

Abstract
Interleukin-17 (IL-17) has emerged as a central player in the mammalian immune system. Although this cytokine exerts a host-defensive role in many infectious diseases, it promotes inflammatory pathology in autoimmunity and other settings. A myriad of studies have focused on how IL-17-producing cells are generated. However, the means by which IL-17 achieves its effects, either for the benefit or the detriment of the host, are due in large part to the induction of new gene expression. Whereas many IL-17 _target genes are common to different disease states, in some cases the effects of IL-17 differ depending on the _target cell, infectious site or pathogen. Gene products induced by IL-17 include cytokines (IL-6, granulocyte-colony-stimulating factor, tumour necrosis factor-α), chemokines (CXCL1, CXCL2, CCL20, among many others), inflammatory effectors (acute-phase protesins, complement) and antimicrobial proteins (defensins, mucins). Different cell types appear to respond differently to IL-17 in terms of _target gene expression, with notable differences seen in mesenchymal and epithelial cells compared with cells of haematopoietic origin. Here, we summarize the major IL-17 _target genes that mediate this cytokine’s activities in both autoimmune and chronic diseases as well as during various types of infections.
Keywords: cytokine, gene _target, interleukin-17, inflammation, signal transduction
Introduction
The interleukin-17 (IL-17) family is the most recently described subclass of cytokines.1 Since 2000, we have started to gain an understanding of IL-17 family members and their corresponding receptors, which has led to new insights into how immunity to infections and autoimmunity are governed. To date, there are six IL-17-family ligands [IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25) and IL-17F], and five receptors (IL-17RA, IL-17RB/IL-25R, IL-17RC, IL-17RD/SEF and IL-17RE).2 Interleukin-17A (hereafter referred to as IL-17) is the most intensively studied, but interest in the rest of the family is growing.
Originally IL-17 was thought to be produced exclusively by T cells,3 but it is now known to be secreted by a variety of innate cells including macrophages, dendritic cells (DC), natural killer, natural killer T, lymphoid tissue inducer and γδ-T cells.4 A major development in this field occurred with the recognition that IL-17-producing CD4+ T cells arise as a population distinct from the classic T helper type 1 (Th1) and Th2 cells.5–7 Whereas it was known for decades that IL-12 induces Th1 cells [interferon-γ (IFN-γ) producers] and IL-4 induces Th2 cells (IL-4, IL-5 and IL-13 producers), it was only recently demonstrated that Th17 cells differentiate upon exposure to combinations of IL-1, IL-6 and transforming growth factor-β (TGF-β). Interleukin-23, although not critical for the induction of the Th17 lineage, is required in vivo for the stabilization and proliferation of Th17 cells.4,8 In addition to IL-17, Th17 cells and most ‘Th17-like’ innate cells produce IL-17F, tumour necrosis factor-α (TNF-α), IL-22 and IL-21; accordingly, in this review we will discuss the involvement of these related Th17 cytokines as they pertain to co-operative _target gene regulation. We refer the reader to numerous reviews outlining mechanisms underlying Th17 differentiation.8–12
Interleukin-17 and other Th17 cytokines are linked to the pathogenesis of diverse autoimmune and inflammatory diseases (Table 1, Fig. 1). Conversely, IL-17 is essential for host defence against many microbes, particularly extracellular bacteria and fungi.13 The IL-17 receptor is expressed ubiquitously, and hence most cells can potentially respond to this cytokine.14 The _target cell types best analysed are of non-immune origin, particularly epithelial and mesenchymal cells within diseased or inflamed tissues.15 Studies have revealed IL-17-dependent activities in immune cells, particularly B lymphocytes and antigen-presenting cells (APC). Here, we aim to describe how IL-17 exerts its beneficial and its harmful properties via specific _target gene regulation in the context of disease (Fig. 1).
Table 1.
_target cells and genes of IL-17. IL-17 acts on a variety of cells due to its ubiquitous receptor. Shown are representative _target cell types, the role of IL-17 (adverse or beneficial) and key _target genes
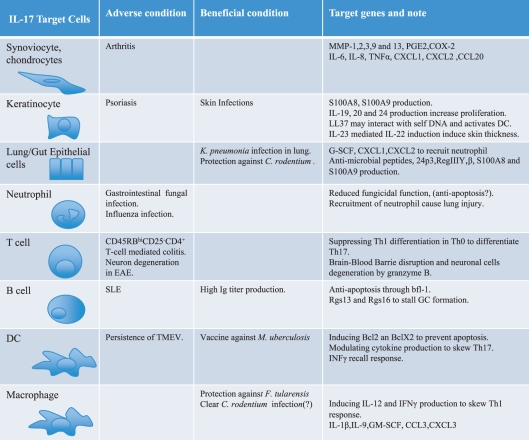 |
Figure 1.

IL-17 signaling and _target genes in various disease settings. In blue are representative mucosal infections where IL-17 plays a key role, along with key _target genes involved in each. In pink are autoimmune diseases and the role of IL-17 and particular _target genes therein.
