

Abstract
Every year almost 500,000 new patients are diagnosed with hepatocellular carcinoma (HCC), a primary malignancy of the liver that is associated with a poor prognosis. Numerous experimental models have been developed to define the pathogenesis of HCC and to test novel drug candidates. This review analyses several mouse models useful for HCC research and points out their advantages and weaknesses. Chemically induced HCC mice models mimic the injury-fibrosis-malignancy cycle by administration of a genotoxic compound alone or, if necessary, followed by a promoting agent. Xenograft models develop HCC by implanting hepatoma cell lines in mice, either ectopically or orthotopically; these models are suitable for drug screening, although extrapolation should be considered with caution as multiple cell lines must always be used. The hollow fibre assay offers a solution for limiting the number of test animals in xenograft research because of the ability for implanting multiple cell lines in one mouse. There is also a broad range of genetically modified mice engineered to investigate the pathophysiology of HCC. Transgenic mice expressing viral genes, oncogenes and/or growth factors allow the identification of pathways involved in hepatocarcinogenesis.
Keywords: Hepatocellular carcinoma, mouse models, hepatology, xenograft, hollow fibre assay, transgenic mice
Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver. Worldwide, approximately 500,000 new patients are diagnosed with HCC each year, often associated with poor prognosis (Motola-Kuba et al. 2006). It is the fifth most frequent neoplasm and ranks third on the list of most lethal cancers (Parkin et al. 2001). The highest incidence of HCC is found in Asia and sub-Saharan Africa. Most cases of HCC develop in a background of chronic liver damage such as cirrhosis or hepatitis (Tsukuma et al. 1993).
Hepatocarcinogenesis is a multistep process involving different genetic alterations that ultimately lead to malignant transformation of the hepatocytes (Farber 1984). Several rodent models have been used in defining the pathogenesis of HCC and have contributed to the current knowledge of HCC. Because of the physiologic and genetic similarities between rodents and humans, the short lifespan and the breeding capacity, rodents are often used for cancer research. Many chemically induced experiments have been conducted on rats (Rattus norvegicus), but mice (Mus musculus) are also a favourite model for cancer because of the availability of gene _targeting methods and the possibility of xenograft implantation. Woodchuck or groundhog (Marmota monax) is often used for studies concerning hepatitis B infection (HBV) induced HCC (Tennant et al. 2004; Cullen et al. 2008; Lu et al. 2008). The woodchuck hepatitis causes liver inflammation, injury and repair process similar to those in HBV-patients (Gudima et al. 2008).
This review focuses only on the available mouse models for HCC research, because of the existence of a broad range of (i) chemically induced models, (ii) xenograft models and (iii) genetically modified models, in mice. The advantages and disadvantages of the three types of experimental mice models will be discussed in this review.
Chemically induced models
Several chemical reagents induce tumour formation when administered in sufficient high doses and time span (Table 1). There are two types of carcinogenic compounds: (i) genotoxic compounds which are characterized by their capacity to induce structural DNA changes and (ii) promoting compounds which lack direct genotoxic capability, but enhance tumour formation after initiation by a hepatotoxic compound (Pitot & Dragan 1991). Treatment with a tumour-promoting agent facilitates the clonal expansion of the preneoplastic cells.
Table 1.
Summary of chemically induced HCC-models
| Time to develop tumours | % of mice with HCC | Metastasis | Remarks | References | |
|---|---|---|---|---|---|
| DEN | |||||
| Single administration | 45–104 weeks (dose dependent) | 80–100% in male | / | Poorly reproducible | Chen et al. (1993); Frey et al. (2000); Hacker et al. (1991); Park et al. (2008); Teoh et al. (2008); Zimmers et al. (2008) |
| Short-term administration | 40–60 weeks | 100% in male | / | Shiota et al. (1999) | |
| Long-term administration | 20–35 weeks | 100% in male 30% in female | Yes | Very agressive tumours (H-ras mutations) | Finnberg et al. (2004) |
| + PB | 20–40 weeks | / | B-catenin activation | Goldsworthy and Fransson-Steen (2002); Klaunig et al. (1988); Weghorst and Klaunig (1989) | |
| + PH | 4–8 weeks (in rats) | / | Farber et al. (1977); Klinman and Erslev (1963) | ||
| Peroxisome proliferators | 50–100 weeks (dose dependent) | Depends on strain, dose and PP-agent | Yes | PP tumorigenicity in humans not known | Hays et al. (2005); Reddy et al. (1976); Takashima et al. (2008) |
| Aflatoxine | Early HCC in 52 weeks high grade HCC 92–110 weeks | Considerable interstrain differences: DBA/2J: 90% C57BL/N: 25–66% | Yes | Suitable model for AFB-induced HCC in humans | McGlynn et al. (2003); Ghebranious and Sell (1998) |
| CCl4 | 104 weeks | 50–94% (dose dependent) in male and female | Yes | Confer and Stenger (1966); Farazi et al. (2006); Weisburger (1977) | |
| Choline deficient diet | 50–52 weeks | 100% | Steatohepatitis | de Lima et al. (2008); Knight et al. (2000); Liquori et al. (2009) | |
| Thioacetamide | 50–80 weeks | 70–100% | Palacios et al. (2008) | ||
The advantage of chemically induced models is the similarity with the injury-fibrosis-malignancy cycle seen in humans. This makes them the favourite models for HCC research.
Diethylnitrosamine
N-nitrosodiethylamine (DEN) is often used as a carcinogenic reagent. The _target organ in which DEN induces malignant tumours is species specific. Mice mainly develop not only liver tumours, but also gastrointestinal (Binato et al. 2008), skin, respiratory (Wang et al. 1992) and haematopoietic tumours (Gray et al. 1991). The carcinogenic capacity of DEN is situated in its capability of alkylating DNA structures. In the first step, DEN is hydroxylated to α-hydroxylnitrosamine (Verna et al. 1996). This bioactivation step is oxygen- and NADPH-dependent and is mediated by cytochrome P450, an enzyme which has its highest activity in the centrilobular hepatocytes. After cleavage of acetaldehyde, an electrophilic ethyldiazonium ion is formed. This ethyldiazonium ion causes DNA damage by reacting with nucleophiles such as DNA-bases. DEN works in a dose-dependent manner (Williams et al. 2000); a single low initiation dose does not lead to the formation of neoplasms, administration of a high dose induces HCC after a period of latency. Several DNA-repair mechanisms, such as non-homologous end joining, recombinational repair, base excision repair and many more, prevent the mutagenity of DNA-adducts when DEN is administered in a single low dose (Pegg 1990; Teoh et al. 2008).
Furthermore, oxidative stress caused by DEN can contribute to hepatocarcinogenesis (Kolaja & Klaunig 1997; Qi et al. 2008). Reactive oxygen species (ROS) generated by the P450-dependent enzymatic system might induce oxidative stress by the formation of hydrogen peroxide and superoxide anions. Production of ROS is known to cause DNA, protein and lipid damage; therefore, oxidative stress can play an important role in carcinogenesis (Kawanishi et al. 2002; Valko et al. 2006).
Mouse tumours induced by DEN harbour activating mutations in the H-ras proto-oncogene (Chen et al. 1993; Stahl et al. 2005). These mutations are rarely seen in humans and are correlated with metastasis and poor prognosis (Tada et al. 1990; Wang et al. 2001). However, Ras-activation that does not involve mutations of the ras-oncogene itself is considered a relative frequent event in hepatocarcinogenesis (Calvisi et al. 2006). Genetically, the DEN-model proves to be a good representation of HCC associated with poor prognosis (Lee et al. 2004).
The time needed after a single DEN-injection to develop HCC does not only depend on the administered dose, but also on sex, age and strain of mice (Rao & Vesselinovitch 1973). The younger the mice, the faster HCC will occur because of the high hepatocyte proliferation rates of juvenile animals (Vesselinovitch & Mihailovich 1983). After a long-term repetitive administration of DEN, HCC develops in 100% of the male mice and in 30% of females. The gender-related difference in HCC incidence is due to the inhibitory effect of estrogens and the stimulating effect of androgens on hepatocarcinogenesis (Nakatani et al. 2001). The methylation status of certain genes that contribute to the susceptibility to hepatocarcinogens might be one of the genetic factors that differs between tumour sensitive and tumour resistant strains (Buchmann et al. 1991). Tumour sensitive mice, such as C3H/HE-strain are characterized by hypermethylation in promoter regions of tumour suppressor genes after hepatocarcinogen exposure, a high degree of methylation inhibits the DNA-transcription (Bird & Wolffe 1999).
A dose-dependent formation of carcinomas after a single injection (5–90 mg/kg) of DEN in 15-day-old mice is observed after 45–104 weeks (Chen et al. 1993; Park et al. 2008). When B6C3F1 mice are exposed to a single dose of 5.0 mg/kg DEN, it takes approximately 64 weeks to develop HCC (Hacker et al. 1991). HCC occurs either within existing adenomas, which are visible after 24 weeks, or within the hepatocellular parenchyma. Metastasis to the lungs happens quite fast after the development of HCC (Vesselinovitch et al. 1978). The time and percentage of tumour development differ between strains, for example sv129 (Park et al. 2008; Teoh et al. 2008) (20 mg/kg DEN, 80–100% HCC after 30–55 weeks), C3H/HE (Frey et al. 2000) (90 mg/kg DEN, 100% HCC after 45–75 weeks), C57BL6/J (Zimmers et al. 2008) (5 mg/kg DEN, 84% HCC after 40–70 weeks). When adult mice are subjected to a short time weekly administration of DEN, it leads to a higher tumour incidence in a shorter time span. Six weeks of intraperitoneal injections (3 weeks with 75 mg/kg followed by 3 weeks with 100 mg/kg) lead to 100% tumour incidence after 52 weeks and the first tumours begin to occur after 30 weeks in male mice (Shiota et al. 1999). Long-term, weekly administration of 35 mg/kg DEN to mice leads to HCC after 20–35 weeks (Finnberg et al. 2004).
A two-stage model in which the initiation by a genotoxic compound is followed by a promotion phase is often used for inducing HCC. DEN can be used as an initiator and phenobarbital (PB) as a promoting agent. Several mechanisms might be responsible for the tumour promoting effect of PB. First, PB can increase the expression of cytochrome P450 a 100-fold, leading to an enhanced effect of DEN (Waxman & Azaroff 1992). Second, the increased cytochrome P450 activity induces oxidative stress (Imaoka et al. 2004). Third, PB can cause hypermethylation in promoter regions of tumour suppressor genes, therefore, enhancing hepatocarcinogenesis (Watson & Goodman 2002). Fourth, PB also interferes with the intracellular communication and might influence cell proliferation. Promotion with PB leads to tumours that exhibit mutations in the β-catenin proto-oncogene (Aydinlik et al. 2001; Loeppen et al. 2002). Mutant nuclear β-catenin expression is associated with non-invasive tumours and a significantly higher 5-year survival rate in humans (Hsu et al. 2000; Mao et al. 2001).
The effects of PB promotion on DEN-initiated mice also vary considerably depending upon strain, sex and age of the mice. While the tumour promoting activity of PB is generally accepted a tumour inhibiting effect is seen when juvenile (15-days-old) B6C3F1 mice are treated with DEN followed by a long-term (36 weeks) exposure to PB (Diwan et al. 1984; Lee et al. 1998). The inhibitory effect is not seen in juvenile female B6C3F1 mice (Weghorst & Klaunig 1989) or other mice strains, such as Balb/c (Klaunig et al. 1988) and C3H (Goldsworthy & Fransson-Steen 2002), initiated with DEN promoted with PB.
