

Abstract
Background:
Pre-exposure of brain to isoflurane, a commonly used anesthetic, induces ischemic tolerance. This phenomenon is called isoflurane preconditioning. However, it is not known whether isoflurane application after ischemia will provide neuroprotection.
Methods:
Corticostriatal slices (400 μm) freshly prepared from adult male Sprague-Dawley rats were subjected to a 15-min oxygen-glucose deprivation (OGD, to simulate ischemia in vitro). Isoflurane was applied after OGD. Brain slices were harvested 2 hr after OGD for measuring 2,3,5-triphenyltetrazolium chloride (TTC) conversion to quantify cell injury. Adult male Sprague-Dawley rats were also subjected to middle cerebral arterial occlusion for 90 min and then treated with or without 2% isoflurane for 60 min started at the onset of reperfusion. The infarct volumes, neurological deficit scores and performance on rotarod were evaluated at 24 hr after the onset of reperfusion.
Results:
Isoflurane applied immediately after the 15-min OGD for 30 min dose-dependently reversed the OGD-induced decrease of TTC conversion. The TTC conversion was 34 ± 16% and 58 ± 28% of the control, respectively, for OGD alone and OGD plus 2% isoflurane (P < 0.05, n = 12). Application of 2% isoflurane for 30 min started at 10 min after the OGD also reduced the OGD-decreased TTC conversion. The presence of 0.3 μM glybenclamide, a general adenosine 5′-triphosphate-sensitive potassium channel blocker, or 500 μM 5-hydroxydecanoic acid, a mitochondrial adenosine 5′-triphosphate-sensitive potassium channel blocker, during the application of 2% isoflurane abolished the isoflurane preservation of TTC conversion. Application of isoflurane during reperfusion also improved neurological outcome after brain ischemia.
Conclusions:
Our results suggest that isoflurane administrated after OGD or brain ischemia provides neuroprotection. Mitochondrial adenosine 5′-triphosphate-sensitive potassium channels may be involved in this protection.
INTRODUCTION
Stroke and brain trauma are leading causes of mortality and morbidity in the United States.1 The underlying pathophysiology for stroke and brain trauma is ischemic brain injury. Despite of intensive research for many years, clinically effective and safe interventions to reduce ischemic brain injury have not been established. Thus, it is an urgent need to identify such interventions.
The concept “ischemic preconditioning” was developed to describe a phenomenon that may be used to reduce ischemic injury in various organ systems, including brain, heart, liver, intestine, kidney, and lung.2-4 Ischemic preconditioning involves applying brief episodes of sub-lethal ischemia to induce a robust protection against the deleterious effects of subsequent, prolonged, lethal ischemia.2-4 In addition to short episodes of ischemia, many stimuli including volatile anesthetics, such as isoflurane and sevoflurane, also can induce preconditioning effects in the brain.4-6 However, preconditioning is clinically feasible only when the occurrence of brain ischemia is predictable, for example, during intracranial vascular procedures requiring temporary occlusion of the blood vessels. In reality, the onset of brain ischemia in patients with stroke and brain trauma often occurs outside the hospital and is not predictable.
Interventions that can be applied after the onset of brain ischemia will have broad applications. It was observed for the first time in 1986 that gentle reperfusion after cardiac ischemia reduced cardiac infarct size in dogs.7 The concept “ischemic postconditioning” was introduced in 2003 by a study in which brief episodes of ischemia were applied at the onset of reperfusion to improve cardiac outcome after ischemia.8 The ischemic postconditioning-induced neuroprotection was shown in rats in 2006.9,10 We hypothesize that postconditioning can be induced by volatile anesthetics. To test this hypothesis, we performed in vitro study by applying oxygen-glucose deprivation (OGD) to rat corticostriatal slices and in vivo study by subjecting rats to transient middle cerebral arterial occlusion (MCAO) and then postconditioned the slices or rats with isoflurane, a commonly used volatile anesthetic.
Materials and Methods
The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Virginia, Charlottesville, Virginia, United States. All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 80-23) revised in 1996. All efforts were made to minimize the number of animals used and their suffering. All reagents unless specified below were obtained from Sigma (St. Louis, MO).
Preparation of brain slices
Similar to the method reported before,11 corticostriatal brain slices were prepared from 2- to 3-month-old, 200- to 250-g, male Sprague-Dawley rats (Hilltop, Scotdale, PA). Rats were anesthetized with isoflurane and then decapitated. Brains were removed rapidly and placed in ice-cold artificial cerebrospinal fluid (aCSF) bubbled with 5% CO2 and 95% O2. The aCSF contained 116 mM NaCl, 26.2 mM NaHCO3, 5.4 mM KCl, 1.8 mM CaCl2, 0.9 mM MgCl2, 0.9 mM NaH2PO4 and 5.6 mM glucose, pH 7.4. The forebrain slices (400 μm thick) were prepared using a vibrating tissue slicer in ice-cold cutting solution containing 260 mM sucrose, 26.2 mM NaHCO3, 3 mM KCl, 1.2 mM NaH2PO4, 5 mM MgCl2 and 9 mM glucose, pH 7.4 and bubbled with 5% CO2 and 95% O2. After sectioning, slices were wrapped in tissue paper bag and placed into a tissue holder (made of plastic, with small holes in it to allow free diffusion of gases and water; this holder also helps to avoid direct gas bubbling on slices). These slices were immersed in circulating aCSF continuously bubbled with 5% CO2 and 95% O2 (oxygenated aCSF) at room temperature for at least 1 hr for recovery of the synaptic function12 and were then transferred to oxygenated aCSF at 37°C for 15 min before they were used for experiments.
In vitro OGD
Ischemia was simulated in vitro by OGD. As described previously,11,12 corticostriatal slices were transferred into a glass beaker containing glucose-free aCSF (also containing 1 mM dithionite, an oxygen absorbent) bubbled with 5% CO2 and 95% N2. Bubbling with these gases for 30 min before the placement of brain slices was allowed to reduce the oxygen content in the solution. Under these conditions, the PO2 in the aCSF was lower than 0.1 mmHg as measured by a Clark oxygen electrode (Cameron Instrument Co., Port Aransas, TX). The beaker containing the slices was immersed in a water bath to keep the temperature of glucose-free aCSF in the beaker at 37°C as monitored by a thermometer. In the time-course experiments, the OGD was maintained for 10, 20 or 30 min. After the OGD, slices were recovered in circulating oxygenated aCSF at 37°C for 2 h to allow cell injury and death that may not be evident immediately after the OGD episode to become apparent.
