

Abstract
Phosphatase and tensin homologue deleted from chromosome 10 (Pten) is expressed aberrantly in non-small cell lung cancer cells, but the role of Pten in lung neoplasia has not been fully elucidated. In this study, we used a genetic approach to inactivate Pten in the bronchial epithelium of mice. Although, by itself, Pten inactivation had no discernible effect on bronchial epithelial histology, it accelerated lung tumorigenesis initiated by oncogenic K-ras, causing more rapid lethality than that induced by oncogenic K-ras alone (8 weeks versus 24 weeks of median duration of survival, respectively). Lung tumors arose in K-ras mutant, Pten-deficient mice that rapidly obstructed bronchial lumina and replaced alveolar spaces. Relative to K-ras mutant tumors, the K-ras mutant, Pten-deficient tumors exhibited more advanced histologic severity and more prominent inflammation and vascularity. Thus, Pten inactivation cooperated with oncogenic K-ras in promoting lung tumorigenesis.
Introduction
A better understanding of the pathogenesis and risk factors for lung cancer, the most common cause of cancer-related death in Western countries, should contribute not only to improvements in the early detection and prevention of this disease but also to the development of more effective therapies for it. Approximately 30% of lung adenocarcinomas carry activating mutations in K-ras (1). Studies have also identified K-ras mutations in atypical adenomatous hyperplasia (AAH) lesions, histologic changes that are thought to precede the development of lung adenocarcinoma (2). A growing body of evidence indicates that K-ras mutations are important in the initiation of lung adenocarcinoma development. The introduction of mutant K-ras into immortalized human bronchial epithelial cells confers certain neoplastic features (3). Mouse models that express mutant K-ras conditionally, somatically, or inducibly have been developed (4-8). These mice develop lung lesions rapidly and with high penetrance. However, only a fraction of the lesions progress into adenocarcinomas and require several months to do so. Thus, findings from studies on cellular and mouse models suggest that mutant K-ras is sufficient to initiate but not to complete the development of lung adenocarcinomas.
In 1997, phosphatase and tensin homologue deleted from chromosome 10 (Pten) was identified as a tumor suppressor gene located on chromosome 10q23.3 (9). It encodes a lipid phosphatase that functions in the phosphatidylinositol 3-kinase (PI3K) signaling cascade. Pten inactivation induces spontaneous tumors in mice, supporting its role as a tumor suppressor gene (10-14). A broad spectrum of somatic Pten deletions in primary human tumors has been reported, including missense mutations, null mutations, and truncations (15). In non-small cell lung cancer (NSCLC) biopsy samples and cell lines, Pten expression is frequently silenced because of Pten promoter methylation or binding of the nuclear factor-κB transcription factor, which suppresses Pten transcription (16-18). Although they occur infrequently, inactivating Pten somatic mutations have been detected in NSCLC (16, 19). Despite this evidence implicating aberrant Pten expression in NSCLC, its role in lung tumorigenesis has yet to be defined.
Tumorigenesis initiated by K-ras mutations can be promoted or suppressed by genetic events that activate PI3K-dependent signaling. In colorectal cancer, K-ras mutations coexist with activating mutations in PIK3CA (20), which encodes the catalytic subunit of PI3K. Similarly, NSCLC cell lines that express no detectable PTEN frequently have K-ras mutations (1, 17), suggesting that alteration in both genes confers a selective advantage in these cells. However, Ras and Pten mutations are mutually exclusive in other tumor types (21-24), suggesting that these events are genetically redundant and that alteration in both genes does not confer a further advantage or is in fact disadvantageous when these events occur together in the same cell. Findings from mouse model studies (21, 25) support the conclusion that these mutations have cooperative or antagonistic effects, depending upon tissue-specific factors.
Whether oncogenic K-ras mutations and Pten inactivation interact to promote or inhibit the development of NSCLC has not yet been defined, an important question given the frequency of these genetic events in NSCLC and the need for a better understanding of the mechanisms driving malignant progression in the bronchial epithelium. Using mouse models in which conditional oncogenic K-ras and Pten null alleles can be _targeted specifically in the bronchial epithelium, we show here that, by itself, Pten inactivation had no discernible effect, but it accelerated lung neoplasia initiated by oncogenic K-ras. We concluded that Pten inactivation is a crucial event in the progression of K-ras-initiated lung tumorigenesis and that strategies to _target PI3K-dependent signaling should be considered in the prevention and treatment of this disease.
Materials and Methods
Mouse experiments
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at The University of Texas M. D. Anderson Cancer Center. KrasLSL, Ptenflox, and CCSPCre strains were interbred to evaluate the effects of oncogenic K-ras and Pten loss alone and in combination in the lung. All of these mouse strains were on a C57/BL6 background.