Timing of initiation with DEN is a critical determinant for the paradoxical effect of PB (Lee et al. 1998). DEN initiation alone leads to the formation of lesions that express the Bcl-2 gene (Bcl-2+), regardless of the moment of initiation. When adult male B6C3F1 mice are initiated with DEN between 6 and 10 weeks of age followed by exposure to PB in drinking water for 36 weeks, PB serves as a tumour promoting agent. Ninety per cent of the tumours consists of eosinophilic lesions that lack the Bcl-2 expression (Bcl-2−), indicating that PB offers a selective growth advantage for the latter. When juvenile male B6C3F1 mice are initiated with DEN at 15 days of age and submitted to long-term PB exposure, PB serves as a tumour inhibiting agent. Only 4% of the lesions are Bcl-2−, even after PB promotion (which normally suppresses the proliferation of Bcl-2+ tumour cells and promotes the proliferation of Bcl-2− cells). The decrease in the number of tumours is explained by the lack of Bcl-2− lesions in juvenile mice and the growth-inhibiting effect of PB on the Bcl-2+ lesions. Juvenile livers are less differentiated because hepatocytes actively proliferate in juvenile mice (Kanamura et al. 1990). These qualitative differences between infant and adult hepatocytes might be the reason for the age-dependent initiation properties.
There is also a gender-related difference in the response to PB promotion (Weghorst & Klaunig 1989). Female mice initiated with DEN at 15 days of age followed by long-term exposure to 0.05% PB show an increase of carcinomas compared with the DEN-only group, instead of the decrease that is seen in male mice.
When male BALB/c mice are initiated with DEN, a long-term treatment of PB promotes the development of liver tumours, regardless of the age of the mice during DEN administration. Genetic differences between strains of mice are likely to contribute to the variational response to PB (Goldsworthy & Fransson-Steen 2002). Because PB induces an increase in methylation of GC-rich region, genetic differences in the methylation status might be one of the causes of the differential response to PB (Counts & Goodman 1995; Watson & Goodman 2002).
Another two-step hepatocarcinogenesis model is known as the Solt-Farber protocol (Farber et al. 1977). In this model, initiation by a hepatocarcinogenic compound is followed by a partial hepatectomy (PH). Partial hepatectomy induces hepatic cell proliferation of the liver, leading to a fast expansion of the initiated cells (Klinman & Erslev 1963). There is a fast occurrence of altered hepatic foci and visible nodules after PH. Unfortunately, PH is a difficult procedure in mice associated with high mortality and has been conducted mostly in rats.
Peroxisome proliferators
Peroxisome proliferators (PPs) induce hepatomegaly, peroxisome proliferation in hepatocytes and induction of several hepatic enzymes. The development of HCC in mice fed a diet containing a PP was first reported in 1976 (Reddy et al. 1976). Currently it is widely accepted that all PPs are capable of inducing HCC after a latency period as a response to a long-term repetitive exposure to these xenobiotics. Methyl clofenapate (Lefevre et al. 1994), ciprofibrate (Ledda-Columbano et al. 2003; Calfee-Mason et al. 2008), fenofibrate (Nishimura et al. 2007), clofibrate (Nagini & Nagarajan 1988; Keller et al. 1990) and Wy-16,643 (Iida et al. 2003) are some examples of peroxisome proliferators that induce liver tumours in rodents (Rao & Reddy 1996). PPs activate the peroxisome proliferator activated receptor α (PPARα) (Hasmall et al. 2000). PPARα is a receptor protein that regulates the expression of several genes, including those involved in cell proliferation and apoptosis (Roberts et al. 1995; Zhao et al. 2007). It is essential for regulating lipid homeostasis and is known to mediate hepatocarcinogenesis (Peters et al. 1997; Hays et al. 2005). Most of the hepatic tumours induced by PPs are well-defined HCCs characterized by a trabecular histological pattern. In 20 to 40% of the cases metastasis occurs. One of the mechanisms of the carcinogenic effect of PPs is the induction of gene mutations due to an increased intracellular H2O2 concentration, because PPs induce transcriptional activation of peroxisomal oxidases, the foremost source of ROS in the liver (Yeldandi et al. 2000). In addition, PP inhibits apoptosis of hepatocytes and stimulates hepatocyte growth; exposure to PP, therefore, leads to hyperplasia and hepatomegaly (Ma et al. 1997). The dual ability of PPs to induce both cellular proliferation and oxidative stress results in cell transformation and cancer.
When sv129 mice were given a diet containing 0.01 or 0.05% di (2-ethylhexyl) phthalate (DEHP) for 100 weeks, all mice developed hepatic tumours (Takashima et al. 2008). After 52 weeks treatment with 0.5% benzafibrate in the diet, the average liver weight is approximately four times higher than in controls (Hays et al. 2005). All mice have grossly visible lesions which imply the presence of adenomas or carcinomas. The same exposure does not induce HCC in PPARα knock out mice, neither does a 1-year dietary supply of benzafibrate lead to hepatic carcinomas or adenomas in C57BL/N-mice, even though preneoplastic foci are found in 75% of the mice (Hays et al. 2005).
It is not known if a long-term exposure to PPs known to be carcinogenic for rodents represents a hazard to humans. When PPARα humanized mice are treated with the experimental PP Wy 16,643 on a long-term basis, there seems to be no hepatocarcinogenic effect (Cheung et al. 2004; Morimura et al. 2006). Extrapolation to the human model should be considered with caution because the PP induced hepatocarcinogenesis might be a species-specific process and PP models do not have much in common with human HCCs from the genetic point of view.
Aflatoxin B1
The hepatotoxin aflatoxin B1 (AFB), produced by certain fungi of the Aspergillus genus such as Asparagillus flavum, is known to be a hepatic carcinogen. These fungi thrive in corn, rice and peanuts stored in moist conditions. In China and Western Africa, the combined high prevalence of AFB and HBV contributes to the high rates of HCC in these regions (Autrup & Wakhisi 1988; Wang & Liu 2007). Experimental models involving AFB administration allow research of the mechanisms involved in AFB-induced hepatocarcinogenesis. AFB is metabolized by the liver microsomal system to the exo-8,9-epoxide intermediate that binds selectively to guanine residues in cellular DNA (Gallagher et al. 1994). Cytochrome P450 is essential in this process (Ramsdell et al. 1991). Exo-8,9-epoxide binds selectively to guanine, transforming it to thymine, hence causing DNA mutations. AFB induces chromosomal aberrations, sister chromatide exchange, chromosomal strand breaks, DNA-adducts, micronuclei and uncontrolled DNA synthesis (Wang & Groopman 1999).
When 7-day-old mice are exposed to 6 mg/kg body weight of AFB, in a bolus injection, HCC is developed after 52 weeks (McGlynn et al. 2003). In 90% of the DBA/2J mice (susceptible strain for HCC) HCC occurs after AFB exposure, while only 25% of the C57BL/N mice (relatively resistant strain for HCC) develop HCC after AFB exposure. The difference in susceptibility is a result of differences in several AFB detoxification loci. Ghebranious et al. injected C57BL/N mice with 10 mg/kg of AFB and had tumours in 66% of the male mice after 52 weeks, while high grade HCC was not developed until 92–110 weeks of age (Ghebranious & Sell 1998).
Carbon tetrachloride
Carbon tetrachloride (CCl4) is one of the most potent hepatotoxins (Weisburger 1977). The hepatotoxicity of CCl4 involves two phases (Avasarala et al. 2006). First, CCl4 is metabolized by cytochrome P450 to form trichloromethyl radicals (Sheweita et al. 2001). These radicals are highly energetic and cause lipid peroxidation and membrane damage. Second, the Kupffer cells induce an inflammatory response, which leads to the secretion of cytokines, chemokines and other proinflammatory factors. These factors not only have a direct cytotoxic effect, but also attract and activate monocytes, neutrophils and lymphocytes which contribute to the tissue damage. The repeated cycle of injury, inflammation and repair leads to fibrosis and eventually HCC. Weekly injections of CCl4 accompanied with alcohol administration through drinking water lead to HCC after 104 weeks (Confer & Stenger 1966; Weisburger 1977; Farazi et al. 2006).
Choline deficient diet
Mice subjected to a long-term choline deficient diet (CDD) develop tumours after 50–52 weeks (Knight et al. 2000). A CDD induces steatosis in all mice, nevertheless fat accumulation can differ considerably between species and is not necessarily strain-dependent (Knight et al. 2000; Liquori et al. 2009). The proposed mechanism of carcinogenicity by CDD is through the formation of oval cells (Tarsetti et al. 1993), a result of oxidative DNA damage and chromosomal instability because of the depletion of hepatic antioxidant mechanisms. It has been assumed that oval cells, either directly or indirectly through the generation of hepatocytes, function as tumour progenitors. CDD can be combined with the administration of a hepatotoxic compound, such as DEN or CCL4, and serves as a good model for steatohepatitis with further development to HCC (de Lima et al. 2008).
Thioacetamide
Thioacetamide (TAA) is a hepatotoxin that can be administered either in drinking water (0.02–0.05%) or by intraperitoneal injections. Repeated administration leads to fibrosis in mice over a period of 10–15 weeks (Palacios et al. 2008). The hepatotoxic action is a result of oxidation properties of the compound, leading to hepatic oxidative stress and liver damage.
Xenograft models
In xenograft models, the tumours are formed by injecting human cancer cells from a lab culture in immune deficient mice. Athymic (nude) or severe combined immune deficient (SCID) mice are often used as hosts. Tumour xenografts can be established either by direct implantation of biopsy material or by inoculation of human tumour cell lines. Several kinds of xenograft models can be distinguished (Table 2). First, the ectopic model, in which human tumour cells are injected subcutaneously in the flank of mice. Second, the orthotopic model, where tumour cells are injected intrahepatically into the mice. The orthotopic xenograft model is more suitable for extrapolation to humans and gives information about the metastatic spread of the tumour. The advantage of xenograft mouse models is the short time span needed for the development of tumours and the fact that it is an efficient way to demonstrate proof-of-principle when enough cell lines are used. This technique provides a model that allows investigation of aspects as in vivo toxicity, absorption and pharmacokinetics of a compound in a pre-clinical trial. Tumour phenotypes can vary remarkably between lines; it is therefore important to use different cell lines when using the xenograft model.
Table 2.
Summary of xenograft models
| Pro | Contra | Use in research | |
|---|---|---|---|
| Ectopic implantation | Fast occurrence of tumours (5–20 weeks) Easy to perform | Considerable differences between cell lines, multiple cell lines need to be tested No direct interaction with liver tissue Weakly extrapolatable to humans Complex tumour–host interactions (metastasis, angiogenesis, etc.) cannot be tested | Proof-of-principle Drug screening |
| Orthopic implantation | Fast occurrence of tumours Possibility to test in fibrotic livers Complex tumour–host interactions can be tested | Difficult procedure Considerable differences between cell lines, multiple cell lines need to be tested | Proof-of-principle Drug screening Tumour–host interactions |
| Hollow fibre model | Fast results (1–2 weeks) Ability for multiplexing reduces the amount of test animals needed Minimal effect on animal welfare Retrieval of tumour cells after experiment for subsequent analysis Angiogenesis can be semi-quantified | No direct interaction with liver tissue Complex tumour–host interactions such as metastasis can not be tested Occurrence of neovascularization around fibres influences results on prolonged periods | Proof-of-principle Drug screening |
Human tumour progression is a complicated process in which the interaction of neoplastic cells and the surrounding tumour environment plays an important role. A micro-evolutionary process takes place in which advantaged tumour cells expand rapidly, a process that is altered when using cell cultures. Therefore, the resemblance between xenograft tumours and human tumours is rather poor (Kelland 2004). Due to the heterogeneity of cell lines, multiple cell lines should be used for drug screening, which often leads to different outcomes depending on the cell line’s phenotype. Cell lines are often used for chemotherapeutic drug screening with common chemotherapeutic agents. Significant differences in tumour growth inhibition were noted between cell lines and discrepancies between previous studies were found. For example, while some studies have shown that EGFR-inhibitor inhibits tumour growth and intrahepatic metastasis by approximately 50% (Matsuo et al. 2003), this is not seen in either of the seven cell lines that was used by Huynh (Huynh et al. 2006). The lack of predictability of results obtained from xenograft mice models has convinced many researchers to use models in which tumours arise in a background that resembles the natural history of HCC.