Isoflurane postconditioning and study groups of brain slices
Isoflurane postconditioning was performed by exposing brain slices to isoflurane after a 15-min OGD. Isoflurane was delivered by the carrier gases (5% CO2 and 95% O2) through an isoflurane vaporizer and the aCSF was pre-gassed with isoflurane for 30 min prior to the introduction of the slices. The solution was continuously gassed with the same isoflurane containing gases during the incubation. Isoflurane concentrations in the aCSF were determined by gas chromatography as we described before13 and the aqueous isoflurane concentration was about 0.44 mM for 2 % isoflurane in the gas phase under the current experimental conditions. After the desired period of isoflurane exposure, slices were recovered in circulating oxygenated aCSF 37°C for 2 h (including isoflurane exposure time). For the time-course experiments, 2% isoflurane was applied for 20, 30, or 60 min immediately after the OGD. For the isoflurane dose-response study, 1, 1.5, 2, 2.5 or 3 % of isoflurane was applied for 30 min immediately after the OGD. For the time-window study, 2% isoflurane was applied for 30 min started at 0, 10, 30, 60, 80 min after the 15-min OGD.
Application of adenosine 5′-triphosphate-sensitive potassium (KATP) channel blockers
To determine whether KATP channels are involved in isoflurane postconditioning-induced neuroprotection, corticostriatal slices were treated with or without 2% isoflurane in the presence or absence of 0.3 μM glybenclamide, a general KATP channel blocker, or 500 μM 5-hydroxydecanoic acid (5-HD), a mitochondrial KATP channel blocker, for 30 min immediately after a 15-min OGD. The selection of the glybenclamide and 5-HD doses was based on previous studies.11,14
Quantification of cell injury in brain slices
Cell injury was quantified by the conversion of 2,3,5-triphenyltetrazolium chloride (TTC) to formazan as we described previously.5 TTC is normally colorless and can be reduced by succinate dehydrogenase in mitochondria of living cells to formazan that is red. TTC was freshly dissolved to make 2% concentration in solution containing 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 10 mM Hepes and 3 mM glucose. Slices were placed in 3 ml fresh TTC solution contained in each well of 12-well plate for 80 min at 37 ± 0.2°C. The TTC solution was removed and the slices were rinsed twice by 3 ml normal saline. The formazan in the brain slices was extracted by 2.5 ml 50:50 mixture of ethanol/dimethylsulfoxide. The 12-well plate was sealed and placed in dark for 24 h. The red solvent extract (1 ml) in duplicates was placed in cuvettes and the absorbance was read at 485 λ in a spectrophotometer. The absorbance of the duplicates was averaged to reflect the amount of formazan in the sample. The slices were then kept in a clean 12-well plate for 2 day and the dry weight of each slice was measured. The absorbance was normalized by the dry weight of the slice and the results under experimental conditions were then converted to the percentage of control slices included in each experiment.
Morphological examination of brain slices
After a 2-hr recovery, slices were fixed in 4% paraformaldehyde in buffered saline overnight at 4°C. The slices then were paraffin embedded. Four micrometer thick sections were obtained from an interior region of the brain slices (approximately 100 μm from the edge) to avoid areas subjected to slicing trauma during slice preparation.11 The sections were stained with hematoxylin and eosin. The sections were examined by an observer blinded to the group assignment to determine the percentage of non-damaged neurons (survival rate) in the cortex. Damaged neurons were identified if the cells had one of the following characteristics: cell swelling, vacuolization, or presence of shrunken, darkened nuclei.15 At least 30 neurons in the cortex were counted from each of the sections and cells in total 3 sections were counted for each experimental condition (> 90 neurons) to compute the percentages.
Transient middle cerebral arterial occlusion
The rats were anesthetized with isoflurane and then intubated and mechanically ventilated with 60% O2-40% N2 containing 2% isoflurane. The tail artery was cannulated for monitoring arterial blood pressure and blood sampling for measuring blood gases and glucose. As we described previously,4 the MCAO was achieved by advancing a 3-0 monofilament nylon suture with a rounded tip to the right internal carotid artery via the external carotid artery until slight resistance was felt. The common carotid artery was not ligated. Isoflurane anesthesia was stopped once the suture was in place. After recovery from anesthesia, rats were placed back in their cages with ad libitum access to food and water. Rats were reanesthetized with isoflurane at 90 min after the onset of MCAO. The nylon suture was removed. The rats in the MCAO only group were allowed to awaken immediately and the reanesthesia time was about 1 min for each animal. The rats in the MACO plus isoflurane postconditioning group were maintained under anesthesia with 2% isoflurane via an endotracheal tube for 60 min. During anesthesia for achieving MCAO and postconditioning, temporalis muscle temperature was strictly maintained at 37 ± 0.2°C by warming blanket and lamps. The inhaled and exhaled gases were also monitored with a Datex infrared analyzer (Capnomac, Helsinki, Finland) to maintain normal end-tidal carbon dioxide concentrations.