Immunohistochemical analysis
Four-micrometer sections of mouse lung tissues were placed on coated slides. After deparaffinization and rehydration, antigen unmasking was performed using heat treatment with 1× DAKO _target retrieval solution (DAKO Corporation) for 15 min. Endogenous peroxidase activity was quenched using 10-min incubation in 3% diluted hydrogen peroxide. After blocking of nonspecific binding for 20 min with DAKO serum-free protein block (DAKO Corporation), the sections were incubated overnight at 4°C with the primary antibodies against factor VIII (rabbit; DAKO Corporation) at a dilution of 1:700, PTEN (Cell Signaling Technologies) at a dilution of 1:100, phosphorylated p70S6R-Ser235/236 (rabbit; Cell Signaling Technologies) at a dilution of 1:100, p40 (rat; Serotec) at a dilution of 1:50, F4/80 (rat; Serotec) at a dilution of 1:50, and pan-cytokeratin (clone MNF116; DAKO) at a dilution of 1:100. After washing with PBS, slides were incubated with a biotin-labeled secondary antibody (antirabbit and antirat) for 30 min at a dilution of 1:300. Primary antibody binding was localized in tissues using peroxidase-conjugated streptavidin (DAKO Corporation). After staining with 3′,3-diaminobenzidine tetrahydrochloride, the slides were counterstained with hematoxylin, dehydrated, and mounted. Negative controls included omission of the primary antibody and preincubation of the primary antibody with a blocking peptide.
For evaluation of the immunohistochemical expression of cell markers, two trained pathologists (G.R. and I.I.W.) independently quantified expression. Factor VIII, p40, and F4/80 staining expression were quantified according to the number of positive cells in a 1-mm2 area within the lesions.
Immunofluorescence analysis
Antigens were retrieved with 0.01 mol/L citrate buffer (pH 6; DakoCytomation) for 25 min in a steamer. The slides were quenched in 1.5% hydrogen peroxide in TBS for 10 min and blocked in DAKO serum-free protein block (DakoCytomation) for 1 h at room temperature. Subsequently, the slides were incubated with anti-surfactant protein C (SPC; Seven Hills Bioreagents, WRAB-SPC, 1:1,200), anti-clara cell-specific protein (CCSP; Upstate Biotechnology, 07-623, 1:3,000), or anti-PTEN (Santa Cruz, 1:100) at 4°C overnight. The immunofluorescence was developed using TSA kits 25 and 22 (Invitrogen) based on the manufacturer’s instructions. As negative controls to determine the specificity of the immunostaining results, we used blocking peptides and omitted the primary antibodies.
For detection of mucin glycoproteins, tissues were stained using procedure periodic acid-Schiff (PAS) staining. Briefly, slides were dewaxed, rehydrated, and oxidized in periodic acid (1% w/v in double-distilled water; Sigma). After washing, slides were stained with a modified Schiff’s reagent in which pararosaniline (basic fuchsin) is substituted by acriflavine. Slides were washed, dehydrated in graded ethanol solutions, air dried, and coverslipped with Canada balsam mounting medium (50% Canada balsam resin, 50% methyl salicylate; Fisher Chemicals). PAS-stained slides were imaged on an Olympus IX-81 fully automated inverted microscope fitted with excitation and emission filter wheels, a Xe-arc lamp, a Hamamatsu ORCAII BT 1024 camera, and an Olympus spinning disc unit to allow for confocal imaging. PAS-stained images were acquired with fluorescence using a single-excitation, double-emission filter combination (490/531,628; Ex/Emgreen,Emred). Data acquisition and image processing were performed using ImagePro Plus 4.1.5 (Media Cybernetics). Using this method, mucin granules appear red and cytoplasmic and nuclear compartments appear green.
Transcriptional profiling
Five micrograms of total RNA extracted from whole mouse lungs were reverse-transcribed in a 20-μL reaction with 200 units of SuperScript II (Invitrogen) and 100 pmol of T7-(dT)24 primer (5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGC GG-(dT)24-3′) in 1× first-strand buffer (Invitrogen) at 42°C for 1 h. The second-strand synthesis was performed at 16°C for 2 h in the presence of Escherichia coli enzymes, DNA polymerase I (40 units), DNA ligase (10 units), RNase H (2 units), and 1× second-strand buffer (Invitrogen). Double-stranded cDNA was blunt-ended using 20 units of T4 DNA polymerase, purified using phenol/chloroform extraction, and transcribed in the absence of biotin-labeled ribonucleotides, resulting in unlabeled cRNA, which was then used as the starting material for the second cycle of PCR. In the second cycle, the first and second cDNA strands were synthesized as described above. The second transcription was performed in the presence of biotin-labeled ribonucleotides, resulting in labeled cRNA. The cRNA was fragmented as described above and added to the hybridization cocktail. The Affymetrix GeneChip system (Affymetrix) was used for hybridization, staining, and imaging of the probe arrays. Hybridization cocktails of 300 μL each containing 15 μg of fragmented cRNA and exogenous hybridization controls were prepared and hybridized to Affymetrix Mouse Genome 430 2.0 GeneChips overnight at 42°C. Hybridized fragments were detected using streptavidin linked to phycoerythrin (Molecular Probes). GeneChips were scanned and imaged using the Affymetrix Gene Chip Operating System (version 1.4.0.036).
Estimated gene expression values were log-transformed. Two-sample t tests were performed as criteria for determining significant differences in mean gene mRNA levels between groups of samples. Fold changes in expression between groups (test versus control, for which we used Ptenf/f;CCSP+/+ mice) were estimated by determining the ratio of the average of the (nonlogged) expression values in the test group with that in the control. Supervised clustering analysis of differentially expressed genes was carried out as follows: (a) each pattern of interest (e.g., genes whose expression was increased or repressed in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice compared with controls) was represented as a series of 1s and -1s; (b) for each gene, the Pearson correlation coefficient was computed for its expression values and each of the predefined patterns; (c) the pattern that best correlated with the expression of each gene was determined; and (d) genes were manually sorted according to the patterns assigned to them. Expression values were visualized as color maps using the Cluster and Java TreeView software programs (26, 27). For color map display, the logs of the expression intensity values were first centered on the centroid means of the values in the four experimental groups.