An interesting setup consists of orthotopic implantations of HCC cells in fibrotic livers (Kornek et al. 2008). Fibrosis is established by intraperitoneal injection of thioacetamide or subcutaneous injection of CCL4 and oral alcohol intake. Tumours in fibrotic livers not only grow significantly larger and more rapidly than those in normal liver, but also have the capacity to metastasize and form satellite nodules. This model provides a useful tool for testing drug efficacy in orthotopic xenografts within the context of liver fibrosis, but there is also variable responsiveness to drugs between cell lines.
Metastatic HCC xenograft models are used for the research on the mechanism of metastasis and relapse. By orthotopic implantation of preserved metastatic tumour tissues of 30 surgical specimens, the first highly metastatic model of HCC (LCI-D20) in nude mice was developed (Sun et al. 1996). Orthotopic implantation of LCI-D20 leads to highly metastatic HCC that exhibits several characteristics of human tumour behaviour and the tumour tissue histologically resembles human HCC. HBV-DNA has been integrated in the cellular DNA of the LCI-D20 tumour cells. Metastasis to the lungs occurs in 100% of the mice 15 days after orthotopic inoculation.
Genetically comparing these highly metastatic cell lines to less metastatic cell lines, such as LCI-D35, leads to the identification of several genes involved in the invasive potential of tumours (Shao et al. 1999).
To limit the number of mice for drug screening, a low-cost, rapid and efficient method was developed, called the hollow fibre assay (HFA) (Hollingshead et al. 1995). This assay involves placing cells from tumour cell lines into small semi-permeable tubes (Figure 1). Tumour cells are inoculated into hollow (1 mm internal diameter) polyvinylidene fluoride fibres which are heat-sealed and cut at 2 cm intervals (Decker et al. 2004; Suggitt et al. 2006). The fibres are cultured for 24–48 h in vitro. Multiple fibres can be implanted subcutaneously or intraperitoneally in athymic mice; therefore, using only one mouse to test several cell lines. In comparison with the traditional tumour xenograft model, the HFA has some important advantages. The ability for multiplexing reduces the number of laboratory animals; this combined with the shorter evaluation time and reduced consumption of test agents makes it an economically interesting method (Shnyder et al. 2006). Li et al. used HFA to test the therapeutic efficacy of tripeptide tyroserleutide (YSL) in five different human HCC-cell lines (Li et al. 2008). The mice were implanted with three hollow fibres containing BEL-7402, SMMC-7721 and Hep3B or two hollow fibres with HepG2 and SK-HEP-1, and were treated with YSL. Because of this multiplex setup, only 30 mice were needed instead of 150 mice if the same experiment was conducted in a classic xenograft model. Other advantages are that the HFA has minimal influence on the animal welfare and it allows retrieval of tumour cells uncontaminated by host cells, which can be used for subsequent analysis. Retrieval of the tumour cells leads to the possibility of using several optical imaging methods to quantify the effect of treatment ex vivo. Tumour cell lines can be genetically engineered with constitutive bioluminescent (i.e. luciferase) or fluorescent (i.e. green fluorescent protein) reporter vectors, which allows in vivo or ex vivo optical imaging (Zhang et al. 2007). Although HFA is not suitable for studying complex host–tumour interactions (Hollingshead et al. 1995), it is possible to study angiogenesis (Phillips et al. 1998). Angiogenic activity around the tumour is measured after 6 days and it takes about 1 month to develop an extensive vascular network towards the tumour (Zhang et al. 2007). The presence of a vascular supply to the hollow fibres has a significant influence on drug delivery and chemo sensitivity, which should be taken into account when using the HFA over a prolonged period (Phillips et al. 1998). Therefore the HFA is only suitable for experiments that acquire a limited time span, which restricts the usefulness for long-term toxicity studies. In addition, it is not possible to assess data regarding the tumour size, an important factor for the evaluation of chemotherapeutic compounds. Only the effect of the drug on a restricted amount of cells can be examined, leading to a reduced inter-cell and cell-host interaction. Furthermore, the small quantity of cells does not resemble the physiology found in HCC. All these factors should be taken into account when using the HFA technique.
Figure 1.

Tumour cells derived from human (or mouse) hepatic tumours (1) are placed in small semi-permeable tubes known as hollow fibres (2). The fibres are cultured for 24-48 hours in vitro before subcutaneous or intraperitoneal implantation in nude mice. The cells can be retrieved after the in vivo assay and used for subsequent analysis (4).
Genetically modified models
Genetically modified mouse models (GMM) are engineered to mimic pathophysiological and molecular features of HCC (Frese & Tuveson 2007). It is a unique model for assessing the effects of oncogenes either alone or in combination with other oncogenes or carcinogenic agents. GMMs facilitate detailed investigation of carcinogenic pathways and allow the assessment of pathway cooperativity and dependency in vivo (Tuveson & Jacks 2002). Tissue-specific expression can be achieved by designing cDNA-constructs that contain promoter elements that are restricted to certain tissues. Although the effect of the mutation is constitutive, its expression is limited by the use of tissue-specific promoters. For hepatic GMMs the albumin promoter is often used. An alternative to the constitutive tissue-specific expression is the induction of specific genes with molecules. This approach allows research towards the role of oncogenes in tumour maintenance and the influence of age on the carcinogenesis.
Because of the large number of transgenic mice, only a small selection of representative models for HCC research will be discussed in this review (Table 3).
Table 3.
Summary of transgenic models
| Promotor | Time to develop tumours | % of mice with HCC | Research | References | |
|---|---|---|---|---|---|
| Viral genes | |||||
| HBx | X-gene promotor, mouse albumin or metallothionein | 52–104 weeks | 70–85 | Hepato-carcinogenesis in a background of hepatitis B or C | Araki et al. (1989); Burk et al. (1988); Koo et al. (2005); Lakhtakia et al. (2003); Sell et al. (1991); Takada et al. (1995); Xiong et al. (2003); Zheng et al. (2007) |
| +DEN: 30 weeks | 85 | ||||
| HCV | Albumin | 90–100 weeks | 15 | Lerat et al. (2002) | |
| HCV core | HBV | 80–105 weeks | 32 | Moriya et al. (1998); Tanaka et al. (2008) | |
| HCV core E1-E2 | Albumin | +DEN: 32 weeks | 100 | Kamegaya et al. (2005) | |
| HBV | 60 weeks | 23 | Naas et al. (2005) | ||
| Oncogenes | |||||
| c-myc | Albumin | 65–90 weeks | 55 | Investigation of carcinogenic pathways Cooperativity of oncogenes | Thorgeirsson and SantoniRugiu (1996) |
| c-myc | WHV | 36 weeks | 100 | Merle et al. (2005) | |
| c-myc + E2F1 | Albumin | 26–35 weeks | 100 | Conner et al. (2003) | |
| β-catenin + H-ras | Cre-Lox | 8 weeks (early HCC) 26 weeks (high grade HCC) | 100 | Harada et al. (2004) | |
| Growth factors | |||||
| TGF-α | Metallothionein 1 | +Zinc: 40–70 weeks | 50 | Investigation of carcinogenic pathways Cooperativity of growth factors with oncogenes Influence of growth factors on sensitivity for hepatocarcinogens | Jhappan et al. (1990) |
| ELF (TGF signalling) | Knock out | 58 weeks | 40–70 | Baek et al. (2008) | |
| TGF-α + c-myc | Albumin | 40 weeks | 100 | Murakami et al. (1993); Ohgaki et al. (1996); Thorgeirsson and SantoniRugiu (1996) | |
| Albumin | + Zinc: 16 weeks | 100 | |||
| EGF | Albumin | 24–36 weeks | 100 | Borlak et al. (2005); Tonjes et al. (1995) | |
| EGF + c-myc | Albumin | 12–18 weeks | 100 | Tonjes et al. (1995); | |
| FGF19 | No liver-specific expression | 52 weeks | 50 | Nicholes et al. (2002) | |
| SV40 T-antigen | Albumin, α1 antitrypsin, serum amyloid P component or antithrombin III | 4–12 weeks | 100 | Fast tumour occurrence due to uncontrolled DNA replication | Araki et al. (1991); Cullen et al. (1993); Sepulveda et al. (1989) |
| Cre-Lox | 20 weeks | 100 | Lou et al. (2005) | ||
| Creating tumour environment | |||||
| AAT | α1 antitrypsin | 52–90 weeks | 100 | Effect of AAT-deficiency on the liver | Geller et al. (1994) |
| PTEN | Albumin/Cre | 40–44 weeks | 66 (male) 30 (female) | Effect of steatohepatitis | Watanabe et al. (2007) |
| PDGF | Albumin | 52 weeks | 100 | Borkham-Kamphorst et al. (2007); Campbell et al. (2005); Czochra et al. (2006); Thieringer et al. (2008); Schnur et al. (2004),Martinez-Chantar et al. (2008) | |
| TGF-β1 | +AAT: >18 weeks | ||||
| GMNT | 40 weeks | 100 | |||
Transgenic models expressing viral genes
Hepatitis B virus
The main cause of HCC worldwide is the hepatitis B virus (HBV). HBV is an enveloped hepatotrophic DNA virus causing several liver diseases such as acute to chronic hepatitis, cirrhosis and HCC. During prolonged infection, viral DNA sequences integrate into the host genome, causing mutations, chromosomal instability and general genomic rearrangements. The genome of HBV is a circular, partially double-stranded DNA molecule and it is characterized by its four overlapping open reading frames (ORF) that encode for surface (S), core (C), polymerase (P) and X proteins (HBx). There is increasing evidence that the expression of viral genes, in particular genes encoding for HBx, might deregulate the control of cellular growth and viability and sensitize hepatocytes to exogenous and endogenous carcinogens (Wu et al. 2001). The oncogenic capacity of HBx is a result of the fact that some of the genes activated by HBx, such as ICAM-1 (Hu et al. 1992), c-myc (Terradillos et al. 1997) and c-fos (Avantaggiati et al. 1993), are important for cell adhesion and proliferation.
Many aspects of HBV-induced hepatocarcinogenesis have been analysed by generating transgenic mice expressing complete fragments of the HBV genome, under control of either the HBV promoter, or constitutive (mouse albumin) or inducible (mouse metallothionein) liver-specific promoters. The first HBV-related transgenic mice model was produced in 1985 by two independent research groups (Babinet et al. 1985; Chisari et al. 1985).
Most of the HBV-related transgenic animals express the HBx genes, which are associated with altered hepatocellular functions and HCC development (Burk et al. 1988; Araki et al. 1989; Xiong et al. 2003). The livers of HBx transgenic mice exhibit megalocytosis, nuclear pleomorphism, hyperchromatism and a slightly increased nuclear/cytoplasmic ratio after approximately 15 weeks (Lakhtakia et al. 2003; Koo et al. 2005). The birthrate of the HBx transgenic mice is a bit lower than that of other transgenic mice, indicating an interaction with the prenatal development of mice (Xiong et al. 2003). The progression to HCC takes approximately 52–104 weeks (Takada et al. 1995; Koo et al. 2005). Transgenic lineages with lower HBx copy number showed lower tumour incidence comparable with that of wild-type mice (Koike et al. 1994), while other experiments did not encounter any tumours in HBx transgenic mice (Lee et al. 1990; Zheng et al. 2007). HBx transgenic mice seem to be more sensitive for HCC development after a single DEN-injection when compared with their non-transgenic counterparts (Sell et al. 1991; Zhu et al. 2004; Zheng et al. 2007).