IL-17-mediated pathogenesis in autoimmune disease
Interleukin-17 mediates adverse effects in many autoimmune diseases. Before the discovery of the Th17 subset as a distinct CD4+ effector population, it was considered that Th1, Th2 and B cells were the main mediators of pathology in autoimmunity. For example rheumatoid arthritis (RA) was widely accepted to be Th1-mediated, supported by the presence of IFN-γ and TNF-α (then thought to be a Th1 cytokine) in synovial lesions and peripheral blood.16 Similarly, inflammatory bowel disease (IBD) was described as a ‘mixed’ Th1 and Th2 pathology, with both IL-4 and IFN-γ implicated.17 However, during the late 1990s, studies pointed to IL-17 as a possible effector in RA and other diseases, despite the prevailing confusion as to whether IL-17 was a Th1 or ‘Th0’ cytokine.18,19 The discovery of the Th17 cell as a bona fide T-cell subset led to a re-kindling of interest in this cytokine in the context of autoimmunity. Indeed, pre-clinical studies supporting a role for IL-17 in disease (outlined herein) led to current clinical trials designed to block IL-17, the IL-17 receptor (IL-17R) or its inducers (i.e. IL-23, IL-6) in autoimmunity.20–22
Rheumatoid arthritis
Many, if not most, autoimmune diseases are now connected in some manner to IL-17 or the Th17 pathway. In particular, RA has been intensively studied, starting even before the recognition of the Th17 subset. The key features of inflamed arthritic joints are proliferating synovial fibroblasts, joint and cartilage erosion, infiltrating CD4+ T cells and autoantibody-producing plasma cells. In addition, increased numbers of innate immune cells (DC, granulocytes and macrophages), in some cases ectopic germinal centres (GC) are found within joints. Early studies showed that high levels of IL-17 were found in the rheumatoid synovium of patients with RA but not of controls or of patients with osteoarthritis. Consistently, adding IL-17 to an in vitro culture system stimulated bone resorption and collagen destruction.23 Furthermore, neutralizing IL-17 or its receptor in collagen-induced arthritis mouse models resolved RA symptoms, IL-17A-deficient mice are protected from collagen-induced arthritis, and adding IL-17 ectopically by gene therapy exacerbated disease.24–26 Consequently, IL-17 appears to promote both inflammation and bone destruction in RA.
These findings pose the question of how IL-17 mediates its pathogenic activities. Interleukin-17 induces pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 from cartilage, synoviocytes, macrophages and bone cells.27 Collectively, these pro-inflammatory cytokines contribute to RA flare-ups and also establish a chronic inflammatory state by a self-reinforcing positive feedback loop wherein IL-17-induced IL-6 maintains the Th17 T-cell population.28 The IL-17 also stimulates the production of multiple chemokines, including IL-8/CXCL8, CXCL1 (KC/Groα), CXCL2 (MIP2α/Groβ), CCL20 (MIP-3α), CCL2 (MCP1) and CCL7 (MCP3).3,5,27,29 These serve to recruit neutrophils, macrophages and lymphocytes to the synovium, thereby enhancing inflammation. Secondary lymphoid GC formations are found ectopically in RA synovial tissue.30,31 Although it has been reported that synovial lymphoid neogenesis is not a major determinant of RA-specific autoantibody responses, this phenomenon is indicative of ongoing inflammation.32
Irreversible deformities in joints are a key feature of advanced RA, caused by extensive cartilage and bone erosion (Table 1). Interleukin-17 contributes to this process by inducing expression of matrix metalloproteinases (MMP) 1, 2, 3, 9 and 13, which drive degradation of extracellular matrix within the joint.33–39 It also induces prostaglandin E2 via cyclooxgenase-2, which enhances inflammation by many mechanisms including vasodilatation.40 Furthermore, IL-17 induces expression of receptor activator of nuclear factor-κB ligand (RANKL) in osteoblasts;23 RANKL is a membrane-bound receptor of the TNF superfamily that promotes differentiation of osteoclasts. Strikingly, Th17 cells also express elevated levels of RANKL, suggesting that they may be particularly adept at promoting bone turnover.41 Accordingly, IL-17 not only enhances inflammation, but stimulates osteoclast differentiation leading to subsequent bone and cartilage damage.42
Although IL-17 alone has the capacity to induce pro-inflammatory factors, its activities are vastly increased when combined with other cytokines, particularly TNF-α (Table 2). This is probably the situation in inflamed joints, where multiple inflammatory cytokines are over-expressed.43 Interleukin-17 synergizes with TNF-α to promote induction of nearly all its _target genes, and in many cases synergy has also been observed with IFN-γ and IL-1β (reviewed in ref. 27). As a consequence, IL-17 alone or together with other inflammatory cytokines in the inflamed joint mediates adverse events in RA.
Table 2.
