in the yolk sac but not in differentiated endothelial cells (55).
PDGFRSKO results in incomplete yolk sac capillary bed reorganization.
Embryonic lethality between E9.5 and E10.5 is often a result of cardiovascular failure, but αSMA staining for cardiomyocytes and PECAM staining for endothelial cells revealed that cardiac and embryonic vascular development appeared normal in E10.5 PDGFRSKO embryos (Fig. 3A to D). No cardiovascular defects were observed in the embryo proper. This lack of a cardiomyocyte phenotype is in agreement with gene deletion analysis of PDGF receptors using an early mesoderm-expressed Cre line, MesP1Cre (27). By contrast, whole-mount views and histological sections of PDGFRSKO embryos at E10.5 revealed an apparent cessation of blood vessel maturation within the yolk sac (Fig. 3E and F). While endoderm and mesothelial layers appeared normal, endothelial vessels were distended and disorganized compared to vessels in control embryos. At this time point, we also observed efficient deletion of both PDGFRα and PDGFRβ in PDGFRSKO yolk sacs (Fig. 3G and H). Analyses of E9.5 and E10.5 mutant yolk sacs indicated no abnormalities in proliferation by bromodeoxyuridine incorporation, and at the same time points, we observed no increase in apoptotic cells, as determined by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling in situ (data not shown).
FIG. 3.

PDGFRSKO mutants demonstrate normal cardiac and embryonic vascular development but disrupted yolk sac vasculature. (A) Whole-mount PECAM staining of intersomitic vessels (isv) in E10.5 control and PDGFRSKO embryos. (B to D) HE (B), αSMA (C), and PECAM (D) staining of E10.5 control and PDGFRSKO hearts. (E) E10.5 whole-mount images of yolk sac blood vessels on the side opposite the vitelline vessel. The embryos were imaged while attached to the placenta to prevent red blood cell loss. (F) HE-stained section of yolk sac demonstrating yolk sac vascular disruptions in PDGFRSKO mutants. (G) Immunohistochemistry of whole-mount yolk sacs demonstrating loss of PDGFRβ in PDGFRSKO mutants by E9.5 compared to the control. (H) Deletion of both PDGFRα and PDGFRβ in PDGFRSKO mutants compared to the control at E10.5 by Western blot analysis. The asterisks indicate blood vessels. d, dorsal; v, ventral; at, atria; ven, ventricle; en, endocardial cushions; tr, trabeculations; ee, extraembryonic endoderm; mes, mesothelium. Scale bars = 10 μm (B) and 20 μm (C and D, F and G).
To assess yolk sac vascular development in our mutants, we performed whole-mount PECAM staining. Vasculogenesis of the yolk sac begins at E7.5, and a number of signaling proteins have been implicated in the process (1). After E8.5, yolk sac vessels undergo a dramatic remodeling event in which the primitive polygonal structure of the vasculature is converted to stable and defined vessels (15). In both control and mutant yolk sacs, we observed the typical honeycomb pattern of endothelial cells at E9.5 (Fig. 4A and B), but at E10.5, PDGFRSKO yolk sacs retained the characteristics of an immature yolk sac and failed to reorganize into the normal hierarchical array of large and small vessels (Fig. 4G).
FIG. 4.

Vascular development is disrupted in PDGFRSKO yolk sacs but not in single mutants. (A to E) Whole-mount E9.5 yolk sac staining for PECAM on the indicated genotypes. A normal vascular plexus was observed in control, PDGFRSKO, PDGFRMKO, and PDGFRβ−/− yolk sacs, but it was disrupted in PDGFRMeox2 yolk sacs. (F to K) Yolk sac vascular remodeling progression to large defined vessels visualized by PECAM staining. PDGFRSKO yolk sacs fail to undergo vascular remodeling, resembling the E9.5 yolk sac vascular plexus. (L to Q) αSMA staining was used to detect VSMC at E10.5 on the indicated genotypes. Recruitment of VSMC to developing vasculature present in control E10.5 yolk sacs is severely reduced in PDGFRβSKO, PDGFRβMKO, and PDGFRMKO yolk sacs and absent in PDGFRSKO and PDGFRβ−/− yolk sacs. The images represent similar regions of the yolk sac adjacent to, but not including, the vitelline vessels. The images are representative of a minimum of three yolk sacs of each genotype. Scale bars = 10 μm.
To establish if these yolk sac defects were caused primarily by loss of one of the receptors, we analyzed yolk sac vessels by PECAM staining in single PDGF receptor mutants. Yolk sacs from PDGFRα−/−, PDGFRβ−/−, PDGFRαSKO, and PDGFRβSKO mutants developed normally, although the remodeled vessels in the E10.5 PDGFRβ−/− yolk sacs were slightly more disorganized than control vessels (Fig. 4D, I, and J and data not shown). We next analyzed embryos with complete deletion of both receptors to determine if this genotype would phenocopy the PDGFRSKO embryos. To increase the probability of obtaining double-mutant embryos, we crossed our conditional animals with Meox2Cre mice. Meox2Cre expresses Cre in all embryonic tissues and the extraembryonic mesoderm (65). Many of the mutant embryos were being resorbed by E9.5, consistent with the phenotype of PDGF receptor double-homozygous embryos (M. D. Tallquist, unpublished observation), but a few embryos were recovered. They exhibited a complete failure in yolk sac remodeling (PDGFRMeox2) (Fig. 4E) that resembled the hyperfusion phenotype described previously (12, 13), providing further evidence that loss of both PDGF receptors results in yolk sac vascular abnormalities.