Evaluation of infarct volume, neurological deficit scores and motor coordination
The evaluation of infarct volume at 24 hr after the transient MCAO was performed after TTC staining as described previously.16 Briefly, brain hemispheres were cut into 2 mm thick coronal brain slices (usually total 6 slices) in the rostral-caudal direction. The slices were incubated with TTC for 30 min at 37°C. The infarct areas were quantified using the NIH Image 1.60 (Bethesda, MD). To account for the cerebral edema and differential shrinkage resulting from brain ischemia and tissue processing and to correct for the individual difference in brain volume, the percentage of infarct volume in the ipsilateral hemisphere volume was calculated.17
Neurological deficit scores were evaluated 24 hr after the transient MCAO based on an eight-point scale. Rats were scored as follows: 0, no apparent deficits; 1, failure to extend left forepaw fully; 2, decreased grip of the left forelimb; 3, spontaneous movement in all directions, contralateral circling only if pulled by the tail; 4, circling or walking to the left; 5, walking only if stimulated; 6, unresponsiveness to stimulation and with depressed level of consciousness; 7, dead.18
Motor coordination was evaluated 24 hr before and 24 hr after the transient MCAO. The rats were placed on an accelerating rotarod. The speed of the rotarod was increased from 4 rpm to 40 rpm in 5 min. The latency and the speed of rat's falling off the rotarod were recorded. Each rat was tested three times. The speed-latency index (latency in seconds × speed in rpm) of each of the three tests was calculated and the mean index of the three trials was used to reflect the motor coordination function of each rat before or after the MCAO. All rats were trained for 3 continuous days before the formal tests.
Statistical analysis
All results except for the neurological deficient scores are presented as means ± S.D. Brain slices for various experimental conditions in each set of experiments were from the same rat brains. Each experimental condition was repeated at least 8 times with brain slices from different rats. Statistical analysis for the brain slice data was performed by one-way repeated measures analysis of variance followed by the Dunn's method or by one-way repeated measures analysis of variance on ranks followed by the Dunn's method as indicated in the figure legends for figure 1 - 5. The infarct volume and speed-latency index were analyzed by t test. Neurological deficient scores were analyzed by Mann-Whitney rank sum test. A P < 0.05 was accepted as significant. All statistical analyses were performed with the SigmaStat (Systat Software, Inc., Point Richmond, CA).
Fig. 1. Time-course of oxygen-glucose deprivation (OGD)-induced cell injury.
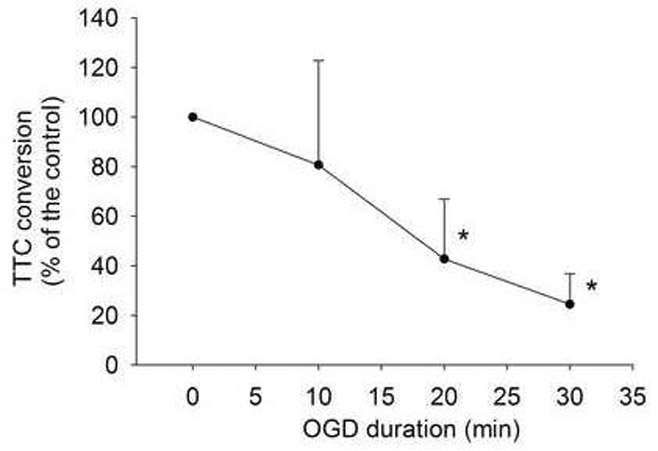
Corticostriatal slices from adult male rats were subjected to various lengths of OGD. Cell injury was quantified by 2, 3, 5-triphenyltetrazolium chloride (TTC) conversion 2 h after OGD. Results are means ± S.D. (n = 8 per time point). Statistical analysis was performed by one-way repeated measures analysis of variance on ranks followed by the Dunn's method. * P < 0.05 compared with control.
Fig. 5. The inhibition of adenosine 5′-triphosphate-sensitive potassium channel blockers on isoflurane postconditioning-induced neuroprotection.
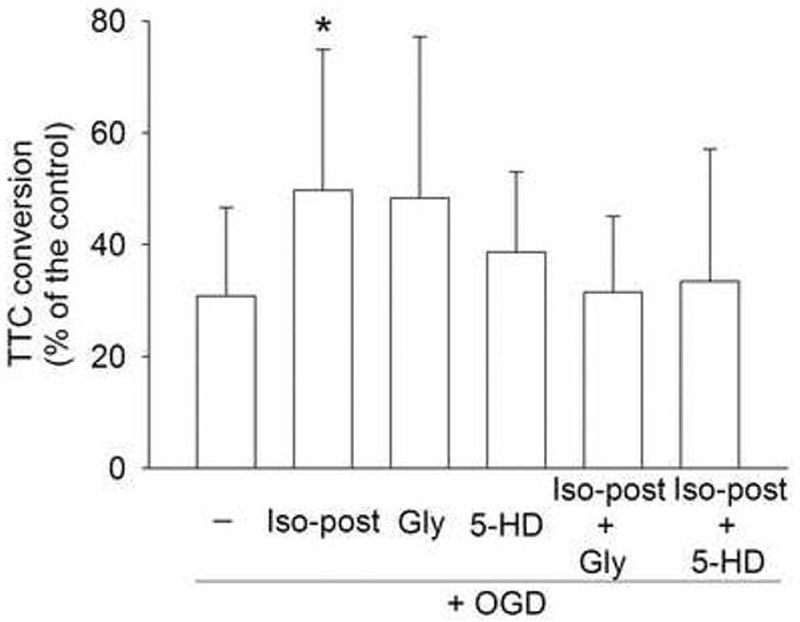
Corticostriatal slices were subjected to a 15-min oxygen-glucose deprivation (OGD) and then were immediately postconditioned with or without 2% isoflurane for 30 min in the presence or absence of 0.3 μM glybenclamide or 500 μM 5-hydroxydecanoic acid (5-HD). Cell injury was quantified by 2,3,5-triphenyltetrazolium chloride (TTC) conversion 2 h after OGD. Results are means ± S.D. (n = 10). Statistical analysis was performed by one-way repeated measures analysis of variance followed by the Dunn's method. * P < 0.05 compared with OGD only. Gly: glybenclamide; Iso-post: isoflurane postconditioning.
RESULTS
TTC staining has been used as a classical method to quantify brain infarct size. TTC staining and then measuring the extracted formazan have been used in recent years to quantify brain injury after ischemia.19-21 This method may be particularly suitable for objectively assessing brain injury that is diffused in nature. Consistent with our previous study,5 the longer the OGD duration, the greater the cell injury in the corticostriatal slices as assessed by TTC conversion (Fig. 1). Histology study showed that a 15-min OGD induced cell injury as evidenced by condensed nuclei (Fig. 2).
Fig. 2. Representative sections of corticostriatal slices stained with hematoxylin and eosin.