Hepatitis C virus
Viral Hepatitis C (HCV) infection is an important risk factor for HCC (Gerber 1993; ElRefaie et al. 1996). Chronic HCV can cause cirrhosis and increases the risk of HCC by approximately 1–3%. The HCV genome is a RNA molecule of approximately 9500 nucleotides. It contains an ORF that counts 9000 nucleotides and encodes for a large glycoprotein that is prone to several post-translational modifications, leading to the final viral proteins and enzymes. A number of HCV proteins, such as core (Feng et al. 2007), NS3 (Deng et al. 2006) and NS5A (Lan et al. 2002) play an important role in the hepatocarcinogenesis. The proteins encoded by the HCV genome, especially core, interrupt the intracellular signal transduction pathways. Unlike HBV, the RNA genome of HCV does not integrate into the host chromosome. Therefore, HCV-related hepatocarcinogenesis is not likely to involve insertional mutagenesis. Numerous transgenic mouse models are made expressing different HCV proteins have been developed and differences occur in the development of tumours. When the complete viral protein is expressed, steatosis and HCC occur in 15% of the mice after approximately 90–100 weeks (Lerat et al. 2002). Other researchers did not encounter the tumour phenotype, indicating that genetic background of mice and/or oncogene expression level play an important role in the outcome of transgenic mice models (Kawamura et al. 1997).
The core protein interferes with the lipid metabolism by activating PPARα, leading to lipid accumulation in the hepatocytes (Tanaka et al. 2008). The expression of HCV core leads to progressive hepatic steatosis in several lines of constitutive transgenic mice, followed by HCC after 80–105 weeks in 32% of the male mice (Moriya et al. 1998; Tanaka et al. 2008). Inducible expression of core protein by tetracycline and Dox administration leads to a peak in steatosis after 2 months but no HCC occurs (Chang et al. 2008). Transgenic mice expressing core, E1 and E2 structural proteins developed HCC after 60 weeks in 23% of the male mice (Naas et al. 2005). An accelerated tumour progression occurs after DEN injection; after merely 32 weeks HCC is found in 100% of the male mice. HCV proteins and chemical carcinogens such as DEN have a synergistic influence on HCC development, because HCV proteins blocks apoptosis in the hepatocytes, leading to an accelerated expansion of the neoplastic hepatocytes (Kamegaya et al. 2005).
Transgenic mice over-expressing oncogenes
An oncogene is a protein encoding gene which participates in the onset and development of cancer. Genetic alterations resulting in the activation or over-expression of oncogenes increase the chance of tumour development.
Myc protein
The Myc protein is a transcription factor that activates the expression of several genes through binding on consensus sequences and recruiting histone acetyltransferases (Figure 2) (Dang & Lewis 1997; Nilsson & Cleveland 2003). When myc is mutated or over-expressed, it is associated with a variety of tumours. The cooperation of myc proteins with other oncogenes and growth factors is critical in the evolution of the malignant phenotype. Transgenic mice over-expressing c-myc develop liver tumours after a long period of latency (35–90 weeks) (Thorgeirsson & SantoniRugiu 1996; Merle et al. 2005). Five to 10% of the tumours show increased β-catenin expression. Co-expression of E2F1/c-Myc accelerates liver cancer development (Conner et al. 2003). Myc transgenic mice are genetically close to human HCC of good prognosis (Lee et al. 2004).
Figure 2.

c-myc is one of the key elements in the malignang transformation. By (1) preventing differentiation and/or (2) cell cycle arrest, deregulated Myc forces cells to remain in a (3) proliferative state and causes (4) genomic instability. (5) Apoptosis is induced by activating death receptor pathways, in which myc has several points of regulation. c-Myc over-expression results in elevated expression of genes involved in regulating the (6) cellular metabolism, allowing tumour cells to switch to an anaerobic metabolism when oxygen is depleted).
β-catenin
β-Catenin, a subunit of the cadherin complex, plays an important role in the development and regeneration of the liver (Figure 3). β-Catenin is one of the key downstream effectors of the Wnt-signalling pathway, a network of proteins that is known for its involvement in tumour development (Schwarz et al. 2003; Brembeck et al. 2006). β-Catenin mutations are considered to be an early event in the hepatocarcinogenesis (Devereux et al. 1999; Laurent-Puig & Zucman-Rossi 2006). Approximately 30% of the human hepatic tumours harbour activated β-catenin mutations. A Wnt activating β-catenin mutation alone causes hepatomegaly, but to induce hepatocarcinogenesis additional mutations or epigenetic changes are required (Harada et al. 2002). When mutations in both the β-catenin and H-ras genes are introduced by adenovirus-mediated Cre expression, early HCC is found in all the mice killed 8 weeks after induction (Harada et al. 2004). High grade HCC was established after approximately 26 weeks.
Figure 3.

Beta-catenin is an integral componant of the Wnt-signaling pathway, a pathway that is one of the central players in maintaining liver health. The association between beta-catenin and E-cadherin is found in the hepatocyte membrane and has significant implications in (1) cell-cell adhesion. Phosphorylation of beta-catenin inhibits the beta-catenin-E-cadherin association, leading to disruption of adherens junctions and loss of intracellular adhesion. The dissociation of this complex induces nuclear translocation of beta-catenin, leading to _target gene expression. Nuclear translocation of beta-catenin is associated with hepatocyte proliferation during development and after partial hepatectomy. Beta-catenin regulates the expression of genes involved in (3) hepatocyte maturation and in (4) biliary specification. The interaction between the protein adenomatous polyposis coli and beta-catenin, and the involvement of beta-catenin in the regulation of proteins important in ammonia metabolism, play an important role in the (5) zonation of the liver, dividing the liver in several structural and (6) metabolic regions.
Transgenic mice models over-expressing growth factors
During HCC an unregulated expression of hepatocyte mitogens occurs, leading to an uncontrolled expansion of the hepatocytes. Over-expression of growth factors that mediate hepatocyte growth induces HCC.
Transforming growth factor-α
Transforming growth factor-α (TGF-α) is a potent hepatotrophic mitogen synthesized in hepatocytes during regeneration (Figure 4). Transgenic mice over-expressing human TGF-α under the inducible methallothionein 1 promoter develop liver tumours as well as an abnormal development of the pancreas and mammary glands (Jhappan et al. 1990). These tumours are genetically similar to human HCC associated with poor prognoses (Lee et al. 2004). Zinc administered through drinking water enhances the tumour formation and after 40–70 weeks, HCC occurs in 50% of the mice. Heterozygotic mice with a defect in embryonic liver fodrin (elf+/−), a stem cell adaptor protein that plays a pivotal role in TGF-signalling, develop HCC in 40–70% of the mice after 58 weeks (Baek et al. 2008). Double transgenic mice carrying both c-myc (albumin promoter) and TGF-α (methallothionein promoter) lead to a tremendous acceleration of the neoplastic development compared to the single transgenic mice overexpressing either c-myc or TGF-α (Murakami et al. 1993; Ohgaki et al. 1996; Thorgeirsson & SantoniRugiu 1996). After approximately 17 weeks, 20% of the mice is diagnosed with HCC that consists of multiple foci of carcinomas and adenomas, with 100% HCC at 40 weeks. The faster occurrence of HCC in the double transgenic model, compared with the parental lines, suggests that the interaction of c-myc and TGF-α increases the malignant conversion by the selection and expansion of preneoplastic cells.
Figure 4.
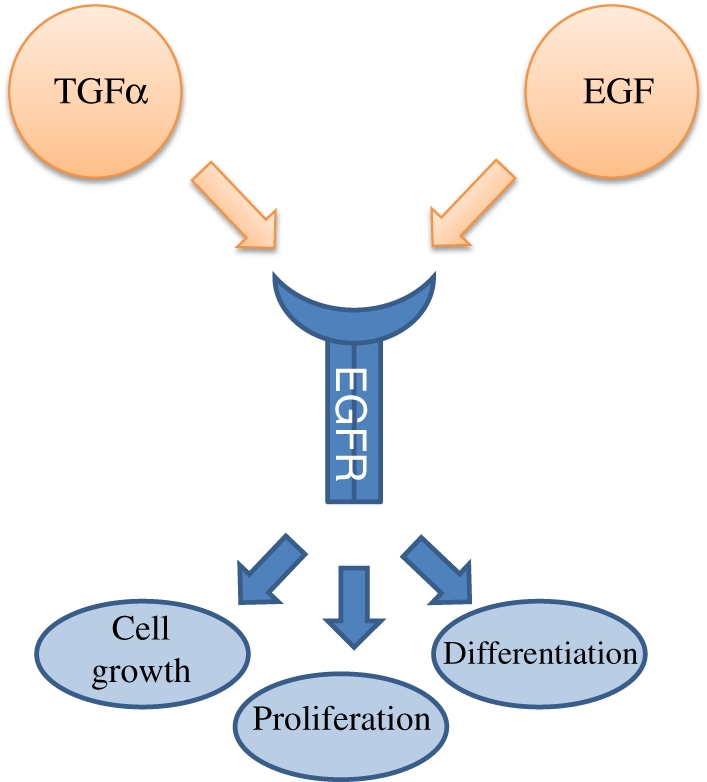
Transforming growth factor alpha (TGF-alpha) and epidermal growth factor (EGF) are important hepatic mitogens that both bind on the epidermal growth factor receptor (EGFR). Upon activation, EGFR undergoes dimerisation which stimulates its intrinsic intracellular protein-tyrosine kinase activity. This leads to the initiation of several signal transduction cascades, leading to cell growth, proliferation and differentiation.
Epidermal growth factor
Epidermal growth factor (EGF) is a growth factor that plays an important role in the regulation of cell growth, proliferation and differentiation (Figure 4). Its over-expression is associated with hepatocellular carcinoma. Over-expression of the secreted form of EGF results in multiple highly malignant hepatic tumours after 24–36 weeks (Tonjes et al. 1995; Borlak et al. 2005). In double transgenic mice expressing EGF and myc, tumour development and mortality are accelerated (Tonjes et al. 1995).
Fibroblast growth factor 19
While most transgenic mouse models use tissue-specific promoters to ensure liver-specific expression, there is one model that does not require liver-specific expression for the development of HCC (Figure 5). A transgenic mouse model over-expressing fibroblast growth factor 19 (FGF19) in skeletal muscle leads to the occurrence of HCC in 50% of the mice after approximately 52 weeks (Nicholes et al. 2002). In contrast to the other mice models, this model produces a higher HCC incidence in female mice than in male. All tumours in the female mice exhibit an increased β-catenin expression, which is not found in tumours in the male mice. The exact mechanism of the FGF19-induced hepatocarcinogenesis is still unclear, but might be a result of the increased metabolic rate which leads to an increase in ROS production.
Figure 5.

The fibroblas growth factor 19 (FGF19) is a high affinity ligand for the fibroblas growth factor receptor 4 (FGFR4) expressed in hepatocytes. Binding of FGF19 to FGFR4 leads to an increased cellular metabolism, associated with the production of reactive oxygen species (ROS). Wether HCC formation is an indirect effect of altered metabolism or a direct affect of FGF19 is unknown.