Loss of PDGF receptors results in an increase of endothelial gene expression.
Vascular remodeling is controlled by activities of multiple cell populations, including endothelial (15) and mural (57) cells. To examine the differentiation status of these cell populations, we analyzed yolk sac gene expression by real-time PCR. First, expression analysis demonstrated an expected decrease in PDGFRα and PDGFRβ in the SM22α-CreTg mutants compared to wild-type yolk sacs (Fig. 5A). Consistent with results from Hand1 mutants (45), another yolk sac-remodeling mutant, we observed enhanced expression of endothelial receptors, such as VEGFR1 (Flt1), VEGFR2 (Flk1), and Tie2 (Fig. 5B). Levels of endothelial-cell-specific genes, VEGF, PDGFB, and VE cadherin, were not significantly different between mutant and control samples (Fig. 5B). Nonetheless, elevated levels of endothelial receptors in PDGFRSKO yolk sacs suggested that impaired vascular development might be inducing an enhanced but nonproductive angiogenic response.
FIG. 5.

Gene expression analysis in E10.5 yolk sacs. (A) PDGFRSKO mutants demonstrate reduced expression of PDGFRα and PDGFRβ. (B) Endothelial cell gene and growth factor expression. (C) Mesothelial and VSMC-specific genes SM22α, αSMA, and MRTFB suggest a slight decrease in expression in PDGFRSKO yolk sacs. Each symbol represents real-time PCR analysis of one sample analyzed in triplicate. A minimum of three samples were analyzed for each gene, and mean bars are shown for each set of samples analyzed. Student t test: *, <0.2; **, <0.07; ***, <0.02. WT, wild type.
To rule out a direct requirement for PDGF receptors in endothelial cells, we generated embryos that lacked all PDGF receptor expression in endothelial cells using a Tie2CreTg mouse line (PDGFREKO) (30). We recovered viable PDGFREKO mutants at E12.5 and E15.5, and no obvious defects were observed in yolk sac development (data not shown). Together, these data imply that PDGF receptor signaling is not employed by endothelial cells and that the remodeling phenotype is caused by loss of the receptors in either VSMC or the mesothelium.
The remodeling defect is not caused by loss of PDGF receptor signaling in VSMC.
We next examined the expression of VSMC genes in the PDGFRSKO yolk sacs. Consistent with the lack of VSMC we observed by lineage tracing (Fig. 1D), VSMC gene expression was reduced. Expression of the gene encoding myocardin-related transcription factor B (MRTFB), a yolk sac smooth muscle transcription factor (70), was reduced in PDGFRSKO yolk sacs (Fig. 5C). Similarly, expression of the smooth muscle cell cytoskeletal gene αSMA was also reduced (Fig. 5C). SM22α expression, by contrast, exhibited only a partial reduction. This result was anticipated, because mesothelial cells also express SM22α, and they did not appear to be reduced in number (Fig. 1D and data not shown).
These expression data suggested a disruption in the VSMC population. Therefore, we examined E10.5 control, PDGFRSKO, and PDGFRMKO yolk sacs for the presence of VSMC. While the control yolk sacs had extensive networks of vessels that contained αSMA-positive cells, in both mutant yolk sac genotypes, few αSMA-expressing cells were present next to endothelial vessels compared to control yolk sacs (Fig. 4L to N). This result suggested that PDGF receptor signaling was required for VSMC formation. To identify if a specific receptor was important for VSMC formation, we examined αSMA staining in null and conditional mutants for PDGFRα and PDGFRβ individually (Fig. 4 and data not shown). In PDGFRβ−/− and PDGFRβ conditional deletion lines, loss of PDGFRβ led to a dramatic reduction in yolk sac VSMC (Fig. 4O to Q). However, despite the lack of VSMC in these mutants, the yolk sac vasculature was organized into vessels of different calibers. These data suggest that PDGFRβ may be the essential PDGF receptor involved in VSMC development in the yolk sac. However, the presence of normal vessel remodeling in the absence of VSMC indicated that the yolk sac phenotype we observed in PDGFRSKO yolk sacs was not caused by a failure of VSMC association. The mesothelial cells were therefore implicated as the primary cell population responsible for this phenotype, as they were the only yolk sac cells that expressed Cre in SM22α-CreTg conditional mutants that did not express Cre in myocardinCre conditional mutants.
PDGF receptor signaling affects ECM deposition.