Quantitative PCR
Quantitative PCR was performed using the SYBR-Green-based system to quantify the expression of selected genes found to be differentially expressed in the gene expression arrays. For this, 2 μg of RNA were converted to cDNA, which was then subjected to PCR using primers designed by using Primer Express (Applied Biosystems). Primer sequences were as follows: Arg1 F 5′-CACTCCCCTGACAACCAGCT-3, 5′-AAGGACACAGGTTGCCCATG-3; IGFtv1 5′-ACTATATCTCCTAGTCCCTGCCTCTAAAG-3′, 5′-CAACATCCATGCATTTTCGG-3′; PCDH21 5′-CTGACGGTGGTTGCCATAGA-3, 5′-GAGGATGTGGTTGGGTTTGC-3′; DMBT1 5-CATGCAAGCCAGCGTGAG-3′, 5′-GCAGAATAGCCCATGGACTGA; Adamdec 5′-CCTGGGACTTCTCGGCTACC-3′; 5′-AAGCAGGACCCAAGACATGC-3′. The cDNA (7 μL per sample) was amplified using SYBR Green PCR Mater Mix (Applied Biosytems) according to manufacturer’s instructions. PCR amplification was performed (95°C for 15 s and 60°C for 15 s for 40 cycles). Afterwards, two additional cycles were performed to generate a dissociation curve, which was used to verify whether the signal generated was from a single _target amplicon or other nonspecific templates, such as primer dimers or contaminating genomic DNA. Serially diluted cDNA (1:1,000) samples were amplified to measure the efficiencies of PCR and to draw the standard curve for each sample, which was used to calculate relative concentrations of _target message. The PCR products and their dissociation curves were detected using the 7500 Fast Real-Time PCR System (Applied Biosystems). The level of L32 in each sample was used as an internal control. The values were expressed relative to the levels of internal control and compared between mouse genotypes by performing t tests.
Results
Conditional mouse model of Pten deficiency in the lung
Pten was genetically inactivated in the bronchial epithelium by interbreeding Ptenflox/+ mice, which have a Pten allele that contains LoxP sites surrounding the PTEN phosphatase domain encoded by exon 5 (28), with CCSPCre/+ mice, which express Cre under the control of the CCSP gene. Expression of this Cre knock-in allele was bronchial specific, based on LacZ expression in CCSPCre;R26RCre reporter mice.9 PCR analysis of PtenΔ5/Δ5;CCSPCre/+ mice showed that recombination of the conditional Pten allele was specific to the lung with no leakage to other tissues (Fig. 1). Clara cells expressed PTEN as shown by the colocalization of CCSP and PTEN in control (PtenΔ5/Δ5;CCSP+/+) mice (Fig. 2). PTEN was not detectable in clara cells after Cre-induced recombination based on the fact that CCSP and PTEN did not colocalize in PtenΔ5/Δ5;CCSPCre/+ mice (Fig. 2).
Figure 1.
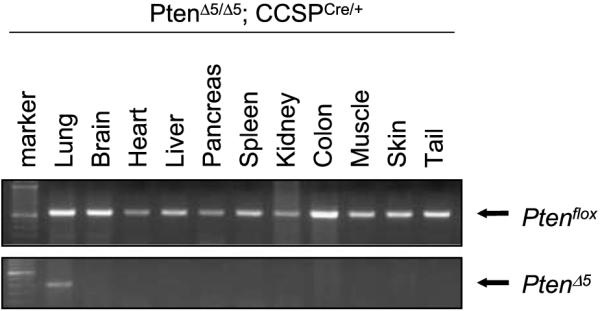
Specific recombination of the conditional Pten allele in lung tissues of Ptenf/+;CCSPCre/+ mice. DNA was extracted from the indicated tissues and subjected to PCR using primers that detect the conditional Pten allele in the germline (Ptenflox) or recombined (PtenΔ5) state. The PCR products were subjected to gel electrophoresis and photographed, revealing bands of the appropriate molecular weights.
Figure 2.

Specific inactivation of Pten in clara cells. Lung tissue samples from mice with the indicated genotypes were stained immunofluorescently with antibodies against CCSP (green) or PTEN (red), which were merged. The signals overlapped in the bronchial epithelia of mice that had not undergone Pten inactivation (Ptenf/f;CCSP+/+) but not in those that had (PtenΔ5/Δ5;CCSPCre/+). Representative bronchi are shown in longitudinal sections and cross-sections. H&E stains illustrate the bronchial histology.
PtenΔ5/Δ5;CCSPCre/+ mice were viable, exhibited no outward phenotypic abnormalities, and remained healthy throughout the course of the study (up to 12 months). At the time of autopsy, their lung weights were similar to those of control mice and their bronchial epithelium was histologically normal (n = 18; data not shown). We concluded that CCSPCre-driven Pten deficiency was not sufficient to initiate lung tumorigenesis.