Synergistic _target genes. IL-17 cooperates with many inflammatory stimuli to activate _target gene expression. Listed are representative gene _targets identified to be induced cooperatively by IL-17 in combination with the indicated stimuli
| Cytokines/stimuli that synergize with IL-17 | Representative _target genes |
|---|---|
| Tumour necrosis factor-α | Cytokines (IL-6, G-CSF, OSM), chemokines (CXCL1, CXCL2, CXCL5, CCL2, CCL7), transcription factors (C/EBPβ, δ,IκBζ), bone remodelling (RANKL, MMP13), antimicrobial peptides (Lcn2, BDs) |
| Interleukin-1β | IL-6, CXCL8, LIF |
| CD40 | IL-6, IL-8, RANTES |
| Oncostatin M | MMP13 |
| Interleukin-22 | Cytokines (IL-19, -20, -24, G-CSF), chemokines (CXCL1), antimicrobial peptides (BDs, S100A7, 8, 9) |
| Vitamin D3 (1,25D3) | LL-37 (cathelicidin) |
| B-cell-activating factor | Twist1 |
| Interferon-γ | IL-6, IL-8 |
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a multi-organ systemic autoimmune disease characterized by autoantibody production. Although traditionally considered mainly a B-cell disease, recent reports indicate that there is likely to be a role for IL-17 in lupus (reviewed in ref.44). Interleukin-17 is elevated in the serum of many SLE patients.45,46 The BXD2 mouse strain develops SLE-like features and spontaneous erosive arthritis with age.45 These mice produce pathogenic autoantibodies because of increased somatic hypermutation and enhanced class-switch recombination, mediated by over-expressed activation-induced cytidine deaminase in GC.47 In studies to elucidate the underlying mechanism of this phenotype, Hsu et al.45 unexpectedly discovered IL-17-dependent signalling in BXD2 B cells that led to enhanced autoantibody production and increased numbers of GCs. CD4 T cells from BXD2 mice were also more prone to differentiate into Th17 cells. The IL-17 functions in this setting by inducing genes encoding Regulator of G-protein signalling 13 and 16 (Rgs13 and Rgs16), which inhibit G-coupled protein receptors such as CXCR4. Hence, IL-17 disrupts trafficking of B cells within lymph nodes, which appears to promote spontaneous generation of autoreactive GC and hence elevated autoantibody production.45
Another animal model with lupus-like features is the Ets-1−/− mouse, characterized by increased high-titre autoantibodies that deposit in kidney.48 In these mice, more naive T cells differentiate into a Th17 phenotype.49 Ets-1 is transcription factor expressed in various immune cells and it skews towards a Th17 phenotype by interfering with the ability of IL-2 to inhibit Th17 differentiation. The Ets1−/− mice have low IL-2 production, but T cells from these mice also appear to be refractory to the inhibitory effects of IL-2 on Th17 development. Interestingly RORγt expression and IL-2-induced signal transducer and activator of transcription 5 (STAT5) were comparable with wild-type and Ets-1−/− T cells, and so the specific pathway by which Ets-1 exerts its effects is down-stream or independent of STAT5.
Interestingly, IL-17 has also been recently shown to synergize with another TNF superfamily member, B-cell activating factor (BAFF), to protect B cells from apoptosis, thereby increasing the number of autoantibody-producing cells.46 Increased BAFF expression is found in ∼ 22–25% of SLE serum samples, and BAFF transgenic mice have a lupus-like phenotype.50 Both IL-17R and BAFFR use a common adaptor molecule Act1; however, Act1 is a positive regulator of IL-17R signalling, whereas it is a negative regulator of BAFFR down-stream pathways.51 Following IL-17 and BAFF stimulation of B cells, Act1 is preferentially recruited to IL-17RA, providing an intriguing mechanism for synergistic signalling between these two systems. Subsequently, IL-17 and BAFF together enhance expression of the transcription factor Twist1, which initiates a cascade of gene expression leading to induced expression of the anti-apoptotic genes Twist-2 and Bfl-1, ultimately promoting B-cell differentiation to autoantibody-producing plasma cells46 (Tables 1 and 2). Hence, the impact of IL-17 on B cells may explain its role in contributing to SLE pathogenesis.
Inflammatory bowel disease
Inflammatory bowel disease, encompassing both Crohn’s disease and ulcerative colitis, is a chronic relapsing inflammatory disorder in the gastrointestinal tract, caused in part by an unregulated immune response to intestinal bacteria. The gut immune system is composed of heterogeneous cell populations, which differ along the gastrointestinal tract (reviewed in ref.52). A notable feature of the gut mucosa is specialized epithelial cells called microfold (M) cells, which sample the gut lumen and transport bacteria and transport antigens to APC on the basolateral surface. In healthy individuals, intestinal DC typically induce T-cell unresponsiveness, which is needed to maintain tolerance to commensal organisms and food antigens.52 Recognition of gut microbes is mediated by Toll-like receptors (TLR) and also intracellular pattern recognition receptors of the nucleotide oligomerization domain (NOD) family. Strikingly, genome-wide association studies (GWAS) identified NOD2 (CARD15) as an IBD-associated gene,53,54 leading to the current paradigm that IBD results from unregulated immune responses against commensal bacteria. In addition to NOD2, additional genes identified in GWAS studies have implicated the Th17 pathway, most notably the IL-23R,55 which is expressed primarily on IL-17-producing cells. Other Th17-associated genes implicated in GWAS studies of IBD include JAK2, IL12B, STAT3, CCR6 and TYK2,56–58 all of which point to a major role for this pathway in IBD pathogenesis.