Mesothelial cells have a number of proposed functions, including transport of fluids, production of growth factors, and secretion of ECM. Because the vascular defects we observed resembled retinoic acid and TGF-β signaling pathway mutants that are deficient in ECM production (3, 7, 18), we examined the distribution of ECM in wild-type, PDGFRSKO, and PDGFRMKO yolk sacs. Staining for fibrillar collagen with picrosirius red (Fig. 6A) demonstrated that PDGFRSKO yolk sacs had a decreased intensity and thickness of collagen. In contrast, PDGFRMKO yolk sacs appeared similar to wild-type samples. Immunohistochemistry for collagen 1 and collagen 4 in PDGFRSKO yolk sacs supported the picrosirius red findings (Fig. 6B and C). The reduction in collagens was limited to the mesothelium, as collagen 4 was detected in close proximity to the endothelial cells. In contrast, fibronectin and laminin appeared relatively unperturbed at this stage (Fig. 6D and E). To determine if the reduction in collagen was at the transcriptional level, we performed real-time PCR analysis. We detected only modest changes in transcript levels of collagens and fibronectin (Fig. 6F).
FIG. 6.

ECM deposition disruption along the mesothelium. (A) Picrosirius red staining of paraffin sections of E10.5 yolk sacs demonstrating PDGFRSKO yolk sac reduction in collagen compared to control and PDGFRMKO yolk sacs. (B to E) Immunohistochemistry of E10.5 control, PDGFRSKO, and PDGFRMKO yolk sac sections for detection of ECM molecule expression as indicated. (F) Quantitative PCR gene expression analysis for matrix molecules in control and PDGFRSKO E10.5 yolk sacs. Individual symbols and mean bars represent each sample analyzed in triplicate for the wild type (WT) and SKO mutants. Student t test: *, <0.15. (G) E10.5 fresh-frozen yolk sac sections of control and PDGFRSKO mutants imaged for MMP activity observed through increasing levels of fluorescence detected by in situ DQ gelatin assay. (H) Gelatin zymography demonstrating increased MMP2 activity in PDGFRSKO E10.5 whole yolk sac lysates compared to control lysates. The arrowheads indicate mesothelial loss of collagen 1 and collagen 4. The asterisks indicate blood vessels. ee, extraembryonic endoderm; mes, mesothelium. Scale bars = 40 μm.
Because collagen synthesis was only moderately affected, we investigated other processes that would explain a reduction in collagen protein levels. One candidate mechanism was an increase in MMP activity. MMPs are a family of proteinases that are capable of degrading a wide variety of ECM proteins. When we examined yolk sacs using an MMP in situ assay, we detected increased MMP activity in mutant mesothelium compared to controls (Fig. 6G). Similarly, gelatin zymography consistently demonstrated higher levels of activated MMP-2 in PDGFRSKO mutant yolk sacs than in control yolk sacs (Fig. 6H). Taken together, these data suggest that PDGF receptor signaling from the mesothelium may function to direct blood vessel remodeling in part by controlling the degradation of matrix in the yolk sac.
PDGF receptor signaling controls blood vessel morphogenesis.
To further examine the role of PDGF receptors in vascular development, we used an allantois culture assay. E8.5 allantoides from wild-type embryos develop rudimentary vascular structures, but when stimulated with either 10% FBS or PDGFBB, a ligand that can activate both PDGFRα and PDGFRβ, the vascular plexus expands (Fig. 7A and C). In addition, we showed that PDGF receptor signaling in these cultures was required for stabilization of the vessels. Using a Cre-expressing adenovirus, we were able to induce recombination in allantois explants from embryos homozygous for both PDGFRα and PDGFRβ floxed alleles (PDGFRfl/fl). When both PDGF receptors were deleted using Cre, vascular expansion was severely reduced (Fig. 7B and C). Quantification of vascular expansion demonstrated that PDGF receptor stimulation yielded cultures that were comparable to those produced by serum stimulation (Fig. 7C). Similarly, addition of a PDGF receptor-specific inhibitor (AG1296) to the cultures resulted in a lack of vascular expansion similar to that of unstimulated cultures (Fig. 7D). We could not examine the effects of the PDGF receptor inhibitor on PDGFBB-stimulated cultures, as these cultures did not adhere to the coverslip. To determine the effect of exogenous addition of matrix, we plated allantois cultures on collagen 4 and found that exogenous collagen 4 resulted in increased vascular expansion in 1% serum. Treatment with collagen 4 was even capable of bypassing the effects of PDGF inhibition on cultures stimulated with either 10% FBS or PDGFBB (Fig. 7D and data not shown).
FIG. 7.

PDGF receptor signaling in allantois cultures. (A) PECAM staining of E8.5 wild-type allantoides grown in culture for 24 to 26 h with stimulation: 1% FBS, 20 ng PDGFBB, or 10% FBS. (B) PECAM staining of adenovirus Cre-treated E8.5 wild-type or PDGF receptor conditional (PDGFRfl/fl) allantoides demonstrating that the deletion of PDGF receptor disrupts vasculogenesis in response to PDGFBB and 10% FBS. The insets represent threshold images of PECAM fluorescence that were used to quantify the vascular area. (C) Quantification of vasculogenesis in the allantois assays by measurement of PECAM staining in stimulated and unstimulated samples in the presence or absence of PDGF receptor expression. (D) Quantification of PECAM staining in the presence of a PDGF receptor inhibitor (AG1296) and increasing concentrations of collagen 4 compared to 10% FBS. (E) E10.5 yolk sac lysates immunoprecipitated for integrin β1 and analyzed for activation by Western blot analysis for phosphorylation of integrin β1 at S785 and integrin β1 demonstrating decreased phosphorylation yet similar levels of integrin β1. Student t test: *, <0.07; **, <0.04; ***, <0.005. Scale bars = 20 μm. Red, PECAM; blue, DAPI.