Simian vacuolating virus 40
The Simian vacuolating virus 40 (SV40) is a DNA-virus that has the potential to cause tumours. It suppresses the transcriptional properties of the tumour-suppressing p53, which is responsible for initiating apoptosis and cell cycle arrest. When the SV40 T-antigen is brought to expression under a specific or inducible promoter such as albumin (Cullen et al. 1993), α1 antitrypsin (Sepulveda et al. 1989), serum amyloid P component (Araki et al. 1991) or antithrombin III (Dubois et al. 1991; Lou et al. 2005), liver tumours are found after a short period of latency (4–12 weeks). Metastasis to the lungs can occur. Tumour progression is very rapid in these models and, therefore, differs radically from the development of human tumours which progress more gradually. Transgenic mice expressing regulatory SV40 sequences by activating oncogenic sequences upon Cre-mediated excision develop HCC on a slower pace (20 weeks) (Lou et al. 2005).
Creating a tumour environment
These models aim to create a tumour environment and mimic the injury-fibrosis-HCC sequence found in naturally occurring tumours.
Alpha-1 antitrypsin
Alpha-1 antitrypsin (AAT) is a glycoprotein that is produced in the liver. Transgenic mice expressing a human form of transport-impaired AAT represent a good model for studying the effects of AAT deficiency on the liver. AAT deficiency is an autosomal recessive disorder in which a mutation causes the production of AAT that is unable to be transported. This leads to decreased AAT activity in serum and deposition of excessive AAT in the liver. Both heterozygous and homozygous individuals develop cirrhosis and HCC. AAT-deficient mice develop HCC after 52–90 weeks (Geller et al. 1994).
Phosphatase and tensine homologue
Phosphatase and tensine homolog (PTEN) is a tumour suppressor gene that regulates the serine-threonine kinase protein kinase B (PKB/akt) pathway (Figure 6). PTEN deficiency induces cellular hyperproliferation, anti-apoptosis and oncogenesis (Horie et al. 2004). Liver-specific PTEN-deficient mice develop hepatic steatosis, inflammation, fibrosis and tumours that are very similar to human non-alcoholic steatohepatitis (NASH) (Watanabe et al. 2007). Liver tumours are present in 66% of male and 30% of female PTEN-deficient mice by 40–44 weeks of age.
Figure 6.

Phosphatase and tensin homolog (PTEN) is a tumour suppressor gene involved in the regulation of the threonine kinase protein kinase B (PKB/akt) pathway, by dephosphorylating phosphatidylinositol (3,4,5)-triphosphate (PIP3). The PKB/akt pathway is involved in the regulation of the cell cycle, apoptosis and cell growth.
Platelet-derived growth factor
Members of the platelet-derived growth factor (PDGF) family are known to play an important role in the embryonic development, cell proliferations, cell migration and angiogenesis. Furthermore, they have been associated with several diseases, including hepatic fibrosis (Bonner 2004). Liver cirrhosis induced by over-expression of PDGF-A, PDGF-B, PDGF-C or PDGF-D is associated with hepatic stellate cell activation possibly as a result of the induction of profibrotic genes such as transforming growth factor-beta1 (TGF-β1) (Campbell et al. 2005; Czochra et al. 2006; Borkham-Kamphorst et al. 2007; Thieringer et al. 2008). Long-term over-expression of the PDGF genes will induce HCC after approximately 52 weeks (Campbell et al. 2005).
Transforming growth factor-beta
Transforming growth factor-beta1 is known to be an important factor in the pathogenesis of fibrosis (Williams & Knapton 1996). TGF-β1 enhances the production of several matrix components and decreases their degradation, thereby causing an accumulation of extracellular matrix which is the basic underlying cause of fibrosis (Lechuga et al. 2004; Qi et al. 2006). Transgenic mice over-expressing TGF-β1 present the most extensive liver fibrosis after approximately 10 weeks; nevertheless, fully developed cirrhosis is not seen, probably because of the high mortality rate in the transgenic strains (Sanderson et al. 1995). Repeated administration of AAT can induce cirrhosis after 18 weeks, which would lead to HCC if the experiment would be continued for a prolonged period (Schnur et al. 2004).
Glycine N-methyltransferase
Glycine N-methyltransferase (GMNT) is the main enzyme responsible for catabolism of hepatic S-adenosylmethionine (SAM), leading to the formation of S-adenosylhomocysteine (SAH), an inhibitor of methyltransferases (Mato & Lu 2007). By controlling the ratio of SAM/SAH, GMNT contributes to the genetic stability by regulating DNA methylation. GNMT-knock-out mice have SAH levels that are 71 times higher than that in control mice (Liu et al. 2007). The transgenic mice start to develop steatosis and liver fibrosis after 3 months, a phenomenon that becomes more prominent after 8 months (Martinez-Chantar et al. 2008). At 8 months, 100% of the mice have developed multifocal HCC.
Conclusion
The availability of a wide range of experimental mice models for HCC research has given researchers the opportunity to assess tumour–host interactions, to perform drug screening and to mimic the complex multistep process that leads to the malignant transformation of hepatocytes. When selecting a model for HCC research, one must first understand the limitations and advantages that the specific model possesses. HCC rarely occurs spontaneously in humans, in contrast to certain mouse strains which have a high background incidence of spontaneous hepatic tumour formation (Diwan et al. 1986). The same mouse strains are very susceptible to hepatocarcinogens. The first step in constructing an experimental setup is choosing an optimal mouse strain.
Chemically induced models are favourable for research that requires HCC to develop in a natural background of liver damage. Different hepatocarcinogenic agents are metabolized through diverse metabolic pathways and extrapolation should be considered with caution when it is uncertain if the agents would be metabolized differently in humans. Age of the mice and dose of administration of hepatotoxins might lead to different tumour phenotypes, which might influence experimental outcomes.
Xenograft models offer a fast solution for drug screening and are an easy way for proof-of-principle experiments. Xenograft models are often used for drug screening; nevertheless, it must be noted that the spectacular results often seen in mice models can seldom be repeated in cancer patients. It should also be noted that multiple cell lines have to be used in experiments because of the heterogenity between cell lines. Hollow fibre assays (HFA) offers a model that allows testing of multiple cell lines in one mouse, leading to a tremendous decrease in required lab animals. HFA and ectopic implantation do not allow research about complex tumour–host interactions, but orthotopic implantation makes it possible to investigate the neoplastic hepatocytes in their natural environment.
Genetically modified mouse models are favourable to identify a range of discrete molecular and histological stages during the multistep process of hepatocarcinogenesis. GMMs harbouring multiple mutations allow the investigation of cooperation and dependency between oncogenes, growth factors and viral genes. Use of an inducible promoter offers significant advantages because the mutation can be switched on and off when needed. The biological insight gained from transgenic mice is invaluable. Nevertheless, a mutation in a gene does not always result in the expected phenotype and the phenotypic outcome can be influenced by many environmental and genetic factors. It should also be stated that most models do not demonstrate the advanced neoplastic lesions and that metastasis occurs only seldom. One should also consider the fact that natural tumours consist of a heterogenic group of cells in which multiple mutations have occurred. The interpretation of results from studies using GMMs is not always simple, especially for extrapolation to humans.
References
- Araki K, Miyazaki J, Hino O, et al. Expression and replication of hepatitis B virus genome in transgenic mice. Proc. Natl Acad. Sci. USA. 1989;86:207–211. doi: 10.1073/pnas.86.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Hino O, Miyazaki J, Yamamura K. Development of 2 types of hepatocellular-carcinoma in transgenic mice carrying the Sv40 large T-antigen gene. Carcinogenesis. 1991;12:2059–2062. doi: 10.1093/carcin/12.11.2059. [DOI] [PubMed] [Google Scholar]
- Autrup H, Wakhisi J. Detection of exposure to aflatoxin in an African population. IARC Sci. Publ. 1988;8:63–66. [PubMed] [Google Scholar]
- Avantaggiati ML, Natoli G, Balsano C, et al. The hepatitis B virus (HBV) pX transactivates the c-fos promoter through multiple cis-acting elements. Oncogene. 1993;8:1567–1574. [PubMed] [Google Scholar]
- Avasarala S, Yang L, Sun Y, et al. A temporal study on the histopathological, biochemical and molecular responses of CCl(4)-induced hepatotoxicity in Cyp2e1-null mice. Toxicology. 2006;228:310–322. doi: 10.1016/j.tox.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Aydinlik H, Nguyen TD, Moennikes O, Buchmann A, Schwarz M. Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of beta-catenin-mutated mouse liver tumors. Oncogene. 2001;20:7812–7816. doi: 10.1038/sj.onc.1204982. [DOI] [PubMed] [Google Scholar]
- Babinet C, Farza H, Morello D, Hadchouel M, Pourcel C. Specific expression of hepatitis-B surface-antigen (Hbsag) in transgenic mice. Science. 1985;230:1160–1163. doi: 10.1126/science.3865370. [DOI] [PubMed] [Google Scholar]
- Baek HJ, Lim SC, Kitisin K, et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology. 2008;48:1128–1137. doi: 10.1002/hep.22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binato M, Kruel Schmidt M, Silveira Volkweis B, Behrend Silva Ribeiro G, Isabel Edelweiss M, Ricachenevsky Gurski R. Mouse model of diethylnitrosamine-induced gastric cancer. J. Surg. Res. 2008;148(2):152–157. doi: 10.1016/j.jss.2007.12.748. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression - belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, van Roeyen CRC, Ostendorf T, Floege J, Gressner AM, Weiskirchen R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 2007;46:1064–1074. doi: 10.1016/j.jhep.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Borlak J, Meier T, Halter R, Spanel R, Spanel-Borowski K. Epidermal growth factor-induced hepatocellular carcinoma: gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene. 2005;24:1809–1819. doi: 10.1038/sj.onc.1208196. [DOI] [PubMed] [Google Scholar]
- Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr. Opin. Genet. Dev. 2006;16:51–59. doi: 10.1016/j.gde.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Buchmann A, Bauerhofmann R, Mahr J, Drinkwater NR, Luz A, Schwarz M. Mutational activation of the C-Ha-Ras gene in liver-tumors of different rodent strains - correlation with susceptibility to hepatocarcinogenesis. Proc. Natl Acad. Sci. USA. 1991;88:911–915. doi: 10.1073/pnas.88.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk RD, Deloia JA, Elawady MK, Gearhart JD. Tissue preferential expression of the hepatitis-B virus (Hbv) surface-antigen gene in 2 lines of Hbv transgenic mice. J. Virol. 1988;62:649–654. doi: 10.1128/jvi.62.2.649-654.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfee-Mason KG, Lee EY, Spear BT, Glauert HP. Role of the p50 subunit of NF-kappa B in vitamin E-induced changes in mice treated with the peroxisome proliferator, ciprofibrate. Food Chem. Toxicol. 2008;46:2062–2073. doi: 10.1016/j.fct.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl Acad. Sci. USA. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang ML, Yeh CT, Chen JC, et al. Altered expression patterns of lipid metabolism genes in an animal model of HCV core-related, nonobese, modest hepatic steatosis. BMC Genomics. 2008;9:109. doi: 10.1186/1471-2164-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Liu L, Castonguay A, Maronpot RR, Anderson MW, You M. Dose-dependent Ras mutation spectra in N-nitrosodiethylamine induced mouse-liver tumors and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced mouse lung-tumors. Carcinogenesis. 1993;14:1603–1608. doi: 10.1093/carcin/14.8.1603. [DOI] [PubMed] [Google Scholar]
- Cheung C, Akiyama TE, Ward JM, et al. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 2004;64:3849–3854. doi: 10.1158/0008-5472.CAN-04-0322. [DOI] [PubMed] [Google Scholar]
- Chisari FV, Pinkert CA, Milich DR, et al. A transgenic mouse model of the chronic hepatitis-B surface-antigen carrier state. Science. 1985;230:1157–1160. doi: 10.1126/science.3865369. [DOI] [PubMed] [Google Scholar]
- Confer DB, Stenger RJ. Nodules in livers of C3h mice after long-term carbon tetrachloride administration - a Light and Electron Microscopic Study. Cancer Res. 1966;26:834–843. [PubMed] [Google Scholar]
- Conner EA, Lemmer ER, Sanchez A, Factor VM, Thorgeirsson SS. E2F1 blocks and c-Myc accelerates hepatic ploidy in transgenic mouse models. Biochem. Biophys. Res. Commun. 2003;302:114–120. doi: 10.1016/s0006-291x(03)00125-6. [DOI] [PubMed] [Google Scholar]
- Counts JL, Goodman JI. Hypomethylation of DNA: a nongenotoxic mechanism involved in tumor promotion. Toxicol. Lett. 1995;82–83:663–672. doi: 10.1016/0378-4274(95)03512-5. [DOI] [PubMed] [Google Scholar]
- Cullen JM, Sandgren EP, Brinster RL, Maronpot RR. Histologic characterization of hepatic carcinogenesis in transgenic mice expressing SV40 T-antigens. Vet. Pathol. 1993;30:111–118. doi: 10.1177/030098589303000203. [DOI] [PubMed] [Google Scholar]
- Cullen JM, Lindsey-Pegram D, Cote PJ. Serologic survey of woodchuck hepatitis virus in North Carolina woodchucks (Marmota monax) J. Zoo Wildl. Med. 2008;39:263–265. doi: 10.1638/2007-0119R.1. [DOI] [PubMed] [Google Scholar]
- Czochra P, Klopcic B, Meyer E, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J. Hepatol. 2006;45:419–428. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Dang CV, Lewis BC. Role of oncogenic transcription factor c-Myc in cell cycle regulation, apoptosis and metabolism. J. Biomed. Sci. 1997;4:269–278. doi: 10.1007/BF02258350. [DOI] [PubMed] [Google Scholar]
- Decker S, Hollingshead M, Bonomi CA, Carter JP, Sausville EA. The hollow fibre model in cancer drug screening: the NCI experience. Eur. J. Cancer. 2004;40:821–826. doi: 10.1016/j.ejca.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Deng L, Nagano-Fujii M, Tanaka M, et al. NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 2006;87:1703–1713. doi: 10.1099/vir.0.81735-0. [DOI] [PubMed] [Google Scholar]
- Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene. 1999;18:4726–4733. doi: 10.1038/sj.onc.1202858. [DOI] [PubMed] [Google Scholar]
- Diwan BA, Rice JM, Ward JM, Ohshima M, Lynch PH. Inhibition by phenobarbital and lack of effect of amobarbital on the development of liver tumors induced by N-nitrosodiethylamine in juvenile B6C3F1 mice. Cancer Lett. 1984;23:223–234. doi: 10.1016/0304-3835(84)90157-5. [DOI] [PubMed] [Google Scholar]
- Diwan BA, Rice JM, Ohshima M, Ward JM. Interstrain differences in susceptibility to liver carcinogenesis initiated by N-nitrosodiethylamine and its promotion by phenobarbital in C57BL/6NCr, C3H/HeNCrMTV- and DBA/2NCr mice. Carcinogenesis. 1986;7:215–220. doi: 10.1093/carcin/7.2.215. [DOI] [PubMed] [Google Scholar]
- Dubois N, Bennoun M, Allemand I, et al. Time-course development of differentiated hepatocarcinoma and lung metastasis in transgenic mice. J. Hepatol. 1991;13:227–239. doi: 10.1016/0168-8278(91)90819-w. [DOI] [PubMed] [Google Scholar]
- ElRefaie A, Savage K, Bhattacharya S, et al. HCV-associated hepatocellular carcinoma without cirrhosis. J. Hepatol. 1996;24:277–285. doi: 10.1016/s0168-8278(96)80005-5. [DOI] [PubMed] [Google Scholar]
- Farazi PA, Glickman J, Horner J, Depinho RA. Cooperative interactions of p53 mutation, telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression. Cancer Res. 2006;66:4766–4773. doi: 10.1158/0008-5472.CAN-05-4608. [DOI] [PubMed] [Google Scholar]
- Farber E. The multistep nature of cancer development. Cancer Res. 1984;44:4217–4223. [PubMed] [Google Scholar]
- Farber E, Solt D, Cameron R, Laishes B, Ogawa K, Medline A. Newer insights into pathogenesis of liver cancer. Am. J. Pathol. 1977;89:477–482. [PMC free article] [PubMed] [Google Scholar]
- Feng XY, Zhang HQ, Liu HZ, et al. Cancerogenic effect of different fragments of the hepatitis C virus core protein. Eur. J. Cancer Prev. 2007;16:304–311. doi: 10.1097/01.cej.0000236252.16855.82. [DOI] [PubMed] [Google Scholar]
- Finnberg N, Stenius U, Hogberg J. Heterozygous p53-deficient (+/−) mice develop fewer p53-negative preneoplastic focal liver lesions in response to treatment with diethylnitrosamine than do wild-type (+/+) mice. Cancer Lett. 2004;207:149–155. doi: 10.1016/j.canlet.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat. Rev. Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- Frey S, Buchmann A, Bursch W, Schulte-Hermann R, Schwarz M. Suppression of apoptosis in C3H mouse liver tumors by activated Ha-ras oncogene. Carcinogenesis. 2000;21:161–166. doi: 10.1093/carcin/21.2.161. [DOI] [PubMed] [Google Scholar]
- Gallagher EP, Wienkers LC, Stapleton PL, Kunze KL, Eaton DL. Role of human microsomal and human complementary DNA-expressed cytochromes P4501A2 and P4503A4 in the bioactivation of aflatoxin B1. Cancer Res. 1994;54:101–108. [PubMed] [Google Scholar]
- Geller SA, Nichols WS, Kim S, et al. Hepatocarcinogenesis is the sequel to hepatitis in Z#2 alpha 1-antitrypsin transgenic mice: histopathological and DNA ploidy studies. Hepatology. 1994;19:389–397. [PubMed] [Google Scholar]
- Gerber MA. Relation of hepatitis-C virus to hepatocellular-carcinoma. J. Hepatol. 1993;17:S108–S111. doi: 10.1016/s0168-8278(05)80433-7. [DOI] [PubMed] [Google Scholar]
- Ghebranious N, Sell S. The mouse equivalent of the human p53ser249 mutation p53ser246 enhances aflatoxin hepatocarcinogenesis in hepatitis B surface antigen transgenic and p53 heterozygous null mice. Hepatology. 1998;27:967–973. doi: 10.1002/hep.510270411. [DOI] [PubMed] [Google Scholar]
- Goldsworthy TL, Fransson-Steen R. Quantitation of the cancer process in C57BL/6J, B6C3F1 and C3H/HeJ mice. Toxicol. Pathol. 2002;30:97–105. doi: 10.1080/01926230252824770. [DOI] [PubMed] [Google Scholar]
- Gray R, Peto R, Brantom P, Grasso P. Chronic nitrosamine ingestion in 1040 rodents: the effect of the choice of nitrosamine, the species studied, and the age of starting exposure. Cancer Res. 1991;51:6470–6491. [PubMed] [Google Scholar]
- Gudima S, He YP, Chai N, et al. Primary human hepatocytes are susceptible to infection by hepatitis delta virus assembled with envelope proteins of woodchuck hepatitis virus. J. Virol. 2008;82:7276–7283. doi: 10.1128/JVI.00576-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker HJ, Mtiro H, Bannasch P, Vesselinovitch SD. Histochemical profile of mouse hepatocellular adenomas and carcinomas induced by a single dose of diethylnitrosamine. Cancer Res. 1991;51:1952–1958. [PubMed] [Google Scholar]
- Harada N, Miyoshi H, Murai N, et al. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer Res. 2002;62:1971–1977. [PubMed] [Google Scholar]
- Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-Ras gene mutations. Cancer Res. 2004;64:48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- Hasmall SC, James NH, Macdonald N, Gonzalez FJ, Peters JM, Roberts RA. Suppression of mouse hepatocyte apoptosis by peroxisome proliferators: role of PPAR alpha and TNF alpha. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2000;448:193–200. doi: 10.1016/s0027-5107(99)00236-5. [DOI] [PubMed] [Google Scholar]
- Hays T, Rusyn I, Burns AM, et al. Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis. 2005;26:219–227. doi: 10.1093/carcin/bgh285. [DOI] [PubMed] [Google Scholar]
- Hollingshead MG, Alley MC, Camalier RF, et al. In vivo cultivation of tumor cells in hollow fibers. Life Sci. 1995;57:131–141. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am. J. Pathol. 2000;157:763–770. doi: 10.1016/s0002-9440(10)64590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu KQ, Yu CH, Vierling JM. Up-regulation of intercellular adhesion molecule 1 transcription by hepatitis B virus X protein. Proc. Natl Acad. Sci. USA. 1992;89:11441–11445. doi: 10.1073/pnas.89.23.11441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh H, Soo KC, Chow PKH, Panasci L, Tran E. Xenografts of human hepatocellular carcinoma: a useful model for testing drugs. Clin. Cancer Res. 2006;12:4306–4314. doi: 10.1158/1078-0432.CCR-05-2568. [DOI] [PubMed] [Google Scholar]
- Iida M, Anna CH, Hartis J, et al. Changes in global gene and protein expression during early mouse liver carcinogenesis induced by non-genotoxic model carcinogens oxazepam and Wyeth-14,643. Carcinogenesis. 2003;24:757–770. doi: 10.1093/carcin/bgg011. [DOI] [PubMed] [Google Scholar]
- Imaoka S, Osada M, Minamiyama Y, et al. Role of phenobarbital-inducible cytochrome P450s as a source of active oxygen species in DNA-oxidation. Cancer Lett. 2004;203:117–125. doi: 10.1016/j.canlet.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61:1137–1146. doi: 10.1016/0092-8674(90)90076-q. [DOI] [PubMed] [Google Scholar]
- Kamegaya Y, Hiasa Y, Zukerberg L, et al. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology. 2005;41:660–667. doi: 10.1002/hep.20621. [DOI] [PubMed] [Google Scholar]
- Kanamura S, Kanai K, Watanabe J. Fine-structure and function of hepatocytes during development. J. Electron Microsc. Tech. 1990;14:92–105. doi: 10.1002/jemt.1060140204. [DOI] [PubMed] [Google Scholar]
- Kawamura T, Furusaka A, Koziel MJ, et al. Transgenic expression of hepatitis C virus structural proteins in the mouse. Hepatology. 1997;25:1014–1021. doi: 10.1002/hep.510250437. [DOI] [PubMed] [Google Scholar]