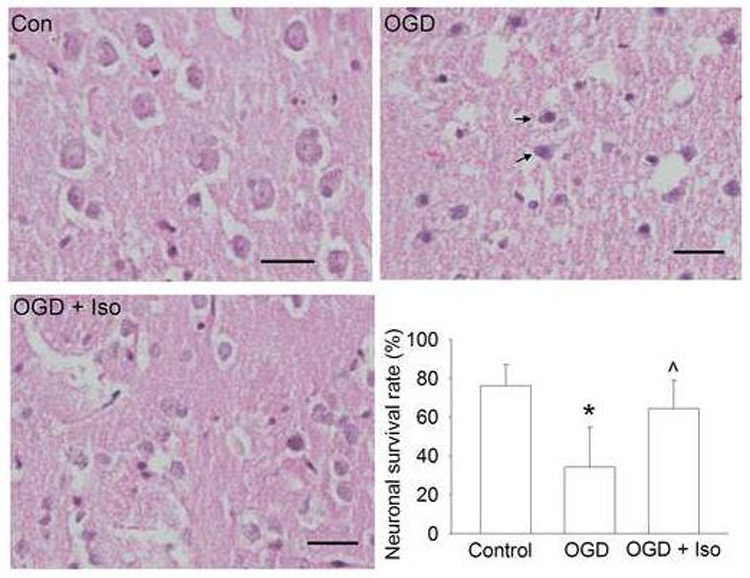
The glucose-oxygen deprivation (OGD) was for 15 min at 37°C. Injured cells are presented with one of the following characteristics: cell swelling, vacuolization (commonly associated with light-staining cytoplasm) or presence of shrunken, darkened nuclei. Arrows in the OGD panel indicated neurons with condensed nuclei. The bar chart presents the quantification data. Results are means ± S.D. (n = 15). Statistical analysis was performed by one-way repeated measures analysis of variance followed by the Dunn's method. * P < 0.05 compared with control. ^ P < 0.05 compared with OGD only. Con: control; Iso: 2% isoflurane applied for 30 min at the onset of simulated reperfusion after the OGD. Scale bar: 20 μm.
The postconditioning of the brain slices with isoflurane immediately after OGD reduced cell injury (Figs 2 and 3). This neuroprotection is time-dependent and the incubation of slices with 2% isoflurane for 30 min significantly reduced the 15-min OGD-induced cell injury (Fig. 3). This neuroprotection was also isoflurane concentration-dependent. Incubation with 1.5% isoflurane for 30 min was sufficient to provide significant neuroprotection (Fig. 3). However, treatment with concentrations higher than 2.5% isoflurane for 30 min did not significantly reduce cell injury. When we applied 2% isoflurane for 30 min started at 0, 10, 30, 60 and 80 min after the 15-min OGD, the incubation of isoflurane at the 0 and 10 min time points significantly reduced cell injury (Fig. 4).
Fig. 3. Time-course and dose-response of isoflurane postconditioning.
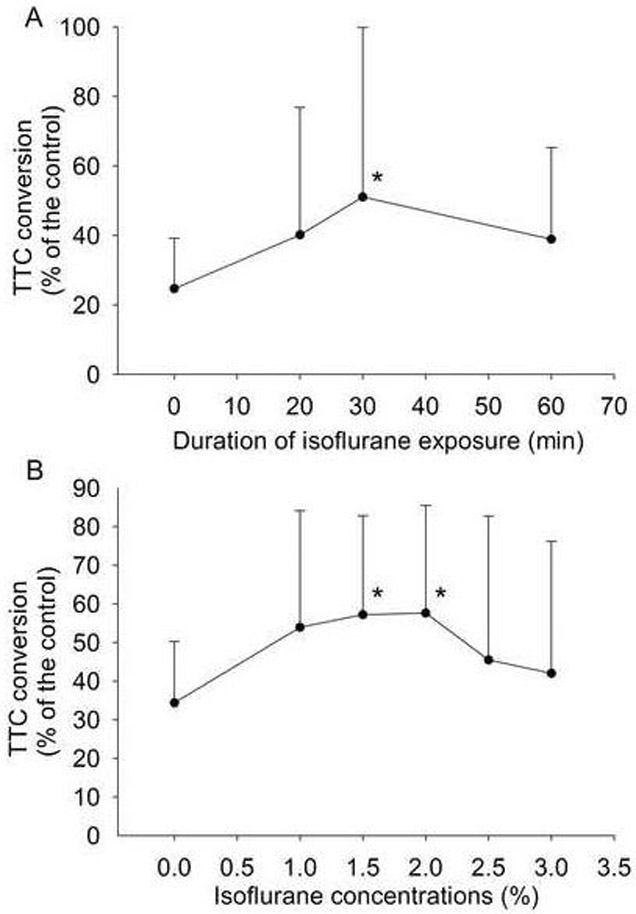
Corticostriatal slices were postconditioned with 2% isoflurane for various durations after a 15-min oxygen-glucose deprivation (OGD) (Panel A) or with various concentrations of isoflurane for 30 min after a 15-min OGD (Panel B). Isoflurane was applied at the onset of simulated reperfusion after the OGD. Cell injury was quantified by 2,3,5-triphenyltetrazolium chloride (TTC) conversion 2 h after the OGD. Results are means ± S.D. (n = 11 for panel A and = 12 for panel B). Statistical analysis was performed by oneway repeated measures analysis of variance on ranks followed by the Dunn's method. * P < 0.05 compared with OGD only.
Fig. 4. Time-window of isoflurane postconditioning.