Finally, to examine how reduced ECM could affect endothelial cell signaling, we determined if integrin function within the yolk sac was disrupted. To accomplish this, we examined the activation status of integrin β1. Integrin β1 is essential for endothelial cell function (5) and is one of the key integrins in endothelial cell morphogenesis. Phosphorylation of integrin β1 on S785 modulates cell adhesion and migration. Using a phosphospecific antibody for S785 of β1 integrin (46), we found that phosphorylation on S785 was reduced, even though levels of β1 integrin were similar in control and PDGFRSKO yolk sacs (Fig. 7E). Taken together, these data suggest that loss of PDGF receptor signaling leads to reduced vascular remodeling that may be related to a loss of matrix integrity.
DISCUSSION
The generation of mature blood vessels requires coordination between endothelial cells and their surrounding tissues. While it is established that growth factor secretion and guidance cues are necessary for appropriate vessel patterning, many of the processes involved in directing vessel remodeling remain a mystery. Here, we have shown that one of the signals required in yolk sac vessel formation derives from PDGF receptor signals in the mesoderm. By deleting the receptors from select cell populations, we showed that PDGF signaling is required for vascular remodeling. Unexpectedly, we observed normal progression of vasculogenesis in the absence of VSMC, suggesting that vascular remodeling is not due to a failure of VSMC formation. Instead, PDGF receptors in the extraembryonic mesoderm provide signals for yolk sac vascular progression.
The origin of VSMC in the yolk sac is currently unclear, but there are two potential sources. One is from embryonic hemangioblasts that arise in the primitive streak and migrate to the yolk sac to form the blood islands. Clonal analysis of brachyury-positive and VEGFR2-positive cells has suggested that a single progenitor can give rise to endothelial and hematopoietic cells and VSMC (14, 24). Another possibility is that the yolk sac VSMC arise from the yolk sac mesothelium. Evidence is accumulating to suggest that the mesothelium can differentiate into components of blood vessels, including VSMC and fibroblasts. The heart was the first tissue identified in which the mesothelium (epicardium) differentiates and contributes to the vascular structures (10, 43, 44, 67). Others have shown more recently that the serosal mesothelium can also contribute to the VSMC of the gut (29, 71). Interestingly, stimulation of either tissue by PDGFBB results in an increase in VSMC differentiation (29, 40). Although the current reagents do not permit us to conclusively prove the origin of yolk sac VSMC, our data are consistent with the possibility that the yolk sac mesothelium can give rise to perivascular cells. Loss of PDGF receptors in the SM22α-Cre transgenic line leads to an early absence of VSMC. Thus, the mesothelium could be a source of perivascular cells that stabilize the vessels, as well as a source of growth factors and ECM to direct vascular remodeling.
In addition to the uncertainty of VSMC origin, there has also been a longstanding debate over the importance of VSMC in the yolk sac. Disruption of myocardin, an SRF transcriptional cofactor and key regulator of VSMC development, leads to embryonic lethality at E9.5 (69), but it is unclear if this phenotype is caused by loss of VSMC in the yolk sac (51). MRTFB mutant embryos (MRTFB is a second member of the myocardin family of transcription factors) display a reduction in yolk sac VSMC, but these embryos survive past E10.5, when many mutants with yolk sac phenotypes perish (47, 70). Often, the difficulty in interpreting known yolk sac phenotypes is that many of the genes are likely to affect yolk sac development by multiple avenues. For example, both endothelial cell differentiation and cardiac function are essential for proper yolk sac vessel maturation. Mutations in multiple components of the TGF-β signaling pathway demonstrate dramatic yolk sac vascular disruptions, but these defects could be caused by a failure in endothelial cell or VSMC function (2, 11, 25, 35, 49, 72).
Studies of PDGFRβ- and PDGFB-null embryos have established that a subset of VSMC, sometimes referred to as pericytes, require PDGFRβ signal transduction (21, 38, 60). The loss of PDGFRβ seems to predominantly affect VSMC of smaller vessels, while VSMC of larger vessels, such as the aorta, are less disrupted. Here, we have shown that yolk sac VSMC are dependent on PDGFRβ expression. In multiple mouse lines, loss of PDGFRβ leads to a severe reduction in VSMC of the yolk sac. Surprisingly, endothelial cells in our mutants continued to mature into hierarchical vessels, and early embryogenesis was undisturbed. The only abnormality caused by loss of VSMC was an increase in vessel tortuousness. Our data demonstrate that the yolk sac vasculature does not require VSMC for stability, possibly because the hemodynamic forces within these vessels are not excessive.