- Kawanishi S, Hiraku Y, Murata M, Oikawa S. The role of metals in site-specific DNA damage with reference to carcinogenesis. Free Radic. Biol. Med. 2002;32:822–832. doi: 10.1016/s0891-5849(02)00779-7. [DOI] [PubMed] [Google Scholar]
- Kelland LR. Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur. J. Cancer. 2004;40:827–836. doi: 10.1016/j.ejca.2003.11.028. [DOI] [PubMed] [Google Scholar]
- Keller BJ, Yamanaka H, Liang D, Thurman RG. Hepatotoxicity due to clofibrate is oxygen-dependent in the perfused-rat-liver. Toxicol. Appl. Pharmacol. 1990;104:259–266. doi: 10.1016/0041-008x(90)90300-j. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Pereira MA, Ruch RJ, Weghorst CM. Dose-response relationship of diethylnitrosamine-initiated tumors in neonatal balb/c mice: effect of phenobarbital promotion. Toxicol. Pathol. 1988;16:381–385. doi: 10.1177/019262338801600310. [DOI] [PubMed] [Google Scholar]
- Klinman NR, Erslev AJ. Cellular response to partial hepatectomy. Proc. Soc. Exp. Biol. Med. 1963;112:338–340. doi: 10.3181/00379727-112-28037. [DOI] [PubMed] [Google Scholar]
- Knight B, Yeoh GC, Husk KL, et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J. Exp. Med. 2000;192:1809–1818. doi: 10.1084/jem.192.12.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike K, Moriya K, Iino S, et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994;19:810–819. [PubMed] [Google Scholar]
- Kolaja KL, Klaunig JE. Vitamin E modulation of hepatic focal lesion growth in mice. Toxicol. Appl. Pharmacol. 1997;143:380–387. doi: 10.1006/taap.1996.8089. [DOI] [PubMed] [Google Scholar]
- Koo JS, Seong JK, Park C, et al. Large liver cell dysplasia in hepatitis B virus X transgenic mouse liver and human chronic hepatitis B virus-infected liver. Intervirology. 2005;48:16–22. doi: 10.1159/000082090. [DOI] [PubMed] [Google Scholar]
- Kornek M, Raskopf E, Tolba R, et al. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro-angiogenic and prometastatic factors in murine liver fibrosis. Liver Int. 2008;28:509–518. doi: 10.1111/j.1478-3231.2008.01670.x. [DOI] [PubMed] [Google Scholar]
- Lakhtakia R, Kumar V, Reddi H, Mathur M, Dattagupta S, Panda SK. Hepatocellular carcinoma in a hepatitis B ‘x’ transgenic mouse model: a sequential pathological evaluation. J. Gastroenterol. Hepatol. 2003;18:80–91. doi: 10.1046/j.1440-1746.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- Lan KH, Sheu ML, Hwang SJ, et al. HCVNS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene. 2002;21:4801–4811. doi: 10.1038/sj.onc.1205589. [DOI] [PubMed] [Google Scholar]
- Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–3786. doi: 10.1038/sj.onc.1209547. [DOI] [PubMed] [Google Scholar]
- Lechuga CG, Hernandez-Nazara ZH, Dominguez Rosales JA, Morris ER, Rincon AR, Rivas-Estilla AM, Esteban-Gamboa A, Rojkind M. TGF-beta1 modulates matrix metalloproteinase-13 expression in hepatic stellate cells by complex mechanisms involving p38MAPK, PI3-kinase, AKT, and p70S6k. Am J Physiol Gastrointest Liver Physiol. 2004;287:G974–987. doi: 10.1152/ajpgi.00264.2003. [DOI] [PubMed] [Google Scholar]
- Ledda-Columbano GM, Perra A, Concas D, et al. Different effects of the liver mitogens triiodo-thyronine and ciprofibrate on the development of rat hepatocellular carcinoma. Toxicol. Pathol. 2003;31:113–120. doi: 10.1080/01926230390173851. [DOI] [PubMed] [Google Scholar]
- Lee TH, Finegold MJ, Shen RF, DeMayo JL, Woo SL, Butel JS. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J. Virol. 1990;64:5939–5947. doi: 10.1128/jvi.64.12.5939-5947.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GH, Ooasa T, Osanai M. Mechanism of the paradoxical, inhibitory effect of phenobarbital on hepatocarcinogenesis initiated in infant B6C3F1 mice with diethylnitrosamine. Cancer Res. 1998;58:1665–1669. [PubMed] [Google Scholar]
- Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- Lefevre PA, Tinwell H, Galloway SM, et al. Evaluation of the genetic toxicity of the peroxisome proliferator and carcinogen methyl clofenapate, including assays using muta(Tm)mouse and Big Blue(Tm) transgenic mice. Hum. Exp. Toxicol. 1994;13:764–775. doi: 10.1177/096032719401301105. [DOI] [PubMed] [Google Scholar]
- Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- Li XL, Liu JY, Lu R, et al. Evaluation of the therapeutic efficacy of tripeptide tyroserleutide (YSL) for human hepatocarcinoma by in vivo hollow fiber assay. Invest. New Drugs. 2008;26(6):525–529. doi: 10.1007/s10637-008-9121-8. [DOI] [PubMed] [Google Scholar]
- de Lima VM, Oliveira CP, Alves VA, et al. A rodent model of NASH with cirrhosis, oval cell proliferation and hepatocellular carcinoma. J. Hepatol. 2008;49:1055–1061. doi: 10.1016/j.jhep.2008.07.024. [DOI] [PubMed] [Google Scholar]
- Liquori GE, Calamita G, Cascella D, Mastrodonato M, Portincasa P, Ferri D. An innovative methodology for the automated morphometric and quantitative estimation of liver steatosis. Histol. Histopathol. 2009;24:49–60. doi: 10.14670/HH-24.49. [DOI] [PubMed] [Google Scholar]
- Liu SP, Li YS, Chen YJ, et al. Glycine N-methyltransferase-/- mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology. 2007;46:1413–1425. doi: 10.1002/hep.21863. [DOI] [PubMed] [Google Scholar]
- Loeppen S, Schneider D, Gaunitz F, et al. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer Res. 2002;62:5685–5688. [PubMed] [Google Scholar]
- Lou DQ, Molina T, Bennoun M, et al. Conditional hepatocarcinogenesis in mice expressing SV 40 early sequences. Cancer Lett. 2005;229:107–114. doi: 10.1016/j.canlet.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Lu MJ, Yao X, Xu Y, et al. Combination of an antiviral drug and immunomodulation against hepadnaviral infection in the woodchuck model. J. Virol. 2008;82:2598–2603. doi: 10.1128/JVI.01613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XF, Stoffregen DA, Wheelock GD, Rininger JA, Babish JG. Discordant hepatic expression of the cell division control enzyme p34(cdc2) kinase, proliferating cell nuclear antigen, p53 tumor suppressor protein, and p21(Waf1) cyclin-dependent kinase inhibitory protein after WY14,643 ([4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid) dosing to rats. Mol. Pharmacol. 1997;51:69–78. doi: 10.1124/mol.51.1.69. [DOI] [PubMed] [Google Scholar]
- Mao TL, Chu JS, Jeng YM, Lai PL, Hsu HC. Expression of mutant nuclear beta-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J. Pathol. 2001;193:95–101. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH720>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Sakurai H, Saiki I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows activity using a hepatocellular carcinoma model. Mol. Cancer Ther. 2003;2:557–561. [PubMed] [Google Scholar]
- McGlynn KA, Hunter K, LeVoyer T, et al. Susceptibility to aflatoxin B-1-related primary hepatocellular carcinoma in mice and humans. Cancer Res. 2003;63:4594–4601. [PubMed] [Google Scholar]
- Merle P, Kim M, Herrmann M, et al. Oncogenic role of the frizzled-7/beta-catenin pathway in hepatocellular carcinoma. J. Hepatol. 2005;43:854–862. doi: 10.1016/j.jhep.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor alpha to Wy-14,643-induced liver tumorigenesis. Carcinogenesis. 2006;27:1074–1080. doi: 10.1093/carcin/bgi329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- Motola-Kuba D, Zamora-Valdes D, Uribe M, Mendez-Sanchez N. Hepatocellular carcinoma. An overview. Ann. Hepatol. 2006;5:16–24. [PubMed] [Google Scholar]
- Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53:1719–1723. [PubMed] [Google Scholar]
- Naas T, Ghorbani M, Alvarez-Maya I, et al. Characterization of liver histopathology in a transgenic mouse model expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2. J. Gen. Virol. 2005;86:2185–2196. doi: 10.1099/vir.0.80969-0. [DOI] [PubMed] [Google Scholar]
- Nagini S, Nagarajan B. Hypolipidemic drug clofibrate promotes hepatic tumor. Biochem. Int. 1988;17:605–610. [PubMed] [Google Scholar]
- Nakatani T, Roy G, Fujimoto N, Asahara T, Ito A. Sex hormone dependency of diethylnitrosamine-induced liver tumors in mice and chemoprevention by leuprorelin. Jpn. J. Cancer Res. 2001;92:249–256. doi: 10.1111/j.1349-7006.2001.tb01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma - Ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 2002;160:2295–2307. doi: 10.1016/S0002-9440(10)61177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- Nishimura J, Nishimura J, Dewa Y, et al. Mechanistic study on fenofibrate-induced hepatocarcinogenesis in a 2-stage hepatocarcinogenesis model of rats: Involvement of oxidative stress. Toxicol. Lett. 2007;172:S173–S173. [Google Scholar]
- Ohgaki H, Sanderson ND, Ton P, Thorgeirsson SS. Molecular analyses of liver tumors in c-myc transgenic mice and c-myc and TGF-alpha double transgenic mice. Cancer Lett. 1996;106:43–49. doi: 10.1016/0304-3835(96)04299-1. [DOI] [PubMed] [Google Scholar]
- Palacios RS, Roderfeld M, Hemmann S, et al. Activation of hepatic stellate cells is associated with cytokine expression in thioacetamide-induced hepatic fibrosis in mice. Lab. Invest. 2008;88:1192–1203. doi: 10.1038/labinvest.2008.91. [DOI] [PubMed] [Google Scholar]
- Park TJ, Kim JY, Oh SP, et al. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology. 2008;47:1533–1543. doi: 10.1002/hep.22212. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int. J. Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119–6129. [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- Phillips RM, Pearce J, Double JA. Temporal characterisation of blood vessel development to hollow fibres: Implications for drug delivery and drug evaluation. Ann. Oncol. 1998;9:72–72. [Google Scholar]
- Pitot HC, Dragan YP. Facts and theories concerning the mechanisms of carcinogenesis. FASEB J. 1991;5:2280–2286. [PubMed] [Google Scholar]
- Qi W, Chen XM, Holian J, et al. Transforming growth factor-beta(1) differentially mediates fibronectin and inflammatory cytokine expression in kidney tubular cells. Am. J. Physiol. Renal. Physiol. 2006;291:F1070–F1077. doi: 10.1152/ajprenal.00013.2006. [DOI] [PubMed] [Google Scholar]
- Qi YT, Chen X, Chan CY, et al. Two-dimensional differential gel electrophoresis/analysis of diethylnitrosamine induced rat hepatocellular carcinoma. Int. J. Cancer. 2008;122:2682–2688. doi: 10.1002/ijc.23464. [DOI] [PubMed] [Google Scholar]
- Ramsdell HS, Parkinson A, Eddy AC, Eaton DL. Bioactivation of aflatoxin B1 by human liver microsomes: role of cytochrome P450 IIIA enzymes. Toxicol. Appl. Pharmacol. 1991;108:436–447. doi: 10.1016/0041-008x(91)90090-2. [DOI] [PubMed] [Google Scholar]
- Rao MS, Reddy JK. Hepatocarcinogenesis of peroxisome proliferators. Peroxisomes: Biol. Role Toxicol. Dis. 1996;804:573–587. doi: 10.1111/j.1749-6632.1996.tb18646.x. [DOI] [PubMed] [Google Scholar]
- Rao KV, Vesselinovitch SD. Age- and sex-associated diethylnitrosamine dealkylation activity of the mouse liver and hepatocarcinogenesis. Cancer Res. 1973;33:1625–1627. [PubMed] [Google Scholar]