CCSPCre-driven oncogenic K-ras expression induces lung lesions
To activate oncogenic K-ras expression in clara cells, CCSPCre/+ mice were interbred with LSL-KrasG12D/+ mice, in which the expression of oncogenic K-ras is controlled by a removable transcription termination STOP element (6). DNA from lung tissues obtained from K-rasLox/+;CCSPCre/+ mice showed recombination of the LSL-K-rasG12D allele (Supplementary Fig. S1, lane 4). The median survival duration in the K-rasLox/+;CCSPCre/+ mice was 24 weeks (n = 16). To examine lung histologic changes, K-rasLox/+;CCSPCre/+ mice (n = 17) were sacrificed at ages 4 to 12 weeks, and the lung lesions were characterized using standardized criteria (29). Lesions were first detected microscopically at 12 weeks of age and included multifocal lesions in alveoli and bronchioles, including atypical AAH lesions and adenomas (solid or papillary; Fig. 3A-C). A distinct epithelial lesion, termed atypical papillary bronchiolar proliferation (APBP), that had papillary proliferations of the bronchiolar epithelium located predominantly at the bronchiolar terminal site was observed and composed of columnar epithelial monomorphous or mildly atypical cells and a central fibrovascular core, which was most common in the larger papillary structures (Fig. 3D and E). No carcinomas were observed by 24 weeks of age.
Figure 3.
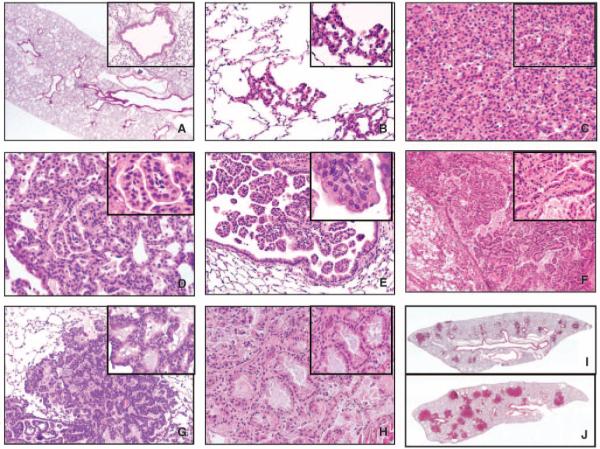
Microphotographs showing representative examples of normal lung histology in a control (PTENf/f;CCSP+/+) mouse (A) and multiple lesions observed in tumor-bearing mice (B-H). The inset pictures show special cell and architectural features of each lesion at higher magnification. B, AAH. C, alveolar adenoma. D, alveolar adenoma with papillary structures. E, APBP. F, extensive proliferation of papillary structures (multiple APBP) filling the lumen of large bronchial airway. Inlet picture depicts a papillae composed of central fibrovascular core lined with cytologic normal cells. The papillary structure depicted in inlet F shows dense fibrous and vascular core compared with papillae in inlet E, which lacks fibrous tissue. G, BAC-like lesion with atypical epithelial cells growing in a lepidic pattern. H, adenocarcinoma with invasion and stromal reaction. I and J, whole lung sections of a KrasLox/+;CCSPCre/+ mouse (I) and a PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mouse (J) that were age-matched (12 wk). Magnifications, 20× and 40× (insets; A-H) and 1.25× (I and J).
Pten inactivation accelerates oncogenic K-ras-induced lung tumorigenesis
Ptenflox/flox;KrasLSL/+ mice were interbred with Ptenflox/flox;CCSPCre/+ mice to generate PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice. DNA from lung tissue samples obtained from these mice exhibited recombination of the conditional K-ras and Pten alleles (Supplementary Fig. S1, lane 6). Although initially healthy, by 2 months of age they began to exhibit weight loss and tachypnea. Their median duration of survival was 8 weeks (n = 13; Fig. 4A). At autopsy, the lungs of PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice were larger and weighed more than those of KrasLox/+;CCSPCre/+ mice (Fig. 4B and C). No extrathoracic metastatic disease was observed.
Figure 4.

Acceleration of lung tumorigenesis in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice. A, Pten inactivation shortened mouse survival. Kaplan-Meier survival curves of mice with the indicated genotypes. B, increase in lung size by Pten inactivation. A photograph of representative lungs obtained from PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice (m) and control (Ptenf/f;CCSP+/+)mice(c). C, increase in lung weights by Pten inactivation. Mean right lung weights of mice killed at the indicated time points (n = 3 mice per group).
To examine lung histologic changes, PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice (n = 18) were sacrificed at ages 4 to 12 weeks. Lesions were detected in these mice as early as 4 weeks of age and were classified as proliferative (AAH, adenomas, or APBP) or frank carcinomas. In two mice, APBP lesions were detected in small and large bronchial structures, which were more proximal than the locations of APBP lesions in KrasLox/+;CCSPCre/+ mice. These mice had extensive papillary proliferation throughout the bronchial and bronchiolar airways, with multiple large papillae and a dense fibrotic core occluding the lumina of the airways (Fig. 3E and F). Another distinct type of epithelial lesion, the bronchioloalveolar cell carcinoma (BAC)-like lesion, was characterized by a proliferation of cells with a cylindrical or cubic appearance, mild nuclear atypia, and cytoplasm filled with mucinous material, which grew along the alveolar septae (Fig. 3G). These tumors expressed cytokeratin (data not shown), supporting their epithelial origin, and were PAS-positive, indicating the presence of mucin, whereas PAS staining was less prominent in tumors from KrasLox/+;CCSPCre/+ mice (Supplementary Fig. S2). Their appearance mimicked the growth pattern of human mucinous BAC, a subtype of lung adenocarcinoma. Another mouse had two lung adenocarcinomas, characterized by two separate nodules that were 7 and 8 mm in diameter, respectively, and consisted of malignant epithelial cells with a high grade of cytologic atypia and increased frequency of mitoses (some of them atypical), with solid, acinar, and papillary patterns of growth and invasive features (Fig. 3H).