Although it is commonly assumed that the two PDGF receptors direct similar cellular responses, there are few in vivo examples of the abilities of these receptors to compensate for each other. This fact is underscored by the disparate phenotypes of the individual receptor knockouts. PDGFRα-null embryos die between E10.5 and E15.5 due to a wide range of defects (62), while PDGFRβ-null embryos die perinatally from vascular defects (60). However, in the yolk sac, we found that if one of the receptors was expressed, blood vessel remodeling occurred normally. The observed redundancy in the yolk sac mesoderm leads to the possibility that the PDGF receptors may contribute to vessel remodeling in other tissues. Another example of receptor cooperativity in vessel remodeling was observed in cardiac neural crest cells. When PDGF receptors are removed from neural crest cells, failure of aortic arch vessel remodeling leads to persistent truncus arteriosus (53). Because PDGFRα/β-null embryos die before E10.5, this general requirement for PDGF receptor signaling and vascular remodeling may have been overlooked.
ECM disruptions have been found in several mouse mutants with yolk sac phenotypes. Mutations in collagen 4, fibronectin, and laminin expression have demonstrated vascular defects (16, 52). TGF-β signaling is considered a key component of ECM deposition, and mutations in several components of TGF-β pathways have also demonstrated defects in yolk sac vascular formation (2, 8, 18, 31, 35, 72). Although disrupted ECM deposition is common to both TGF-β pathway mutant and PDGF receptor mutant yolk sacs, the cell types responsible are different. Recently, it was demonstrated that endothelium-specific deletion of TGF-βR1 or TGF-βR2 recapitulates the knockout yolk sac phenotype (6). Our data now point to a second essential population of cells required for yolk sac vessel growth, the mesothelium, and demonstrate that ECM secretion by endothelial cells is not sufficient to promote normal vascular development.
Control of ECM levels by PDGF receptor-expressing cells via MMP activity is a relatively new concept in vascular remodeling. Currently, the few papers addressing this topic conflict. Some data propose that PDGF receptor stimulation results in increased levels of MMP proteins and activity (54), while others propose that stimulation of PDGF receptors leads to inhibition of MMP activity (28). In the yolk sac, we can imagine two possible mechanisms explaining increased MMP activity. The first is that PDGF receptor interactions with MT1-MMP at the cell membrane attenuate MT1-MMP activity, but in the absence of PDGF receptors MMP activity is increased. Data supporting this scenario are as follows: MT1-MMP can activate MMP-2 (56, 63), MT1-MMP is expressed in mesothelial cell populations (74), and MT1-MMP can physically associate with PDGFRβ (33). An alternative possibility is that PDGF receptor stimulation results in secretion of tissue inhibitors of metalloproteinases.
Our findings suggest that PDGF receptors act coordinately in the extraembryonic mesoderm and are involved in the process of vascular remodeling. We have also shown that VSMC are not required in the establishment of mature endothelial vessels in the yolk sac. These studies open up new questions about the cooperativity of the two receptors in stromal cell populations throughout the developing embryo and potentially during vascular remodeling during the disease process.
Acknowledgments
We thank Ondine Cleaver, Dan Garry, and Eric Olson for providing the Tie2CreTg, Tie2GFPTg, and myocardinCre mice, respectively. We also thank Robert Gerard for generously providing Cre-expressing adenovirus. We thank Rhonda Bassel-Duby, Rolf Brekken, Ondine Cleaver, Rita Perlingiero, and our laboratory colleagues for comments on the manuscript. We also thank Angie Bookout, Michelle Kazi, Haley Newton, Banu Eskiocak, and Holly Mead for excellent technical assistance.
We declare no competing financial interest.
Footnotes
Published ahead of print on 7 July 2008.
REFERENCES
- 1.Argraves, W. S., and C. J. Drake. 2005. Genes critical to vasculogenesis as defined by systematic analysis of vascular defects in knockout mice. Anat. Rec. A 286875-884. [DOI] [PubMed] [Google Scholar]
- 2.Arthur, H. M., J. Ure, A. J. Smith, G. Renforth, D. I. Wilson, E. Torsney, R. Charlton, D. V. Parums, T. Jowett, D. A. Marchuk, J. Burn, and A. G. Diamond. 2000. Endoglin, an ancillary TGFβ receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 21742-53. [DOI] [PubMed] [Google Scholar]
- 3.Bohnsack, B. L., L. Lai, P. Dolle, and K. K. Hirschi. 2004. Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Genes Dev. 181345-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohnsack, B. L., L. Lai, J. L. Northrop, M. J. Justice, and K. K. Hirschi. 2006. Visceral endoderm function is regulated by quaking and required for vascular development. Genesis 4493-104. [DOI] [PubMed] [Google Scholar]