Animal models support a role for the IL-23/IL-17 axis in IBD. For example, the IL-10−/− mouse develops spontaneous colitis, which is prevented when the mice are crossed to IL-23p19−/− animals (lacking Th17 cells) but not when crossed to IL-12p35−/− animals (lacking Th1 cells).59 Similarly, in chemically induced IBD such as trinitrobenzene sulphonic acid (TNBS) -induced IBD, disease severity is higher in mice deficient in IL-12p40−/− (lacking both IL-23 and IL-12) compared with IL-12p35−/− (lacking just IL-12).60 In the TNBS model, IL-17 _target genes such as IL-6 and CXCL2 were reduced in the colon, and IL-17RA−/− mice were protected from severe disease.61 In contrast, results differ in another chemically induced model, dextran sulphate sodium (DSS) -induced colitis. The DSS disrupts the epithelial cell barrier, causing mucosal microflora to activate mucosal macrophages. In this setting, neutralizing IL-17 worsened symptoms, which was associated with increased expression of TNF-α, IFN-γ, IL-6 and CCL5/RANTES.62 A recent study compared cytokine profiles in acute (day 7) and chronic (day 35–75) TNBS- or DSS-induced IBD. Data suggested that various cytokines and effector cells are working in different time-points.63 For example, IL-6, TNF-α, IL-17 and CXCL1 were elevated in the acute DSS model (7 days), which later shifted to a more Th2/Th17-like profile (IL-4, IL-10 as well as IL-17). In the acute TNBS model, IFN-γ, IL-12, IL-17 and CCL2 were elevated, reminiscent of a mixed Th1/Th17 profile. Although detailed cellular mechanisms are still not well defined, the timing of disease and initiation of disease may dictate which Th cell type is most instrumental in establishing disease.
RAG1−/− mice adoptively transferred with CD45RBhi CD25− CD4+ T cells develop aggressive colitis and wasting disease, which is strongly IL-23-dependent but also IFN-γ-dependent. Interestingly, however, IL-17 is protective in this setting, because IL-17−/− CD45RBhi T cells induce a more aggressive disease. Surprisingly, IL-17RA−/− CD45RBhi T cells were also more aggressive, indicating that these cells respond to IL-17.64 Naive Th0 cells do not express IL-17RA, but start to do so upon differentiation to Th1. Interleukin-17 suppresses IFN-γ secretion by repressing T-bet, the master regulator of Th1 cells, which in turn leads to suppression of IFN-γ, IL-12Rβ1 and osteopontin expression (Table 1). Therefore, in this transfer model, IL-17 protects against IBD by limiting Th1 cell activity.65
Psoriasis
Psoriasis is a chronic inflammatory skin disorder characterized by dermal hyperplasia. The key histological features of psoriatic skin are epidermal keratinocyte hyperproliferation, vascular proliferation and infiltration of DCs, macrophages, neutrophils and T cells.66 The critical roles of IL-23/IL-17 were highlighted in a GWAS study that linked IL-23R polymorphisms to psoriasis, similar to IBD.67 Based on this finding, a model of psoriasis was developed using intradermal injection of IL-23. In this model, anti-IL-17 treatment decreased granulocyte colony-stimulating factor (G-CSF) and MMP-13, although it had no effect on erythema, induration and parakeratosis.68 Several IL-10-family cytokines have been found in psoriatic skin, including IL-19, IL-20, IL-22 and IL-24. However, IL-19−/− and IL-24−/− mice still developed skin thickening following IL-23 injection, although mice lacking the common receptor IL-20R2 were resistant.68 In contrast, Zheng et al.69 observed no elevation of IL-19, IL-20 or IL-24 in the IL-23 injection model, whereas IL-22−/− mice exhibited significant reduction in skin thickness. A role for IL-22 is consistent with findings in human microarray studies, which identify IL-22 as well as many typical Th17 genes.70–72 As a consequence, IL-22 from Th17 cells and an IL-20R2-using cytokine have roles in developing psoriasis-like symptoms caused by ectopic IL-23.