- Reddy JK, Rao S, Moody DE. Hepatocellular carcinomas in acatalasemic mice treated with nafenopin, a hypolipidemic peroxisome proliferator. Cancer Res. 1976;36:1211–1217. [PubMed] [Google Scholar]
- Roberts RA, Soames AR, Gill JH, James NH, Wheeldon EB. Nongenotoxic hepatocarcinogens stimulate DNA-synthesis and their withdrawal induces apoptosis, but in different hepatocyte populations. Carcinogenesis. 1995;16:1693–1698. doi: 10.1093/carcin/16.8.1693. [DOI] [PubMed] [Google Scholar]
- Sanderson N, Factor V, Nagy P, et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl Acad. Sci. USA. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur J, Olah J, Szepesi A, Nagy P, Thorgeirsson SS. Thioacetamide-induced hepatic fibrosis in transforming growth factor beta-1 transgenic mice. Eur. J. Gastroenterol. Hepatol. 2004;16:127–133. doi: 10.1097/00042737-200402000-00002. [DOI] [PubMed] [Google Scholar]
- Schwarz M, Wanke I, Wulbrand U, Moennikes O, Buchmann A. Role of connexin32 and beta-catenin in tumor promotion in mouse liver. Toxicol. Pathol. 2003;31:99–102. doi: 10.1080/01926230390173932. [DOI] [PubMed] [Google Scholar]
- Sell S, Hunt JM, Dunsford HA, Chisari FV. Synergy between hepatitis-B virus expression and chemical hepatocarcinogens in transgenic mice. Cancer Res. 1991;51:1278–1285. [PubMed] [Google Scholar]
- Sepulveda AR, Finegold MJ, Smith B, et al. Development of a transgenic mouse system for the analysis of stages in liver carcinogenesis using tissue-specific expression of SV40 large T-antigen controlled by regulatory elements of the human alpha-1-antitrypsin gene. Cancer Res. 1989;49:6108–6117. [PubMed] [Google Scholar]
- Shao DM, Wang QH, Chen C, et al. N-acetylglucosaminyltransferase V activity in metastatic models of human hepatocellular carcinoma in nude mice. J. Exp. Clin. Cancer Res. 1999;18:331–335. [PubMed] [Google Scholar]
- Sheweita SA, El-Gabar MA, Bastawy M. Carbon tetrachloride changes the activity of cytochrome P450 system in the liver of male rats: role of antioxidants. Toxicology. 2001;169:83–92. doi: 10.1016/s0300-483x(01)00473-5. [DOI] [PubMed] [Google Scholar]
- Shiota G, Harada K, Ishida M, et al. Inhibition of hepatocellular carcinoma by glycyrrhizin in diethylnitrosamine-treated mice. Carcinogenesis. 1999;20:59–63. doi: 10.1093/carcin/20.1.59. [DOI] [PubMed] [Google Scholar]
- Shnyder SD, Cooper PA, Scally AJ, Bibby MC. Reducing the cost of screening novel agents using the hollow fibre assay. Anticancer Res. 2006;26:2049–2052. [PubMed] [Google Scholar]
- Stahl S, Ittrich C, Marx-Stoelting P, et al. Genotype-phenotype relationships in hepatocellular tumors from mice and man. Hepatology. 2005;42:353–361. doi: 10.1002/hep.20768. [DOI] [PubMed] [Google Scholar]
- Suggitt M, Cooper PA, Shnyder SD, Bibby MC. The hollow fibre model–facilitating anti-cancer pre-clinical pharmacodynamics and improving animal welfare. Int. J. Oncol. 2006;29:1493–1499. [PubMed] [Google Scholar]
- Sun FX, Tang ZY, Lui KD, et al. Establishment of a metastatic model of human hepatocellular carcinoma in nude mice via orthotopic implantation of histologically intact tissues. Int. J. Cancer. 1996;66:239–243. doi: 10.1002/(SICI)1097-0215(19960410)66:2<239::AID-IJC17>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Tada M, Omata M, Ohto M. Analysis of Ras gene-mutations in human hepatic malignant-tumors by polymerase chain-reaction and direct sequencing. Cancer Res. 1990;50:1121–1124. [PubMed] [Google Scholar]
- Takada S, Tsuchida N, Kobayashi M, Koike K. Disruption of the function of tumor-suppressor gene P53 by the Hepatitis-B Virus X-Protein and Hepatocarcinogenesis. J. Cancer Res. Clin. Oncol. 1995;121:593–601. doi: 10.1007/BF01197776. [DOI] [PubMed] [Google Scholar]
- Takashima K, Ito Y, Gonzalez FJ, Nakajima T. Different mechanisms of DEHP-induced hepatocellular adenoma tumorigenesis in wild-type and Ppar alpha-null mice. J. Occup. Health. 2008;50:169–180. doi: 10.1539/joh.l7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Moriya K, Kiyosawa K, Koike K, Aoyama T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: Implications for HCV-associated hepatocarcinogenesis. Int. J. Cancer. 2008;122:124–131. doi: 10.1002/ijc.23056. [DOI] [PubMed] [Google Scholar]
- Tarsetti F, Lenzi R, Salvi R, et al. Liver carcinogenesis associated with feeding of ethionine in a choline-free diet: evidence against a role of oval cells in the emergence of hepatocellular carcinoma. Hepatology. 1993;18:596–603. [PubMed] [Google Scholar]
- Tennant BC, Toshkov IA, Peek SF, et al. Hepatocellular carcinoma in the woodchuck model of hepatitis B virus infection. Gastroenterology. 2004;127:S283–293. doi: 10.1053/j.gastro.2004.09.043. [DOI] [PubMed] [Google Scholar]
- Teoh NC, Dan YY, Swisshelm K, et al. Defective DNA strand break repair causes chromosomal instability and accelerates liver carcinogenesis in mice. Hepatology. 2008;47:2078–2088. doi: 10.1002/hep.22194. [DOI] [PubMed] [Google Scholar]
- Terradillos O, Billet O, Renard CA, et al. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene. 1997;14:395–404. doi: 10.1038/sj.onc.1200850. [DOI] [PubMed] [Google Scholar]
- Thieringer F, Maass T, Czochra P, et al. Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene. 2008;423:23–28. doi: 10.1016/j.gene.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson SS, SantoniRugiu E. Transgenic mouse models in carcinogenesis: Interaction of c-myc with transforming growth factor alpha and hepatocyte growth factor in hepatocarcinogenesis. Br. J. Clin. Pharmacol. 1996;42:43–52. doi: 10.1046/j.1365-2125.1996.03748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonjes RR, Lohler J, Osullivan JF, et al. Autocrine mitogen Igegf cooperates with C-Myc or with the Hcs locus during hepatocarcinogenesis in transgenic mice. Oncogene. 1995;10:765–768. [PubMed] [Google Scholar]
- Tsukuma H, Hiyama T, Tanaka S, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N. Engl. J. Med. 1993;328:1797–1801. doi: 10.1056/NEJM199306243282501. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Jacks T. Technologically advanced cancer modeling in mice. Curr. Opin. Genet. Dev. 2002;12:105–110. doi: 10.1016/s0959-437x(01)00272-6. [DOI] [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996;71:57–81. doi: 10.1016/0163-7258(96)00062-9. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch SD, Mihailovich N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983;43:4253–4259. [PubMed] [Google Scholar]
- Vesselinovitch SD, Mihailovich N, Rao KVN. Morphology and metastatic nature of induced hepatic nodular lesions in C57bl X C3h F1 mice. Cancer Res. 1978;38:2003–2010. [PubMed] [Google Scholar]
- Wang JS, Groopman JD. DNA damage by mycotoxins. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1999;424:167–181. doi: 10.1016/s0027-5107(99)00017-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu XM. Contamination of aflatoxins in different kinds of foods in China. Biomed. Environ. Sci. 2007;20:483–487. [PubMed] [Google Scholar]
- Wang ZY, Agarwal R, Khan WA, Mukhtar H. Protection against Benzo[a]Pyrene-induced and N-Nitrosodiethylamine-induced lung and forestomach tumorigenesis in a/J mice by water extracts of green tea and licorice. Carcinogenesis. 1992;13:1491–1494. doi: 10.1093/carcin/13.8.1491. [DOI] [PubMed] [Google Scholar]
- Wang Q, Lin ZY, Feng XL. Alterations in metastatic properties of hepatocellular carcinoma cell following H-ras oncogene transfection. World J. Gastroenterol. 2001;7:335–339. doi: 10.3748/wjg.v7.i3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Horie Y, Kataoka E, et al. Non-alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J. Gastroenterol. Hepatol. 2007;22:S96–S100. doi: 10.1111/j.1440-1746.2006.04665.x. [DOI] [PubMed] [Google Scholar]
- Watson RE, Goodman JI. Effects of phenobarbital on DNA methylation in GC-rich regions of hepatic DNA from mice that exhibit different levels of susceptibility to liver tumorigenesis. Toxicol. Sci. 2002;68:51–58. doi: 10.1093/toxsci/68.1.51. [DOI] [PubMed] [Google Scholar]
- Waxman DJ, Azaroff L. Phenobarbital induction of cytochrome P-450 gene expression. Biochem. J. 1992;281(Pt 3):577–592. doi: 10.1042/bj2810577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weghorst CM, Klaunig JE. Phenobarbital promotion in diethylnitrosamine-initiated infant B6c3f1 mice - influence of gender. Carcinogenesis. 1989;10:609–612. doi: 10.1093/carcin/10.3.609. [DOI] [PubMed] [Google Scholar]
- Weisburger EK. Carcinogenicity studies on halogenated hydrocarbons. Environ. Health Perspect. 1977;21:7–16. doi: 10.1289/ehp.77217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AO, Knapton AD. Hepatic silicosis, cirrhosis, and liver tumors in mice and hamsters: studies of transforming growth factor beta expression. Hepatology. 1996;23:1268–1275. doi: 10.1002/hep.510230548. [DOI] [PubMed] [Google Scholar]
- Williams GM, Iatropoulos MJ, Jeffrey AM. Mechanistic basis for nonlinearities and thresholds in rat liver carcinogenesis by the DNA-reactive carcinogens 2-acetylaminofluorene and diethylnitrosamine. Toxicol. Pathol. 2000;28:388–395. doi: 10.1177/019262330002800306. [DOI] [PubMed] [Google Scholar]
- Wu CG, Salvay DM, Forgues M, et al. Distinctive gene expression profiles associated with Hepatitis B virus x protein. Oncogene. 2001;20:3674–3682. doi: 10.1038/sj.onc.1204481. [DOI] [PubMed] [Google Scholar]
- Xiong J, Yao YC, Zi XY, et al. Expression of hepatitis B virus X protein in transgenic mice. World J. Gastroenterol. 2003;9:112–116. doi: 10.3748/wjg.v9.i1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- Zhang GJ, Chen TB, Bednar B, et al. Optical imaging of tumor cells in hollow fibers: evaluation of the antitumor activities of anticancer drugs and _target validation. Neoplasia. 2007;9:652–661. doi: 10.1593/neo.07421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WL, Iskandar S, Kooshki M, Sharpe JG, Payne V, Robbins ME. Knocking out peroxisome proliferator-activated receptor (PPAR) alpha inhibits radiation-induced apoptosis in the mouse kidney through activation of NF-kappa B and increased expression of IAPs. Radiat. Res. 2007;167:581–591. doi: 10.1667/RR0814.1. [DOI] [PubMed] [Google Scholar]
- Zheng YY, Chen WL, Louie SG, Yen TSB, Ou JHJ. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology. 2007;45:16–21. doi: 10.1002/hep.21445. [DOI] [PubMed] [Google Scholar]
- Zhu HZ, Wang Y, Chen JQ, Cheng GX, Xue JL. Transgenic mice expressing hepatitis B virus X protein are more susceptible to carcinogen induced hepatocarcinogenesis. Exp. Mol. Pathol. 2004;76:44–50. doi: 10.1016/j.yexmp.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Jin XL, Gutierrez JC, et al. Effect of in vivo loss of GDF-15 on hepatocellular carcinogenesis. J. Cancer Res. Clin. Oncol. 2008;134:753–759. doi: 10.1007/s00432-007-0336-4. [DOI] [PubMed] [Google Scholar]