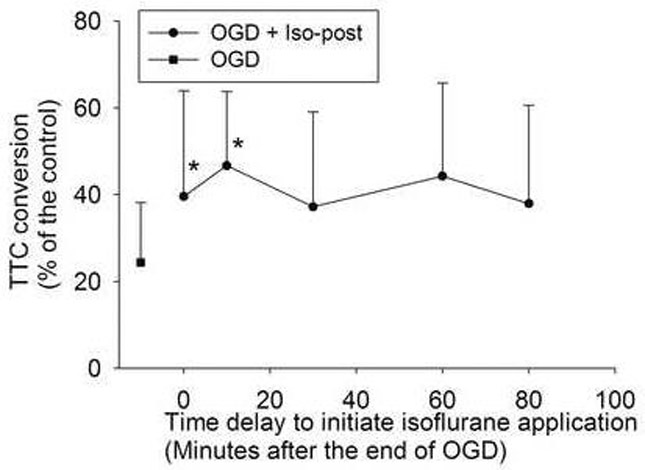
Corticostriatal slices were postconditioned with 2% isoflurane for 30 min at various time points after the end of a 15-min oxygen-glucose deprivation (OGD). Cell injury was quantified by 2,3,5-triphenyltetrazolium chloride (TTC) conversion 2 h after OGD. Results are means ± S.D. (n = 9). Statistical analysis was performed by one-way repeated measures analysis of variance on ranks followed by the Dunn's method. * P < 0.05 compared with OGD only. Iso-post: isoflurane postconditioning.
Glybenclamide, a general KATP channel inhibitor, attenuated the isoflurane postconditioning-induced neuroprotection. The specific mitochondrial KATP channel inhibitor 5-HD also blocked the neuroprotection induced by isoflurane postconditioning (Fig 5). Both glybenclamide and 5-HD did not affect the cell injury induced by OGD only (Fig. 5).
There were no differences in physiological parameters among rats in MCAO and MCAO plus isoflurane postconditioning groups in the in vivo study (Table 1). Application of 2% isoflurane for 60 min started at the onset of reperfusion reduced brain infarction and improved neurological deficient scores and motor coordination after the transient MCAO (Fig. 6). These results suggest that isoflurane induced a postconditioning effect in the brain under in vivo conditions.
Table 1.
Physiological parameters in rats subjected to middle cerebral arterial occlusion (MCAO) with or without isoflurane postconditioning.
| MCAO |
MCAO + Isoflurane |
|||
|---|---|---|---|---|
| Before MCAO | During MCAO | Before MCAO | During MCAO | |
| MABP (mm Hg) | 112 ± 7(7) | 113 ± 7(7) | 112 ± 7(8) | 111 ± 7(8) |
| Heart rate (beat/min) | 340 ± 13 (7) | 341 ± 15 (7) | 331 ± 29 (8) | 344 ± 11 (8) |
| Temperature (°C) | 37.0 ± 0.1 (7) | 37.0 ± 0.1 (7) | 37.0 ± 0.1 (8) | 37.1 ± 0.2 (8) |
| pH | 7.36 ± 0.2 (5) | 7.36 ± 0.2 (5) | 7.41 ± 0.22 (7) | 7.45 ± 0.26 (5) |
| PaCO2 (mm Hg) | 43 ± 18 (5) | 42 ± 16 (5) | 37 ± 18 (7) | 35 ± 20 (5) |
| PaO2 (mm Hg) | 240 ± 76 (5) | 217 ± 44 (5) | 257 ± 80 (7) | 210 ± 40 (5) |
| Glucose (mg/dl) | 166 ± 71 (5) | 179 ± 94 (5) | 172 ± 66 (7) | 196 ± 92 (5) |
Arterial samples were taken 10 min before and after the onset of MCAO. Data are mean ± S.D. (sample sizes). Temporalis muscle temperature was collected and shown here. There were no statistical differences in these physiological parameters among the rats treated with or without 2% isoflurane for 60 min during the reperfusion after the MCAO. MABP: mean arterial blood pressure.
Fig. 6.
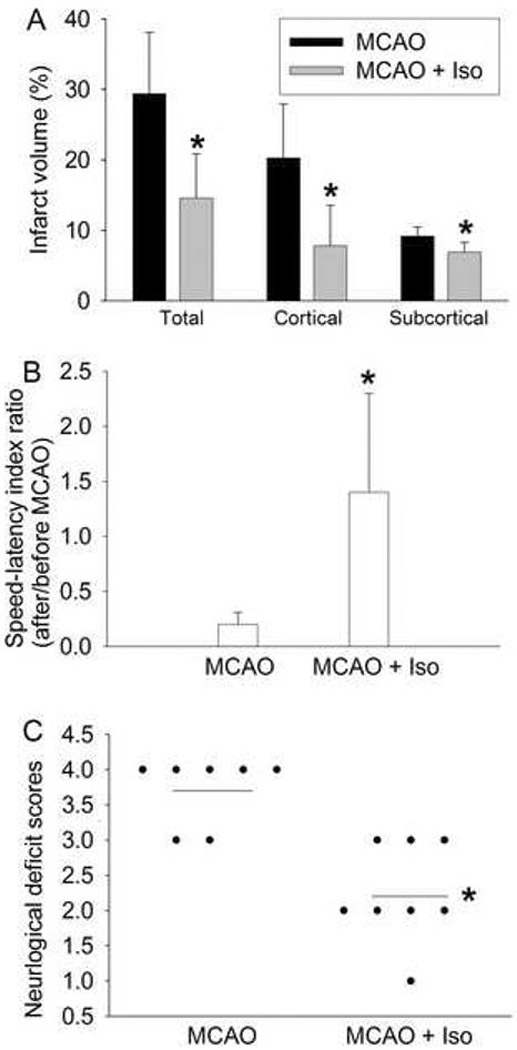
Improvement of neurological outcome by isoflurane postconditioning after transient right middle cerebral arterial occlusion (MCAO). MCAO was for 90 min and isoflurane postconditioning was for 60 min started at the onset of reperfusion. A: Percentage of infarct volume in ipsilateral hemisphere volume. Results are the means ± S.D. (n = 7 for the MCAO only group and n = 8 for the MCAO plus isoflurane postconditioning group). B: The performance on rotarod. Rats were tested before and 24 hr after the MCAO and the speed-latency index ratio of these two tests are presented. Results are the means ± S.D. (n = 7 for the MCAO only group and n = 8 for the MCAO plus isoflurane postconditioning group). C: Neurological deficit scores that were evaluated immediately before the animals were sacrificed for the assessment of infarct sizes (data are presented in Fig. 6A) are shown. Each circle represents the score for a single rat. A horizontal bar indicates the mean value for each group. * P < 0.05 compared with the corresponding MCAO only group. Iso: isoflurane postconditioning.