A mechanism of IL-17 activity in psoriasis likely stems from its co-operative gene regulation IL-22 and other stimuli. Together with IL-17, IL-22 synergistically increases expression of skin antimicrobial peptides, including β-defensin-2 (BD-2), S100A7 (psoriasin) and S100A8/9 (calprotectin).73 Supporting this, S100A7–9 are elevated in psoriasis, correlating with disease onset.74 Interestingly, psoriasis patients are more resistant to skin infections than people without psoriasis, perhaps as the result of elevated antimicrobial peptide production. Another antimicrobial peptide, cathelicidin (LL37), is synergistically increased by treatment with IL-17 in combination with 1,25-dihydroxyvitamin D3.75 LL37-bound self-DNA fragments trigger TLR9 in DC, which induces a potent adaptive immune response, possibly one of the mechanisms by which self-tolerance is broken.76
Experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) is a model of multiple sclerosis, a T-cell-mediated autoimmune disease of the central nervous system. It is elicited by immunization of neuroantigens, such as myelin basic protein and proteolipid protein. As with many other autoimmune conditions, Th1 cells were long thought to be responsible for EAE pathology, despite the fact that IFN−/−, IFNγR−/− and IL-12p35−/− mice were susceptible.77–79 Landmark studies comparing the IL-12p35−/− and IL-23p19−/− mice showed clearly that the Th17 pathway was responsible for pathology.80 Further evidence for the role of Th17 cells in driving EAE was shown in STAT6−/−/T-bet−/− doubly deficient mice, lacking Th1 and Th2 cells.81 Furthermore, EAE in these mice could be ameliorated by treatment with anti-IL-17 antibodies. An elegant study noted that regulation of IL-17 pathogenic activity can be controlled by IL-10. Specifically, Th17 cells derived ex vivo following treatment with TGF-β and IL-6 do not cause EAE upon adoptive transfer, whereas Th17 cells derived ex vivo with TGF-β, IL-6 and IL-23 develop EAE.82 Interleukin-23 suppresses IL-10, correlating with elevated expression of IL-17 _target genes such as CXCL10 (IP10), CCL2, CXCL2 and CCL20.82 Both IL-17 and IL-22 also disrupt the tight junctions that form the blood–brain barrier, enabling Th17 cells to migrate into the central nervous system and cause neuronal damage.83
In summary, increasing numbers of autoimmune conditions implicate the Th17 pathway. Although the specific genes and mechanisms exhibit some variation depending on the location, chronicity and type of disease, there are many common threads in terms of IL-17-mediated gene expression. These findings have made blocking the IL-17 pathway an attractive _target for anti-cytokine therapy.22 On the flip side, the same pathways that promote disease in autoimmunity are beneficial in many infection settings, which will be considered in the following sections.
Infection
In contrast to its adverse effects in autoimmunity, IL-17 plays a vital role in protecting the host from infection.13 This is particularly evident at mucosal sites such as lung, gut and the oral cavity. Interleukin-17-producing cells are enriched at mucosal surfaces, and Th17 cells express the CCR6 receptor that _targets them to mucosal areas.84,85 Pro-inflammatory cytokines such as, IL-6, IL-1β and TNF-α, which mediate defensive responses are induced by IL-17. In particular, IL-6, acts in a positive feedback loop to further amplify Th17 differentiation and activate acute-phase responses and complement.28 Interleukin-17 modulates neutrophils via cytokines that promote polymorphonuclear cell expansion and survival (G-CSF, granulocyte–macrophage CSF)86 as well as neutrophil chemoattractants (CXCL1, CXCL2 and CXCL5). Additionally, CXCL9, CXCL10 and CCL20 are _target genes of IL-17, which have chemotactic activity for lymphocytes, DC and other immune cells, _targeting them to mucosal surfaces; IL-17 induces CCL2 and CCL7, monocyte-recruiting chemokines. Interestingly, IL-17 suppresses CCL5/RANTES, although the significance of this with respect to infection has not been determined.29,87 Anti-microbial peptides, which contribute to host defence by direct killing of invading organisms, are strongly up-regulated by IL-17. Notably, some of the chemokines regulated by IL-17 also exhibit antimicrobial activity (e.g. CCL20, which binds the CCR6 receptor found on DC and Th17 cells).88,89 In the following sections, we will discuss IL-17-induced genes in representative infectious models at various mucosal sites. Not surprisingly, most of the genes identified in the context of infection are the same as those identified from autoimmune studies or cell lines, but some appear to be somewhat specific to a particular pathogen or infected tissue.