Compared with KrasLox/+;CCSPCre/+ mice, PtenΔ5/Δ5;KrasLox/+; CCSPCre/+ mice had more extensive replacement of normal lung by tumor and higher numbers of lesions (Fig. 3I and J; Supplementary Table S1; P < 0.01 for each histologic category). We concluded that the lung lesions in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice developed earlier and in greater numbers and exhibited more severe histologic changes than did those in KrasLox/+;CCSPCre/+ mice.
Bronchial and alveolar tumors have distinct phenotypes
Both of the tumor-bearing genotypes developed tumors in the alveolar and bronchial compartments. In some mice, bronchial tumors were adjacent to alveolar tumors, raising the possibility that adjacent tumors were phenotypically distinct. To investigate this possibility, we examined the expression of CCSP and SPC as markers of bronchial and alveolar epithelial cells, respectively. Indeed, the majority of cells within alveolar lesions were SPCpos CCSPneg, whereas the bronchial tumors were SPCneg CCSPpos (Fig. 5). This pattern of expression was evident in both tumor-bearing genotypes (PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ and KrasLox/+;CCSPCre/+). We conclude that the mice in this study developed phenotypically distinct tumors, exhibiting bronchial or alveolar epithelial markers.
Figure 5.
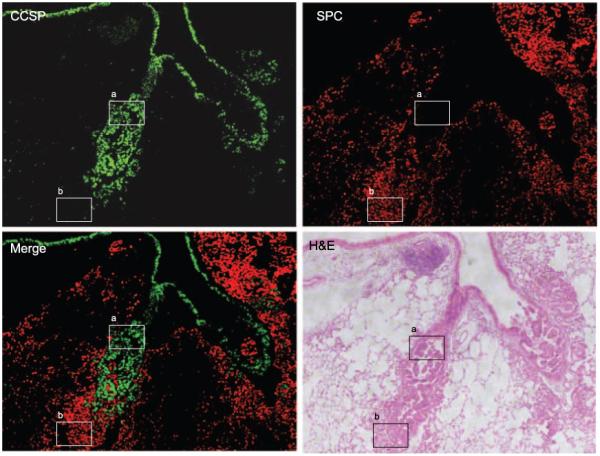
Bronchial and alveolar tumors are phenotypically distinct. Representative images of lung tissues stained immunofluorescently from a KrasLox/+;CCSPCre/+ mouse to detect CCSP (green) and SPC (red), which were merged. Boxed areas indicate a SPCneg CCSPpos bronchial tumor (a) and a SPCpos CCSPneg alveolar tumor (b).
Pten inactivation increases intratumoral vascularity and inflammation
In the K-ras/Pten mutant tumors in our study, PTEN was undetectable whereas Ser235/236-phosphorylated S6 ribosomal protein was highly expressed (Supplementary Fig. S3A-D), which is consistent with unrestrained PI3K activation. One consequence of PI3K activation in cells transformed by oncogenic Ras is increased expression of chemokines that lead to intratumoral inflammation and angiogenesis, which, in turn, promotes tumor cell proliferation (30-36). We postulated that Pten deletion enhances the infiltration of these cell types into tumors and tested this hypothesis by performing immunohistochemical analysis to quantify intratumoral macrophages (F4/80; ref. 36), endothelial cells (factor vIII; ref. 34), and neutrophils (p40; ref. 36; Supplementary Fig. S3E-I).
Correlation of the cell numbers with the tumor genotypes and histologies revealed higher infiltration of neutrophils and endothelial cells in APBP lesions from PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice than in APBP lesions from KrasLox/+;CCSPCre/+ mice (Supplementary Table S2), indicating that Pten inactivation was associated with an influx of neutrophils and endothelial cells in this particular lesion. Comparing lesions at different stages of malignant progression within the K-ras/Pten mutant lungs revealed that neutrophils and endothelial cells were more frequent in BAC-like lesions than they were in APBP lesions (Supplementary Table S2), indicating that the infiltration of these cell types increased with malignant progression. In contrast, the infiltration of macrophages decreased sharply with malignant progression in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice and did not vary with genotype in APBP lesions (Supplementary Table 2), indicating that the effects of Pten inactivation on the tumor microenvironment were cell type specific.
Pten inactivation enriches the lungs in genes encoding growth factors and chemokines
Findings from these models indicated that Pten inactivation accelerated K-ras-initiated malignant progression in the lung and enhanced intratumoral inflammation and vascularity. Based on these findings, we hypothesized that oncogenic K-ras-induced lung tumorigenesis is driven in part by a host response to the presence of transformed alveolar epithelial cells. These cells secrete chemokines that recruit host inflammatory cells, fibroblasts, and endothelial cells, which, in turn, secrete chemokines and growth factors that promote epithelial cell expansion, thereby accelerating tumorigenesis (37). As a first step toward identifying the peptides that might be involved in this process, we examined gene expression in whole lung tissues (containing tumor and surrounding normal lung), recognizing that the critical peptides could be derived from any or all of these cell types.