DISCUSSION
Intensive research efforts have been devoted in the last decades to identifying effective methods to reduce ischemic brain injury. One of such directions is to revascularize/reopen the occluded vessels for timely reperfusion, for example, by using thrombolytic reagents in ischemic stroke22 or by reducing brain edema and intracranial pressure in brain trauma. However, it is now a well-known phenomenon that reperfusion can cause injury.23 Thus, manipulation of the reperfusion process may produce neuroprotection. In 2006, it was shown that short interruptions of reperfusion (3 cycles of 30-sec reperfusion and 10 sec of blood supply interruptions) reduced brain infarct size in rats.24 However, the application of this ischemic postconditioning-induced neuroprotection may be very limited because of the difficulty to achieve well-controlled interruptions of reperfusion and the risks associated with these manipulations. Thus, it would be more applicable if relatively safe drugs are capable to induce the postconditioning effects.
Volatile anesthetics are relatively safe drugs and have been used in clinical practice for many years. Volatile anesthetics have been shown to induce preconditioning effects in the brain.4,11 In this study, we showed that application of isoflurane during the onset of simulated reperfusion reduced cell injury in rat corticostriatal slices. Application of isoflurane during reperfusion after MCAO also improved neurological outcome in rats. These results suggest that isoflurane induces a postconditioning effect in the brain.
To characterize this isoflurane postconditioning-induced neuroprotection, we performed time-course and dose-response studies. Our results showed that concentrations as low as 1.5% isoflurane for 30 min applied at the onset of simulated reperfusion was sufficient to significantly reduce cell injury after a 15-min OGD in rat corticostriatal slices. This protection was reserved when 2% isoflurane was applied. These results suggest that isoflurane at clinically relevant concentrations induces postconditioning effects. However, application of isoflurane concentrations higher than 2% for 30 min or 2% isoflurane for longer than 30 min did not significantly reduce the cell injury. The reasons for the failure of higher isoflurane concentrations or longer incubations to induce a protective effect are not known. One possibility for this phenomenon is that isoflurane at high concentrations or for long incubations may cause/enhance mechanisms to kill cells, which can overwhelm the isoflurane-induced protective effects. For example, isoflurane has been shown to cause mild to moderate increase of free radicals, such as nitric oxide, in the brain.25,26 Production of a large amount of free radicals during the reperfusion phase is considered to be a major process to cause reperfusion injury.27 The additional free radical production stimulated by isoflurane at the onset of simulated reperfusion may then exaggerate the reperfusion injury. Consistent with our findings reported here, it has been shown that prolonged exposure of high concentrations of isoflurane induces neuronal injury.28,29
Many authors have believed that the short interruptions of reperfusion are needed immediately or at a short delay after the onset of reperfusion for the ischemic postconditioning-induced myocardial protection to occur.30,31 We found 4 studies on ischemic postconditioning-induced neuroprotection in the literature: the short interruptions of reperfusion was started within 10 min after the onset of reperfusion in three studies9,32,33 and a short episode of ischemia was applied at 2 days after the initial ischemia in the fourth study.10 Our study showed that the treatment with 2% isoflurane for 30 min should be applied within 10 min after the onset of the simulated reperfusion for isoflurane to induce a postconditioning effect under our current experimental conditions.
Interestingly, a recent study showed that the combination of sevoflurane preconditioning and postconditioning did not provide additional cardioprotection over the sevoflurane preconditioning or postconditioning alone.34 Similarly, the combination of ischemic preconditioning and postconditioning may not induce better neuroprotection than ischemic preconditioning or postconditioning alone.33 These findings suggest that preconditioning and postconditioning may involve shared mechanisms to induce protection.
Multiple intracellular mechanisms including activation of protein kinase C, mitogen-activated protein kinase, adenosine receptors and KATP channels have been proposed to be involved in the anesthetic preconditioning-induced cardioprotection and neuroprotection.4,35,36 We hypothesize that isoflurane postconditioning-induced neuroprotection involves KATP channels. Activation/opening of KATP channels is one of the important mechanisms for ischemic preconditioning-induced protection in various organs and ischemic postconditioning-induced cardioprotection.30,35,37 KATP channels are inhibited by adenosine 5′-triphosphate and opened under energy-depleted conditions. There are two types of KATP channels: those located on the surface of the sarcolemma and those on the inner mitochondrial membrane. It has been proposed that the opening of mitochondrial KATP channels generates an outward current that can stabilize the mitochondrial membrane and reduce the opening of mitochondrial permeability transition pore to block cell death.38,39 Multiple studies have implicated the involvement of mitochondrial KATP channels in the ischemic preconditioning-induced cardioprotection.3,37 Recently, Yang and coworkers have shown that the ischemic postconditioning-induced cardioprotection may be mediated by mitochondrial KATP channels.30 In addition, activation/opening of mitochondrial KATP channels may also be involved in volatile anesthetic preconditioning- and postconditioning-induced cardioprotection.40 Similarly, evidence suggests that volatile anesthetic preconditioning-induced neuroprotection is mediated by mitochondrial KATP channels.35 Our current study showed that the isoflurane postconditioning-induced neuroprotection was abolished by glybenclamide, a general KATP channel blocker, and 5-HD, a specific mitochondrial KATP channel blocker. These results suggest that the isoflurane postconditioning-induced neuroprotection is mediated by the mitochondrial KATP channels.