- 5.Carlson, T. R., H. Hu, R. Braren, Y. H. Kim, and R. A. Wang. 2008. Cell-autonomous requirement for β1 integrin in endothelial cell adhesion, migration and survival during angiogenesis in mice. Development 1352193-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carvalho, R. L., F. Itoh, M. J. Goumans, F. Lebrin, M. Kato, S. Takahashi, M. Ema, S. Itoh, M. van Rooijen, P. Bertolino, P. Ten Dijke, and C. L. Mummery. 2007. Compensatory signalling induced in the yolk sac vasculature by deletion of TGFβ receptors in mice. J. Cell Sci. 1204269-4277. [DOI] [PubMed] [Google Scholar]
- 7.Carvalho, R. L., L. Jonker, M. J. Goumans, J. Larsson, P. Bouwman, S. Karlsson, P. T. Dijke, H. M. Arthur, and C. L. Mummery. 2004. Defective paracrine signalling by TGFβ in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development 1316237-6247. [DOI] [PubMed] [Google Scholar]
- 8.Cohen, T. V., O. Kosti, and C. L. Stewart. 2007. The nuclear envelope protein MAN1 regulates TGFβ signaling and vasculogenesis in the embryonic yolk sac. Development 1341385-1395. [DOI] [PubMed] [Google Scholar]
- 9.Copp, A. J. 1995. Death before birth: clues from gene knockouts and mutations. Trends Genet. 1187-93. [DOI] [PubMed] [Google Scholar]
- 10.Dettman, R. W., W. Denetclaw, Jr., C. P. Ordahl, and J. Bristow. 1998. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 193169-181. [DOI] [PubMed] [Google Scholar]
- 11.Dickson, M. C., J. S. Martin, F. M. Cousins, A. B. Kulkarni, S. Karlsson, and R. J. Akhurst. 1995. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1211845-1854. [DOI] [PubMed] [Google Scholar]
- 12.Dominguez, M. G., V. C. Hughes, L. Pan, M. Simmons, C. Daly, K. Anderson, I. Noguera-Troise, A. J. Murphy, D. M. Valenzuela, S. Davis, G. Thurston, G. D. Yancopoulos, and N. W. Gale. 2007. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc. Natl. Acad. Sci. USA 1043243-3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drake, C. J., and C. D. Little. 1995. Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc. Natl. Acad. Sci. USA 927657-7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ema, M., P. Faloon, W. J. Zhang, M. Hirashima, T. Reid, W. L. Stanford, S. Orkin, K. Choi, and J. Rossant. 2003. Combinatorial effects of Flk1 and Tal1 on vascular and hematopoietic development in the mouse. Genes Dev. 17380-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flamme, I., T. Frolich, and W. Risau. 1997. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J. Cell Physiol. 173206-210. [DOI] [PubMed] [Google Scholar]
- 16.George, E. L., H. S. Baldwin, and R. O. Hynes. 1997. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood 903073-3081. [PubMed] [Google Scholar]
- 17.Gerhardt, H., M. Golding, M. Fruttiger, C. Ruhrberg, A. Lundkvist, A. Abramsson, M. Jeltsch, C. Mitchell, K. Alitalo, D. Shima, and C. Betsholtz. 2003. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 1611163-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goumans, M. J., A. Zwijsen, M. A. van Rooijen, D. Huylebroeck, B. A. Roelen, and C. L. Mummery. 1999. Transforming growth factor-beta signalling in extraembryonic mesoderm is required for yolk sac vasculogenesis in mice. Development 1263473-3483. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton, T. G., R. A. Klinghoffer, P. D. Corrin, and P. Soriano. 2003. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol. Cell. Biol. 234013-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hellstrom, M., H. Gerhardt, M. Kalen, X. Li, U. Eriksson, H. Wolburg, and C. Betsholtz. 2001. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 153543-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellstrom, M., M. Kalen, P. Lindahl, A. Abramsson, and C. Betsholtz. 1999. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1263047-3055. [DOI] [PubMed] [Google Scholar]
- 22.Hogan, B., R. Beddington, F. Costantini, and E. Lacy. 1994. Manipulating the mouse embryo: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Plainview, NY.
- 23.Holtwick, R., M. Gotthardt, B. Skryabin, M. Steinmetz, R. Potthast, B. Zetsche, R. E. Hammer, J. Herz, and M. Kuhn. 2002. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 997142-7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber, T. L., V. Kouskoff, H. J. Fehling, J. Palis, and G. Keller. 2004. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature 432625-630. [DOI] [PubMed] [Google Scholar]
- 25.Jadrich, J. L., M. B. O'Connor, and E. Coucouvanis. 2006. The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 1331529-1541. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, D. W., J. N. Berg, M. A. Baldwin, C. J. Gallione, I. Marondel, S. J. Yoon, T. T. Stenzel, M. Speer, M. A. Pericak-Vance, A. Diamond, A. E. Guttmacher, C. E. Jackson, L. Attisano, R. Kucherlapati, M. E. Porteous, and D. A. Marchuk. 1996. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13189-195. [DOI] [PubMed] [Google Scholar]
- 27.Kang, J., Y. Gu, P. Li, B. L. Johnson, H. M. Sucov, and P. S. Thomas. 2008. PDGF-A as an epicardial mitogen during heart development. Dev. Dyn. 237692-701. [DOI] [PubMed] [Google Scholar]