We transcriptionally profiled RNA from lung tissue samples from the two tumor-bearing genotypes (PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ and KrasLox/+;CCSPCre/+) and the two nontumor-bearing ones (PtenΔ5/Δ5;CCSPCre/+ and Ptenflox/flox;CCSP+/+) sacrificed at 8 weeks of age. Using specific criteria (at least 2-fold increase or decrease relative to that of the control group; P < 0.01), a total of 1,167 genes that were differentially expressed due to Pten inactivation, Kras mutation, or both were identified. Supervised clustering analysis of these 1,167 transcripts was then performed against a set of predefined expression patterns of interest to identify clusters of coexpressed genes in each of the four genotypes (Fig. 6). Four major clusters of genes were identified: genes highly expressed in the K-ras/Pten mutant samples relative to their expression in the other three genotypes (cluster 1), genes expressed at a reduced level in the K-ras/Pten-mutant samples relative to their expression in the other three genotypes (cluster 2), genes highly expressed in both the K-ras/PTEN mutant and K-ras mutant/Pten wild-type samples relative to their expression in the other two genotypes (cluster 3), and genes expressed at a reduced level in both the K-ras/PTEN mutant and K-ras mutant/Pten wild-type samples relative to their expression in the other two genotypes (cluster 4). The genes in these four clusters are listed in Supplementary Table S3. Among the genes in cluster 1, quantitative PCR was performed on five of these, four of which (Arg1, Pcdh21, Dmbt1, and Igf1) were confirmed to be differentially expressed in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice (Supplementary Fig. S4A), validating the accuracy of the array results.
Figure 6.

Mouse genotype-dependent clustering of 1,167 genes. Expression profiling of RNA samples from mice with Pten inactivation (PtenΔ5/Δ5;CCSPCre/+, n = 4), K-ras mutation (KrasLox/+;CCSPCre/+, n = 3), both (PtenΔ5/Δ5;KrasLox/+;CCSPCre/+, n = 3), or neither (Ptenf/f;CCSP+/+, n = 4). Supervised clustering of 1,167 RNA transcripts shows genes that either increased (yellow) or decreased (blue) in the experimental groups compared with controls (Ptenf/f;CCSP+/+, P < 0.01, fold change > 2.0). The genes were evaluated against a set of predefined expression patterns of particular interest (left) to identify clusters of coexpressed genes (right). Each row of the expression matrix represents a gene, and each column represents a profiled sample. Expression values are relative to the centroid mean in the four experimental groups. A partial list of genes in cluster 1 is indicated on the right.
Of potential relevance to the tumor phenotypes in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice were genes in cluster 1 that encode a growth factor (Pdgfc), a growth factor receptor (Ret), a cysteine protease (Cstb), a proligand protease (Adam19), chemokines/cytokines (Ccl9, CXCL2, Il1, and Mif), a chemokine receptor (Ccr1), and other inflammation-associated genes (Arg1, Spp1, and Kng1). CXCL2 is a CXCR2 ligand secreted by tumor cells that recruits inflammatory cells and endothelial cells and has been shown to promote lung tumorigenesis in mice and humans (31, 32, 34). Arg-1 encodes arginine-1, an enzyme involved in L-arginine metabolism, and is considered a relatively specific marker of M2 murine macrophages. M2 macrophages (also called tumor-associated macrophages) have been implicated in tumor promotion through their secretion of protumorigenic growth factors and cytokines (37). Immunohistochemical analysis of arginine-1 revealed localization of this protein to the macrophages infiltrating and surrounding tumors in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice (Supplementary Fig. S4B), indicating that these macrophages have M2 properties. Thus, transcriptional analysis revealed genes highly expressed in the lungs of PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice that promote inflammation and angiogenesis and reflect the presence of tumor-associated macrophages.
Discussion
During the process of lung tumorigenesis, normal lung epithelium progresses to carcinoma in a stepwise fashion, evolving through histologic intermediates and accumulating genetic and epigenetic changes, which is the basis for the premise that lung tumorigenesis is a multistep, stochastic process, a hypothesis that has not yet been rigorously tested. Here, we report that loss of the Pten tumor suppressor gene is a critical secondary event in a mouse model of lung tumorigenesis. Although PTEN expression has, to our knowledge, not been reported on in premalignant lung lesions isolated from patients, PI3K-dependent signaling is frequently activated in bronchial premalignancy as shown by immunohistochemical studies of AKT phosphorylation in bronchial dysplasia (38). Thus, studies from both mouse and human models indicate the relevance of PI3K activation as an early event that drives malignant progression in the lung. These findings support the multistep hypothesis and provide a rationale for investigating the efficacy of strategies _targeting the PI3K-dependent pathway in the prevention and treatment of NSCLC.