Our study has limitations. We used a TTC conversion method as the main method to quantify the cell injury in brain slices. The quantification of cell injury by this method correlates well with that by lactate dehydrogenase release in brain slices.41 However, it is not known with the TTC conversion method what types of cells are injured by the ODG and the simulated reperfusion and are protected by the isoflurane postconditioning. Neurons are more sensitive to ischemic injury and our histological study suggests the injured cells are neurons. Thus, our results may most likely reflect the changes of neurons in the slices. Also, all rats were anesthetized with isoflurane briefly (< 2 min) and then brains were harvested in the brain slice study. The brains might have been preconditioned by isoflurane. However, the preconditioning effects may be very minimal because it took longer than 5 min of isoflurane exposure to induce significant preconditioning effects in brain slices.11 In addition, the brain slices for all experimental conditions in each set of experiments on each day were always from the same rat brain. Finally, the rats in the in vivo study may have benefited from the exposure to isoflurane during the procedure of MCAO. However, animals in both MCAO only and MCAO plus isoflurane postconditioning groups received isoflurane during the procedure. The better neurological functions of rats in the MCAO plus isoflurane postconditioning group should be contributed to the isoflurane exposure during reperfusion because this isoflurane exposure is the only difference in the experimental procedures between MCAO only group and MCAO plus isoflurane postconditioning group. Drug-drug interaction to explain the different neurological outcome between these two groups of animals is unlikely because we used the same drug isoflurane during the MCAO procedure and the early phase of reperfusion.
In summary, we provided evidence to show for the first time that isoflurane at clinically relevant concentrations can induce a postconditioning effect in the brain. The mechanism for this protection may involve the activation/opening of mitochondrial KATP channels.
Acknowledgments
Funding: This study was supported by grants (GM065211 and NS045983 to Z Zuo) from the National Institute of Health, Bethesda, Maryland, United States, by a grant from the International Anesthesia Research Society (Frontiers in Anesthesia Research Award to Z Zuo), Cleveland, Ohio, United States and by a Grant-in-Aid from the American Heart Association Mid-Atlantic Affiliate (0755450U to Z Zuo), Baltimore, Maryland, United States.
Footnotes
The research work was performed in and should be attributed to the Department of Anesthesiology, University of Virginia, Charlottesville, VA22908, U.S.A.
Summary statement: Treatment of brain slices with isoflurane immediately after oxygen-glucose deprivation reduced cell injury. Isoflurane application during reperfusion decreased ischemic brain injury in rats. This isoflurane postconditioning-induced protection may be mediated by mitochondrial adenosine 5′-triphosphate-sensitive potassium channels.
References
- 1.Martin JA, Smith BL, Matthews TJ, Ventura SJ. Births and Deaths: Preliminary Data for 1998. National Vital Statistics Reports. 1999;47:1–45. [PubMed] [Google Scholar]
- 2.Ferrari R, Ceconi C, Curello S, Percoco G, Toselli T, Antonioli G. Ischemic preconditioning, myocardial stunning, and hibernation: basic aspects. American Heart Journal. 1999;138:S61–8. doi: 10.1016/s0002-8703(99)70322-4. [DOI] [PubMed] [Google Scholar]
- 3.Tomai F, Crea F, Chiariello L, Gioffre PA. Ischemic preconditioning in humans: models, mediators, and clinical relevance. Circulation. 1999;100:559–63. doi: 10.1161/01.cir.100.5.559. [DOI] [PubMed] [Google Scholar]
- 4.Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of p38 mitogen-activated protein kinase. Molecular Pharmacology. 2004;65:1172–80. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- 5.Wang C, Lee J, Jung H, Zuo Z. Pretreatment with volatile anesthetics, but not with the nonimmobilizer 1,2-dichlorohexafluorocyclobutane, reduced cell injury in rat cerebellar slices after an in vitro simulated ischemia. Brain Research. 2007;1152:201–8. doi: 10.1016/j.brainres.2007.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Payne RS, Akca O, Roewer N, Schurr A, Kehl F. Sevoflurane-induced preconditioning protects against cerebral ischemic neuronal damage in rats. Brain Res. 2005;1034:147–52. doi: 10.1016/j.brainres.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto F, Allen BS, Buckberg GD, Bugyi H, Leaf J. Reperfusion conditions: importance of ensuring gentle versus sudden reperfusion during relief of coronary occlusion. J Thorac Cardiovasc Surg. 1986;92:613–20. [PubMed] [Google Scholar]
- 8.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 9.Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–21. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- 10.Burda J, Danielisova V, Nemethova M, Gottlieb M, Matiasova M, Domorakova I, Mechirova E, Ferikova M, Salinas M, Burda R. Delayed postconditionig initiates additive mechanism necessary for survival of selectively vulnerable neurons after transient ischemia in rat brain. Cell Mol Neurobiol. 2006;26:1141–51. doi: 10.1007/s10571-006-9036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng S, Zuo Z. Isoflurane preconditioning reduces Purkinje cell death in an in vitro model of rat cerebellar ischemia. Neuroscience. 2003;118:99–106. doi: 10.1016/s0306-4522(02)00767-4. [DOI] [PubMed] [Google Scholar]
- 12.Popovic R, Liniger R, Bickler PE. Anesthetics and mild hypothermia similarly prevent hippocampal neuron death in an in vitro model of cerebral ischemia. Anesthesiology. 2000;92:1343–9. doi: 10.1097/00000542-200005000-00024. [DOI] [PubMed] [Google Scholar]
- 13.Zuo Z, Johns RA. Halothane, enflurane, and isoflurane do not affect the basal or agonist-stimulated activity of partially isolated soluble and particulate guanylyl cyclases of rat brain. Anesthesiology. 1995;83:395–404. doi: 10.1097/00000542-199508000-00020. [DOI] [PubMed] [Google Scholar]