- 28.Karakiulakis, G., E. Papakonstantinou, A. J. Aletras, M. Tamm, and M. Roth. 2007. Cell type-specific effect of hypoxia and platelet-derived growth factor-BB on extracellular matrix turnover and its consequences for lung remodeling. J. Biol. Chem. 282908-915. [DOI] [PubMed] [Google Scholar]
- 29.Kawaguchi, M., D. M. Bader, and B. Wilm. 2007. Serosal mesothelium retains vasculogenic potential. Dev. Dyn. 2362973-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kisanuki, Y. Y., R. E. Hammer, J. Miyazaki, S. C. Williams, J. A. Richardson, and M. Yanagisawa. 2001. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230230-242. [DOI] [PubMed] [Google Scholar]
- 31.Larsson, J., M. J. Goumans, L. J. Sjostrand, M. A. van Rooijen, D. Ward, P. Leveen, X. Xu, P. ten Dijke, C. L. Mummery, and S. Karlsson. 2001. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 201663-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leber, T. M., and R. P. M. Negus. 2001. Detection and quantitation of matrix metalloproteases by zymography. Methods Mol. Med. 39509-514. [DOI] [PubMed] [Google Scholar]
- 33.Lehti, K., E. Allen, H. Birkedal-Hansen, K. Holmbeck, Y. Miyake, T. H. Chun, and S. J. Weiss. 2005. An MT1-MMP-PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 19979-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leveen, P., M. Pekny, S. Gebre-Medhin, B. Swolin, E. Larsson, and C. Betsholtz. 1994. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 81875-1887. [DOI] [PubMed] [Google Scholar]
- 35.Li, D. Y., L. K. Sorensen, B. S. Brooke, L. D. Urness, E. C. Davis, D. G. Taylor, B. B. Boak, and D. P. Wendel. 1999. Defective angiogenesis in mice lacking endoglin. Science 2841534-1537. [DOI] [PubMed] [Google Scholar]
- 36.Li, L., J. M. Miano, P. Cserjesi, and E. N. Olson. 1996. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res. 78188-195. [DOI] [PubMed] [Google Scholar]
- 37.Lindahl, P., M. Hellstrom, M. Kalen, L. Karlsson, M. Pekny, M. Pekna, P. Soriano, and C. Betsholtz. 1998. Paracrine PDGF-B/PDGF-Rβ signaling controls mesangial cell development in kidney glomeruli. Development 1253313-3322. [DOI] [PubMed] [Google Scholar]
- 38.Lindahl, P., B. R. Johansson, P. Leveen, and C. Betsholtz. 1997. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277242-245. [DOI] [PubMed] [Google Scholar]
- 39.Long, X., E. E. Creemers, D. Z. Wang, E. N. Olson, and J. M. Miano. 2007. Myocardin is a bifunctional switch for smooth versus skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 10416570-16575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu, J., T. E. Landerholm, J. S. Wei, X. R. Dong, S. P. Wu, X. Liu, K. Nagata, M. Inagaki, and M. W. Majesky. 2001. Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 240404-418. [DOI] [PubMed] [Google Scholar]
- 41.McAllister, K. A., K. M. Grogg, D. W. Johnson, C. J. Gallione, M. A. Baldwin, C. E. Jackson, E. A. Helmbold, D. S. Markel, W. C. McKinnon, J. Murrell, et al. 1994. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 8345-351. [DOI] [PubMed] [Google Scholar]
- 42.Mercola, M., C. Y. Wang, J. Kelly, C. Brownlee, L. Jackson-Grusby, C. Stiles, and D. Bowen-Pope. 1990. Selective expression of PDGF A and its receptor during early mouse embryogenesis. Dev. Biol. 138114-122. [DOI] [PubMed] [Google Scholar]
- 43.Mikawa, T., A. Borisov, A. M. Brown, and D. A. Fischman. 1992. Clonal analysis of cardiac morphogenesis in the chicken embryo using a replication-defective retrovirus: I. Formation of the ventricular myocardium. Dev. Dyn. 19311-23. [DOI] [PubMed] [Google Scholar]
- 44.Mikawa, T., and R. G. Gourdie. 1996. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 174221-232. [DOI] [PubMed] [Google Scholar]
- 45.Morikawa, Y., and P. Cserjesi. 2004. Extra-embryonic vasculature development is regulated by the transcription factor HAND1. Development 1312195-2204. [DOI] [PubMed] [Google Scholar]
- 46.Mulrooney, J. P., T. Hong, and L. B. Grabel. 2001. Serine 785 phosphorylation of the beta1 cytoplasmic domain modulates beta1A-integrin-dependent functions. J. Cell Sci. 1142525-2533. [DOI] [PubMed] [Google Scholar]
- 47.Oh, J., J. A. Richardson, and E. N. Olson. 2005. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc. Natl. Acad. Sci. USA 10215122-15127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orr-Urtreger, A., M. T. Bedford, M. S. Do, L. Eisenbach, and P. Lonai. 1992. Developmental expression of the alpha receptor for platelet-derived growth factor, which is deleted in the embryonic lethal Patch mutation. Development 115289-303. [DOI] [PubMed] [Google Scholar]
- 49.Oshima, M., H. Oshima, and M. M. Taketo. 1996. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 179297-302. [DOI] [PubMed] [Google Scholar]
- 50.Pietras, K., J. Pahler, G. Bergers, and D. Hanahan. 2008. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological _targeting. PLoS Med. 5e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pipes, G. C., E. E. Creemers, and E. N. Olson. 2006. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 201545-1556. [DOI] [PubMed] [Google Scholar]