Extracellular bacterial infections
Klebsiella pneumoniae (lung mucosa)
The first report to describe the IL-17RA−/− mouse assessed pulmonary infection by an extracellular pathogen, Klebsiella pneumoniae.90Klebsiella pneumoniae is a Gram-negative bacterium that causes intra-abdominal and urinary tract infections, as well as hospital and community-acquired pneumonia. Infection with K. pneumoniae in IL-17RA−/− mice led to reduced survival and elevated bacterial burden. Neutrophil levels in lung were severely impaired, which was linked to reduction of CXC chemokines and G-CSF in bronchoalveolar lavage fluid. The same group demonstrated a requirement for IL-23 in defence against K. pneumoniae.91 Mice that were IL-23−/− showed a significant reduction in CCL3, CXCL2, CXCL1, CXCL5 and IL-6. When it was recognized that IL-22 is also a Th17-derived cytokine, IL-22 was found to be an even more critical player than IL-17 in the setting of K. pneumoniae.92 Although blocking both IL-17 and IL-22 significantly lowered G-CSF and CXCL1 expression in bronchoalveolar lavage fluid, the addition of IL-22 alone did not serve to increase G-CSF and CXCL1 expression. Rather, IL-22 affected mainly production of IL-6 and CCL3. In addition to stimulating chemokine expression, IL-17 and IL-22 induce lipocalin 2 (Lcn2, 24p3) in tracheal epithelial cells. The Lcn2 blocks catecholate-type siderophores of Gram-negative bacteria, preventing them from scavenging free iron,93,94 and is potently regulated by IL-17.95 Surprisingly, however, IL-17 and IL-22 are apparently not necessary for Lcn2 induction in K. pneumoniae infections, whereas TLR4 and Myd88 are essential.96
Citrobacter rodentium (gut mucosa)
Interleukin-17-expressing cells are particularly abundant in the gut, including classic αβ-T cells as well as more innate cells such as γδ-T and natural killer T cells. Citrobacter rodentium is a naturally occurring murine enteric pathogen, and is an extracellular Gram-negative organism considered a model for enteropathic attaching and effacing Escherichia coli infections. In model studies of C. rodentium infection, IL-17 did not play a significant protective role compared with IL-22 at early stages of disease.97 Infection with C. rodentium induces a variety of antimicrobial peptides, such as S100A8, S100A9, RegIIIβ and RegIIIγ. RegIIIβ and RegIIIγ are C-type lectins originally thought to selectively kill Gram-positive bacteria. Not surprisingly, adding back RegIIIγ protected mice significantly. In contrast, direct comparison of IL-17A−/− versus IL-17F−/− mice suggested that IL-17 and IL-17F do contribute to protection from C. rodentium in later stages.98 Here, CXCL1, CXCL2, IFN-γ, IL-1β, IL-6, TNF-α and inducible nitric oxide synthase were regulated normally in the colons of IL-17A−/−, IL-17F−/− and IL-17A/F−/− mice following C. rodentium infection. Similarly, IL-17A−/− and IL-17F−/− mice showed increased expression of BD2, Lcn2, S100A8, S100A9, RegIII and RegIIIγ compared with wild-type mice, yet these mice still had a higher bacterial burden. However, BD1, BD3 and BD4 were impaired in the knockout strains, implying that in this setting they may be the major IL-17 gene _targets.
Porphyromonas gingivalis (oral mucosa)
The oral cavity is another important but often overlooked site of mucosal infection. Studies of a major human periodontal pathogen revealed a strongly protective role for IL-17 receptor signalling, at least in acute oral infections. Porphyromonas gingivalis is a Gram-negative anaerobic microbe that is one of the three major pathogens associated with periodontal disease, characterized by gingival tissue destruction, chronic infection and bone loss in the alveolar bone crest of the jaw. Despite the potential of IL-17 to promote bone destruction as it does in the context of RA, IL-17RA−/− mice are more susceptible to infection than wild-type mice. The major IL-17 gene _targets shown to be impaired were CXCL1, CXCL2 and CXCL5,99,100 which correlated with reduced neutrophil recruitment to the gingival area. Consistently, CXCR2−/− mice (the receptor for CXCL1, CXCL2 and CXCL5) are exquisitely susceptible to P. gingivalis-induced periodontal disease. Interestingly, these studies also revealed a gender bias for IL-17R activity. Regardless of genetic background, female IL-17RA−/− mice were more susceptible than males to P. gingivalis-induced bone loss. Although the female mice showed a more dramatic reduction in CXC chemokine expression, the male mice were mainly impaired in G-CSF. Consequently, although both genders had reduced neutrophil activity, this appeared to be directed through different mechanisms.99
Intracellular bacterial infections
It is commonly considered that IL-17 is primarily important for protection against extracellular pathogens rather than intracellular bacteria. Indeed, in many cases, but not all, IL-17 is dispensable for host defence against intracellular bacteria.13 For example, Listeria monocytogenes induces IL-17 in liver γδ-T cells, which is associated with enhanced neutrophil recruitment;101 however, IL-17RA−/− mice have no defect in survival, arguing against a role for IL-17 in this disease.101Mycobacterium tuberculosis infection studies also revealed critical roles of IFN-γ rather than IL-17 in primary infections, although IL-17 is induced during infection.102,103 Nonetheless, the picture is more complex than this because IL-17 is essential for an effective vaccine-induced response to M. tuberculosis.104 Although the detailed mechanisms need to be defined more carefully, IL-17 may contribute more to the development of memory responses than to initial infections for some intracellular organisms.104 In contrast, infection with Francisella tularensis, another intracellular organism, shows a requirement for IL-17.105 Mechanistically, this is regulated via IL-17-mediated induction of IFN-γ and IL-12 in macrophages, linking Th17 and Th1 responses in vivo. Similarly, responses to Chlamydia infection also involve IL-17.106 Therefore, the concept that IL-17 activity is exclusively linked to protection from extracellular pathogens is overly simplistic.