In contrast to its potency in combination with oncogenic K-ras, Pten inactivation in clara cells, by itself, had no apparent effect on the lung. However, the lung contains multiple epithelial cell types, and we have not excluded the possibility that epithelial cell types, other than clara cells, are sensitive to transformation by Pten loss nor have we excluded the possibility that lung tumors would occur if Pten inactivation were studied on a genetic background other than C57/BL6 that is more susceptible to lung tumorigenesis. Regardless, these findings differ quite strikingly from the effect of Pten inactivation in other tissue types, including, among others, the prostate, breast, kidney, brain, and ovary (15). Emblematic of its tumor suppressive effect, Pten inactivation in the prostate initiates tumorigenesis in a gene dosage-dependent fashion, requiring loss of both Pten alleles in the prostate epithelium to induce invasive prostate cancer with complete penetrance (39). The absence of tumors in Pten-deficient mice in this study and the long latency of tumors (6 months) in the setting of complete Pten loss in the prostate suggest that additional genetic events are required for carcinoma development. Indeed, PTEN maintains genomic integrity through multiple mechanisms, and Pten-deficient cells are highly prone to centromere breakage, chromosomal translocations, and DNA double-strand breaks (40, 41), which are consistent with the conclusion that Pten inactivation drives tumorigenesis in part through increased susceptibility to genomic damage.
Findings presented here and elsewhere (7) show that CCSP-driven expression of oncogenic K-ras rapidly induces lung lesions, indicating its potency as an initiating event in lung tumorigenesis. Researchers have developed at least five oncogenic K-ras-driven mouse models of lung cancer, including models that express the oncogene inducibly, conditionally, or somatically (4-8, 42). In the conditional models, the LSL-KrasG12D allele is activated by adenoviral Cre delivered intranasally or by interbreeding the mice with transgenic mice that express Cre driven by CCSP or cytomegalovirus promoter fragments (5-7, 42). Unlike the findings in KrasLox/+;CCSPCre mice reported in our study, lung adenocarcinomas develop in the other K-ras-driven models. The reasons for this difference are unclear, but possibilities include differences in the genetic backgrounds of the strains involved, the levels or cell type specificity of Cre expression in the conditional models, and the pathologic criteria used for the diagnosis of adenocarcinoma. Regardless, the fact that lung adenocarcinomas were not detected in KrasLox/+;CCSPCre mice and were slow to develop in the other models provides substantial evidence that additional genetic events are required for adenocarcinoma development.
Previous studies in oncogene-transformed cells have uncovered a biochemical barrier to malignant progression that is of potential relevance to the mouse models in this study. Premalignant lesions induced by oncogenic K-ras or other oncogenes typically undergo a transient proliferation followed by programmed senescence, and only a minority of premalignant lesions persists and progresses to a fully transformed state (43-46). Consistent with these findings, none of the lung lesions in KrasLox/+;CCSPCre mice progressed into lung adenocarcinomas within the timeframe of this study. The programmed senescence that follows Ras-induced transformation is mediated by a negative feedback loop that suppresses PI3K, an effect regulated in part by RasGAPs, RasGEFs, Sprouty proteins, and MKPs (47). Although findings in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice indicate that Pten inactivation promoted the development and malignant progression of lung lesions (AAH, adenomas, and APBP), the noncarcinomatous lesions (AAH, adenomas, and APBP) vastly outnumbered the carcinomas (BAC-like lesions and adenocarcinomas), suggesting that, by itself, PI3K activation was not sufficient to overcome the barriers that block the progression of premalignant lesions and that other factors were required for carcinoma development.
We explored the possibility that the carcinomas arose in this model in part through epithelial cell nonautonomous mechanisms. These are crucial factors in the malignant progression of both murine and human lung cancer models (30, 33-35, 48, 49). We found that the tumor microenvironment in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice was enriched in endothelial cells and inflammatory cells and that the lungs of these mice expressed high levels of chemokines and growth factors. Of particular interest in this regard was the high expression of CXCL2 and MIF, which are CXCR2 ligands (50, 51). The ability of Pten inactivation to increase CXCL2 expression is consistent with the evidence that CXCL8, the human functional homologue of CXCL2, is a transcriptional _target of Ras, which increases CXCL8 expression in part through activation of PI3K (30). The lung lesions in KrasLA1 mice, which develop lung adenocarcinomas because of somatic activation of mutant K-ras (8), express CXCL2, are infiltrated with neutrophils and endothelial cells, and regress in response to CXCR2 neutralization, an effect mediated in part by apoptosis of intratumoral endothelial cells (34). Thus, the high expression of CXCR2 ligands may contribute to the enhanced vascularity, inflammation, and accelerated progression of lung lesions in PtenΔ5/Δ5;KrasLox/+;CCSPCre/+ mice.
In summary, we have created a mouse model that develops lung adenocarcinoma due to the combined effects of oncogenic K-ras expression and Pten inactivation. This mouse model may be of value in screening PI3K inhibitors, as well as other novel therapeutic agents and drug combinations, before initiating human clinical trials and provides a platform on which to study mechanisms of cross-talk between K-ras-dependent and PTEN-dependent signaling pathways to better understand how these pathways promote lung tumorigenesis.
Acknowledgments
Grant support: NIH grants U01 CA105352, P50 CA70907, and R01 CA105155.
Footnotes
K. Iwanaga, Y. Yang, M.G. Raso, and L. Ma contributed equally to this work.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Li et al., manuscript submitted.