- 52.Poschl, E., U. Schlotzer-Schrehardt, B. Brachvogel, K. Saito, Y. Ninomiya, and U. Mayer. 2004. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 1311619-1628. [DOI] [PubMed] [Google Scholar]
- 53.Richarte, A. M., H. B. Mead, and M. D. Tallquist. 2007. Cooperation between the PDGF receptors in cardiac neural crest cell migration. Dev. Biol. 306785-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robbins, J. R., P. G. McGuire, B. Wehrle-Haller, and S. L. Rogers. 1999. Diminished matrix metalloproteinase 2 (MMP-2) in ectomesenchyme-derived tissues of the Patch mutant mouse: regulation of MMP-2 by PDGF and effects on mesenchymal cell migration. Dev. Biol. 212255-263. [DOI] [PubMed] [Google Scholar]
- 55.Rolny, C., I. Nilsson, P. Magnusson, A. Armulik, L. Jakobsson, P. Wentzel, P. Lindblom, J. Norlin, C. Betsholtz, R. Heuchel, M. Welsh, and L. Claesson-Welsh. 2006. Platelet-derived growth factor receptor-beta promotes early endothelial cell differentiation. Blood 1081877-1886. [DOI] [PubMed] [Google Scholar]
- 56.Sato, H., T. Takino, Y. Okada, J. Cao, A. Shinagawa, E. Yamamoto, and M. Seiki. 1994. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 37061-65. [DOI] [PubMed] [Google Scholar]
- 57.Saunders, W. B., B. L. Bohnsack, J. B. Faske, N. J. Anthis, K. J. Bayless, K. K. Hirschi, and G. E. Davis. 2006. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J. Cell Biol. 175179-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schatteman, G. C., K. Morrison-Graham, A. van Koppen, J. A. Weston, and D. F. Bowen-Pope. 1992. Regulation and role of PDGF receptor alpha-subunit expression during embryogenesis. Development 115123-131. [DOI] [PubMed] [Google Scholar]
- 59.Schlaeger, T. M., S. Bartunkova, J. A. Lawitts, G. Teichmann, W. Risau, U. Deutsch, and T. N. Sato. 1997. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc. Natl. Acad. Sci. USA 943058-3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soriano, P. 1994. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 81888-1896. [DOI] [PubMed] [Google Scholar]
- 61.Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 2170-71. [DOI] [PubMed] [Google Scholar]
- 62.Soriano, P. 1997. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 1242691-2700. [DOI] [PubMed] [Google Scholar]
- 63.Strongin, A. Y., I. Collier, G. Bannikov, B. L. Marmer, G. A. Grant, and G. I. Goldberg. 1995. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 2705331-5338. [DOI] [PubMed] [Google Scholar]
- 64.Tallquist, M. D., and P. Soriano. 2003. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development 130507-518. [DOI] [PubMed] [Google Scholar]
- 65.Tallquist, M. D., and P. Soriano. 2000. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26113-115. [DOI] [PubMed] [Google Scholar]
- 66.Van den Akker, N. M., L. C. Winkel, M. H. Nisancioglu, S. Maas, L. J. Wisse, A. Armulik, R. E. Poelmann, H. Lie-Venema, C. Betsholtz, and A. C. Gittenberger-de Groot. 2008. PDGF-B signaling is important for murine cardiac development: its role in developing atrioventricular valves, coronaries, and cardiac innervation. Dev. Dyn. 237494-503. [DOI] [PubMed] [Google Scholar]
- 67.Vrancken Peeters, M. P., A. C. Gittenberger-de Groot, M. M. Mentink, and R. E. Poelmann. 1999. Smooth muscle cells and fibroblasts of the coronary arteries derive from epithelial-mesenchymal transformation of the epicardium. Anat. Embryol. 199367-378. [DOI] [PubMed] [Google Scholar]
- 68.Wang, D., P. S. Chang, Z. Wang, L. Sutherland, J. A. Richardson, E. Small, P. A. Krieg, and E. N. Olson. 2001. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105851-862. [DOI] [PubMed] [Google Scholar]
- 69.Wang, Z., D. Z. Wang, G. C. Pipes, and E. N. Olson. 2003. Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. USA 1007129-7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei, K., N. Che, and F. Chen. 2007. Myocardin-related transcription factor B is required for normal mouse vascular development and smooth muscle gene expression. Dev. Dyn. 236416-425. [DOI] [PubMed] [Google Scholar]
- 71.Wilm, B., A. Ipenberg, N. D. Hastie, J. B. Burch, and D. M. Bader. 2005. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 1325317-5328. [DOI] [PubMed] [Google Scholar]
- 72.Yang, X., L. H. Castilla, X. Xu, C. Li, J. Gotay, M. Weinstein, P. P. Liu, and C. X. Deng. 1999. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 1261571-1580. [DOI] [PubMed] [Google Scholar]
- 73.Yoder, M. C., V. E. Papaioannou, P. P. Breitfeld, and D. A. Williams. 1994. Murine yolk sac endoderm- and mesoderm-derived cell lines support in vitro growth and differentiation of hematopoietic cells. Blood 832436-2443. [PubMed] [Google Scholar]
- 74.Zhong, J., M. M. Gencay, L. Bubendorf, J. K. Burgess, H. Parson, B. W. Robinson, M. Tamm, J. L. Black, and M. Roth. 2006. ERK1/2 and p38 MAP kinase control MMP-2, MT1-MMP, and TIMP action and affect cell migration: a comparison between mesothelioma and mesothelial cells. J. Cell Physiol. 207540-552. [DOI] [PubMed] [Google Scholar]