Fungal infections
In the setting of fungal infections, IL-17 plays both protective and destructive roles depending on route of infection and perhaps the morphological form of the organism. The primary organism where this has been examined is Candida albicans, a common commensal that colonizes human mucosal surfaces. Candida albicans causes both systemic and mucosal infections, but the immune responses at these sites are quite different.107 The most severe form of candidiasis is a disseminated disease associated with hospital settings, which is modelled by intravenous injection of C. albicans into mice. The IL-17RA−/− mice are highly susceptible to this form of disease. Although detailed analysis of relevant _target genes was not performed, neutrophil recruitment was severely defective.108 Consistent with this, two reports using mouse models of oropharyngeal candidiasis (‘thrush’) showed that IL-17 and IL-23 play a strongly protective role against mucosal C. albicans.109,110 Microarray analysis showed that immunocompetent wild-type mice infected with C. albicans up-regulate numerous classic IL-17 _target genes in the oral mucosa, including BD3, S100A8/9, MMP-8, G-CSF, CCL20, IL-6, CXCL1 and CXCL5.109 BD3 and S100A8/9, in particular, were strongly impaired in IL-17RA−/− tongue tissue. In contrast, few IFN-γ signature genes were induced in this setting, arguing against a role for Th1 cells.109 Studies of the pattern recognition receptors involved in early activation events indicate that C-type lectin receptors such as Dectin 1, Dectin 2 and the Mannose receptor as well as the NLRP3 inflammasome induce IL-23 from macrophages and DC, hence promoting Th17 cell differentiation (reviewed in ref.111). These findings are supported by studies in humans with mutations in STAT3 (who are selectively Th17-deficient), who are exquisitely susceptible to mucosal candidiasis and staphylococcal infections.112
In contrast, in a gastrointestinal model of C. albicans and Aspergillus fumigatus mucosal infection induced by injection of fungi into the gut, Romani and colleagues113 reported that IFN-γ plays a primary role in protection, whereas IL-23 and IL-17 exacerbate inflammation. However, when IFN-γ is absent, IL-23 plays a protective role via mechanisms involving cross-regulation of IL-12 and IL-23.113 A caveat of this model is that it does not have a true parallel human disease state, but may nonetheless reflect the complexity of immune responses that differ by anatomic location.
Viral infections
Viral host defence depends heavily on Type I IFNs that modulate viral replication, and so IL-17 is considered to be relatively less important. However, there is emerging evidence that IL-17 may participate in viral immune responses, which can be beneficial or detrimental to the host. It is also intriguing that a homologue of IL-17 is encoded in a Herpesvirus saimiri, a T-cell tropic γ herpesvirus.114 The significance of this is unknown, although viral IL-17 promotes positive signalling through IL-17RA.114 Nonetheless, this homologue presumably benefits the virus in some aspect of pathogenesis, by some as yet unknown mechanism.
In poxvirus infections, genetically engineered recombinant vaccinia viruses (VV) encoding IL-12, IL-23 or IL-17 were created to test the roles of these cytokines in viral host defence. Surprisingly, VV-IL-23 and VV-IL-17, but not VV-IL-12, caused reduced virulence in mice.115 Although the mechanism was not well defined, protection was not mediated by enhanced cytotoxic lymphocyte activity. Surprisingly, IL-23-induced viral resistance was also not primarily the result of IL-17, as IL-17−/− mice infected with VV-IL-23 were not significantly compromised.115 Interleukin-22 and other Th17-hallmark cytokines were not evaluated in this study. In a contrasting report, an IL-17-expressing VV was found to be more virulent than its parental virus in mice, associated with altered immunoglobulin G isotype generation.116 The distinctions between these models is unclear, but may reflect the fine line between host defence and immunopathology mediated by IL-17.
Interleukin-17 signalling may be counterproductive in certain viral settings, by contributing to the ‘cytokine storm’ that characterizes disease pathology. In an influenza infection model, IL-17 and IL-17F were induced as soon as 2 days post-infection. The survival rate of IL-17RA−/− mice was higher than WT, associated with reduction in neutroophil chemokines and inflammatory cytokines (G-CSF, CXCL1, IL-6, TNF-α, IL-1β and IFN-γ). Lung injury in this setting may be partly the result of IL-17-mediated oxidization of phospholipids by recruited neutrophils.117
Another report identified a function of IL-17 in maintaining virus persistence in a model using Theiler’s murine encephalomyelitis virus (TMEV),118 which leads to a demyelinating disease. In a susceptible mouse strain, Th17 development was elevated during TMEV infection, and Th17 cells infiltrated into the central nervous system. Viral persistence in brain and spinal cord and production of IL-6, CXCL1 and MCP-1 correlated with the appearance of Th17 cells.118 Importantly, the mechanism by which the virus persists was because of the resistance to apoptosis of infected astrocytes. Interestingly, IL-17F did not mediate an anti-apoptotic effect. Collectively, these findings indicate that infection by viruses often induces Th17 differentiation and so its signature _target genes, yet this is often ineffective in terms of host defence, and may even promote immunopathology.