References
- 1.Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6:1353–62. [PubMed] [Google Scholar]
- 2.Mori M, Tezuka F, Chiba R, et al. Atypical adenomatous hyperplasia and adenocarcinoma of the human lung: their heterology in form and analogy in immunohistochemical characteristics. Cancer. 1996;77:665–74. [PubMed] [Google Scholar]
- 3.Sato M, Vaughan MB, Girard L, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–28. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 4.Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–62. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerra C, Mijimolle N, Dhawahir A, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–20. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 6.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji H, Houghton AM, Mariani TJ, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25:2105–12. doi: 10.1038/sj.onc.1209237. [DOI] [PubMed] [Google Scholar]
- 8.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 10.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–90. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 11.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000;60:3605–11. [PubMed] [Google Scholar]
- 13.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki A, de la Pompa JL, Stambolic V, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–78. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 15.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 16.Marsit CJ, Zheng S, Aldape K, et al. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005;36:768–76. doi: 10.1016/j.humpath.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Soria JC, Lee HY, Lee JI, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002;8:1178–84. [PubMed] [Google Scholar]
- 18.Xia D, Srinivas H, Ahn YH, et al. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NFκB-dependent pathway. J Biol Chem. 2007;282:3507–19. doi: 10.1074/jbc.M610141200. [DOI] [PubMed] [Google Scholar]
- 19.Forgacs E, Biesterveld EJ, Sekido Y, et al. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557–65. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]
- 20.Velho S, Oliveira C, Ferreira A, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649–54. doi: 10.1016/j.ejca.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 21.Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev. 2004;18:1800–5. doi: 10.1101/gad.1213804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tolkacheva T, Chan AM. Inhibition of H-Ras transformation by the PTEN/MMAC1/TEP1 tumor suppressor gene. Oncogene. 2000;19:680–9. doi: 10.1038/sj.onc.1203331. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda T, Yoshinaga K, Suzuki A, Sakurada A, Ohmori H, Horii A. Anticorresponding mutations of the KRAS and PTEN genes in human endometrial cancer. Oncol Rep. 2000;7:567–70. doi: 10.3892/or.7.3.567. [DOI] [PubMed] [Google Scholar]
- 24.Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60:1800–4. [PubMed] [Google Scholar]
- 25.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 26.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saldanha AJ. Java Treeview-extensible visualization of microarray data. Bioinformatics. 2004;20:3246–8. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 28.Lesche R, Groszer M, Gao J, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–9. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 29.Nikitin AY, Alcaraz A, Anver MR, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–16. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 30.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–58. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 31.Arenberg DA, Keane MP, DiGiovine B, et al. Epithelial-neutrophil activating peptide (ENA-78) is an important angiogenic factor in non-small cell lung cancer. J Clin Invest. 1998;102:465–72. doi: 10.1172/JCI3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arenberg DA, Kunkel SL, Polverini PJ, Glass M, Burdick MD, Strieter RM. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J Clin Invest. 1996;97:2792–802. doi: 10.1172/JCI118734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wislez M, Fleury-Feith J, Rabbe N, et al. Tumorderived granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor prolong the survival of neutrophils infiltrating bronchoalveolar subtype pulmonary adenocarcinoma. Am J Pathol. 2001;159:1423–33. doi: 10.1016/S0002-9440(10)62529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wislez M, Fujimoto N, Izzo JG, et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006;66:4198–207. doi: 10.1158/0008-5472.CAN-05-3842. [DOI] [PubMed] [Google Scholar]
- 35.Wislez M, Rabbe N, Marchal J, et al. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res. 2003;63:1405–12. [PubMed] [Google Scholar]
- 36.Wislez M, Spencer ML, Izzo JG, et al. Inhibition of mammalian _target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-ras. Cancer Res. 2005;65:3226–35. doi: 10.1158/0008-5472.CAN-04-4420. [DOI] [PubMed] [Google Scholar]
- 37.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Investig. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsao AS, McDonnell T, Lam S, et al. Increased phospho-AKT (Ser(473)) expression in bronchial dysplasia: implications for lung cancer prevention studies. Cancer Epidemiol Biomarkers Prev. 2003;12:660–4. [PubMed] [Google Scholar]
- 39.Carver BS, Pandolfi PP. Mouse modeling in oncologic preclinical and translational research. Clin Cancer Res. 2006;12:5305–11. doi: 10.1158/1078-0432.CCR-06-0482. [DOI] [PubMed] [Google Scholar]
- 40.Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 41.Gollin SM. Mechanisms leading to chromosomal instability. Semin Cancer Biol. 2005;15:33–42. doi: 10.1016/j.semcancer.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 42.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 43.Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 44.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 46.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 47.Courtois-Cox S, Genther Williams SM, Reczek EE, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellocq A, Antoine M, Flahault A, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol. 1998;152:83–92. [PMC free article] [PubMed] [Google Scholar]
- 49.Bremnes RM, Camps C, Sirera R. Angiogenesis in non-small cell lung cancer: the prognostic impact of neoangiogenesis and the cytokines VEGF and bFGF in tumours and blood. Lung Cancer. 2006;51:143–58. doi: 10.1016/j.lungcan.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 50.Bernhagen J, Krohn R, Lue H, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 51.Strieter RM, Gomperts BN, Keane MP. The role of CXC chemokines in pulmonary fibrosis. J Clin Invest. 2007;117:549–56. doi: 10.1172/JCI30562. [DOI] [PMC free article] [PubMed] [Google Scholar]