- 14.Lim YJ, Zheng S, Zuo Z. Morphine preconditions purkinje cells against cell death under in vitro simulated ischemia-reperfusion conditions. Anesthesiology. 2004;100:562–8. doi: 10.1097/00000542-200403000-00015. [DOI] [PubMed] [Google Scholar]
- 15.Bak IJ, Misgeld U, Weiler M, Morgan E. The preservation of nerve cells in rat neostriatal slices maintained in vitro: a morphological study. Brain Research. 1980;197:341–53. doi: 10.1016/0006-8993(80)91120-8. [DOI] [PubMed] [Google Scholar]
- 16.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. PNAS. 1999;96:12866–9. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. Journal of Cerebral Blood Flow & Metabolism. 1990;10:290–3. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 18.Rogers DC, Campbell CA, Stretton JL, Mackay KB. Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke. 1997;28:2060–5. doi: 10.1161/01.str.28.10.2060. [DOI] [PubMed] [Google Scholar]
- 19.Preston E, Webster J. Spectrophotometric measurement of experimental brain injury. J Neurosci Methods. 2000;94:187–92. doi: 10.1016/s0165-0270(99)00146-6. [DOI] [PubMed] [Google Scholar]
- 20.Jiang SX, Lertvorachon J, Hou ST, Konishi Y, Webster J, Mealing G, Brunette E, Tauskela J, Preston E. Chlortetracycline and demeclocycline inhibit calpains and protect mouse neurons against glutamate toxicity and cerebral ischemia. J Biol Chem. 2005;280:33811–8. doi: 10.1074/jbc.M503113200. [DOI] [PubMed] [Google Scholar]
- 21.Weaver JG, Tarze A, Moffat TC, Lebras M, Deniaud A, Brenner C, Bren GD, Morin MY, Phenix BN, Dong L, Jiang SX, Sim VL, Zurakowski B, Lallier J, Hardin H, Wettstein P, van Heeswijk RP, Douen A, Kroemer RT, Hou ST, Bennett SA, Lynch DH, Kroemer G, Badley AD. Inhibition of adenine nucleotide translocator pore function and protection against apoptosis in vivo by an HIV protease inhibitor. J Clin Invest. 2005;115:1828–38. doi: 10.1172/JCI22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher M, Brott TG. Emerging therapies for acute ischemic stroke: new therapies on trial. Stroke. 2003;34:359–61. doi: 10.1161/01.str.0000054627.69159.c2. [DOI] [PubMed] [Google Scholar]
- 23.Gross GJ, Auchampach JA. Reperfusion injury: does it exist? J Mol Cell Cardiol. 2007;42:12–8. doi: 10.1016/j.yjmcc.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao P, Huang Y, Zuo Z. Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. J Neuropathol Exp Neurol. 2006;65:945–52. doi: 10.1097/01.jnen.0000235123.05677.4b. [DOI] [PubMed] [Google Scholar]
- 25.Loeb AL, Raj NR, Longnecker DE. Cerebellar nitric oxide is increased during isoflurane anesthesia compared to halothane anesthesia: a microdialysis study in rats. Anesthesiology. 1998;89:723–30. doi: 10.1097/00000542-199809000-00024. [DOI] [PubMed] [Google Scholar]
- 26.Baumane L, Dzintare M, Zvejniece L, Meirena D, Lauberte L, Sile V, Kalvinsh I, Sjakste N. Increased synthesis of nitric oxide in rat brain cortex due to halogenated volatile anesthetics confirmed by EPR spectroscopy. Acta Anaesthesiol Scand. 2002;46:378–83. doi: 10.1034/j.1399-6576.2002.460408.x. [DOI] [PubMed] [Google Scholar]
- 27.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–9. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 28.Wei H, Liang G, Yang H. Isoflurane preconditioning inhibited isoflurane-induced neurotoxicity. Neurosci Lett. 2007;425:59–62. doi: 10.1016/j.neulet.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei H, Kang B, Wei W, Liang G, Meng QC, Li Y, Eckenhoff RG. Isoflurane and sevoflurane affect cell survival and BCL-2/BAX ratio differently. Brain Res. 2005;1037:139–47. doi: 10.1016/j.brainres.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by _targeting cell signaling pathways. J Am Coll Cardiol. 2004;44:1103–10. doi: 10.1016/j.jacc.2004.05.060. [DOI] [PubMed] [Google Scholar]
- 31.Vinten-Johansen J, Yellon DM, Opie LH. Postconditioning: a simple, clinically applicable procedure to improve revascularization in acute myocardial infarction. Circulation. 2005;112:2085–8. doi: 10.1161/CIRCULATIONAHA.105.569798. [DOI] [PubMed] [Google Scholar]
- 32.Jiang X, Shi E, Nakajima Y, Sato S. Postconditioning, a series of brief interruptions of early reperfusion, prevents neurologic injury after spinal cord ischemia. Ann Surg. 2006;244:148–53. doi: 10.1097/01.sla.0000217608.08582.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab. 2008;28:232–41. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- 34.Deyhimy DI, Fleming NW, Brodkin IG, Liu H. Anesthetic preconditioning combined with postconditioning offers no additional benefit over preconditioning or postconditioning alone. Anesth Analg. 2007;105:316–24. doi: 10.1213/01.ane.0000267524.71445.e7. [DOI] [PubMed] [Google Scholar]
- 35.Xiong L, Zheng Y, Wu M, Hou L, Zhu Z, Zhang X, Lu Z. Preconditioning with isoflurane produces dose-dependent neuroprotection via activation of adenosine triphosphae-regulated potassium channels after focal cerebral ischemia in rats. Anesthesia & Analgesia. 2003;96:233–7. doi: 10.1097/00000539-200301000-00047. [DOI] [PubMed] [Google Scholar]
- 36.Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:551–65. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- 37.Tomai F, Crea F, Gaspardone A, Versaci F, De Paulis R, Penta de Peppo A, Chiariello L, Gioffre PA. Ischemic preconditioning during coronary angioplasty is prevented by glibenclamide, a selective ATP-sensitive K+ channel blocker. Circulation. 1994;90:700–5. doi: 10.1161/01.cir.90.2.700. [DOI] [PubMed] [Google Scholar]
- 38.Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H649–57. doi: 10.1152/ajpheart.2001.280.2.H649. [DOI] [PubMed] [Google Scholar]
- 39.Gateau-Roesch O, Argaud L, Ovize M. Mitochondrial permeability transition pore and postconditioning. Cardiovasc Res. 2006;70:264–73. doi: 10.1016/j.cardiores.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 40.Obal D, Dettwiler S, Favoccia C, Scharbatke H, Preckel B, Schlack W. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth Analg. 2005;101:1252–60. doi: 10.1213/01.ANE.0000181336.96511.32. [DOI] [PubMed] [Google Scholar]
- 41.Xue QS, Yu BW, Wang ZJ, Chen HZ. Effects of ketamine, midazolam, thiopental, and propofol on brain ischemia injury in rat cerebral cortical slices. Acta Pharmacol Sin. 2004;25:115–20. [PubMed] [Google Scholar]