IL-17 signalling: synergy and mechanisms
Mechanisms of IL-17 signalling are poorly described compared with other cytokine receptor subfamilies. Although reviewed in detail elsewhere,2,27,51 IL-17 binds to both the IL-17RA and IL-17RC subunits to mediate signalling.119 Both receptors encode a conserved signalling motif known as the SEFIR (SEF/IL17R) domain,120 which engages the Act1 adaptor/ubiquitin ligase enzyme through its own SEFIR motif.121–123 Act1 in turn recruits TRAF6, which leads to activation of the nuclear factor-κB (NF-κB) pathway. Act1 is also upstream of the CCAAT/Enhancer Binding Protein (C/EBP)-β and C/EBP-δ and mitogen-activated protein kinase pathways, all of which act in concert to control _target gene expression. Most IL-17 downstream genes have NF-κB and C/EBP binding sites, and in many cases both are necessary for IL-17-mediated promoter activity.95,124
As mentioned previously, a notable feature of IL-17 is its strong co-operative effect with other cytokines in regulating down-stream gene/protein expression. Interleukin-17 has been shown to synergize with IL-1β, IL-22, IFN-γ, TNF-α, Oncostatin M, CD40, BAFF and Vitamin D3 (1,25-dihydroxyvitamin D3), and this list may grow29,73,75,125,126 (Table 2). This synergy is reflected in the fact that IL-17 alone is not a potent inducer of inflammatory pathways such as NF-κB, despite the potent in vivo effects of an IL-17 deficiency (reviewed in ref. 2). The molecular mechanisms underlying synergistic signalling are not fully elucidated, although various pathways are implicated. Many IL-17 _target genes are controlled post-transcriptionally by messenger RNA stabilization. In particular, CXCL1, CXCL2, IL-6, I-κBζ and CXCL5 messenger RNAs are induced by IL-17 and/or TNF-α somewhat weakly and are subject to rapid degradation, but in the presence of both IL-17 and TNF-α their stability is significantly enhanced.127 Mechanistically this is mediated by the mitogen-activated protein kinase pathway and Act1, but surprisingly not by TRAF6. At the promoter level, co-operative induction of C/EBP proteins but not NF-κB by the combination of IL-17 and TNF-α contributes to co-operative induction of the IL-6 promoter.124 The synergy between IL-17 and BAFF, however, is mediated by the NF-κB pathway, where IL-17RA out-competes the BAFFR for Act1, leading to positive rather than suppressive signalling.46 Interleukin-17 has also been shown to regulate the TNF receptor, which may account for enhanced TNF-α signalling capacity in the presence of IL-17.128 Similarly, the synergy between IL-17 and IL-19, IL-20 and IL-24 was dependent on the expression of IL-22 receptor.125 Finally, the mechanisms by which IL-17 and IL-22 synergize to regulate antimicrobial peptides such as S100A8/9 and IL-20 family members still need to be determined. In summary, although IL-17 is generally a weak inducer of _target genes, this cytokine has a major impact in vivo. Therefore, the synergistic effects of IL-17 almost certainly play a significant role in dictating its physiological activities.
Conclusion and perspectives
As suggested by ubiquitous IL-17R expression,14 IL-17 plays a role in multiple cell types and conditions. Initial studies revealed the IL-17 _target genes from mesenchymal and epithelial cells, but recent work has shown important IL-17 _target genes in lymphocytes and other immune cells as well. Defining the complete range of IL-17 effects in vivo will probably never be fully achieved, but it is clear that this cytokine is a central player in numerous disease states.
Acknowledgments
S.L.G. is supported by the National Institutes of Health (AR054389), the Alliance for Lupus Research and Amgen.
Glossary
Abbreviations:
- APC
antigen-presenting cell
- BAFF
B-cell activating factor
- BD
β-defensin
- C/EBP
CCAAT/enhancer binding protein
- DC
dendritic cell
- DSS
dextran sulphate sodium
- EAE
experimental autoimmune encephalomyelitis
- GC
germinal centre
- G-CSF
granulocyte colony-stimulating factor
- GWAS
genome-wide association studies
- IBD
inflammatory bowel disease
- IFN
interferon
- IL
interleukin
- Lcn2
lipocalin 2/24p3
- MMP
matrix metalloproteinase
- NF-κB
nuclear factor-κB
- NOD
nucleotide oligomerization domain
- RA
rheumatoid arthritis
- RANKL
receptor activator of nuclear factor-κB ligand
- SEFIR
SEF/IL17R
- SLE
systemic lupus erythematosus
- STAT5
signal transducer and activator of transcription 5
- TGF
transforming growth factor
- Th
T helper
- TLR
Toll-like receptor
- TMEV
Theiler’s murine encephalomyelitis virus
- TNBS
trinitrobenzene sulphonic acid
- TNF
tumour necrosis factor
- VV
vaccinia virus
Disclosures
SLG has received a research grant and honoraria from Amgen.
References
- 1.Aggarwal S, Gurney AL. IL-17: a prototype member of an emerging family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- 2.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–67. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 5.Park H, Li Z, Yang X, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 7.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–45. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 8.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–6. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 10.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–53. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Bluestone JA, Mackay CR, O’shea JJ, Stockinger B. The functional plasticity of T cell subsets. Nat Rev Immunol. 2009;9:811–6. doi: 10.1038/nri2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–48. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 13.O’Quinn D, Palmer M, Lee Y, Weaver C. Emergence of the Th17 pathway and its role in host defense. Adv Immunol. 2008;99:115–63. doi: 10.1016/S0065-2776(08)00605-6. [DOI] [PubMed] [Google Scholar]
- 14.Yao Z, Spriggs M, Derry J, et al. Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- 15.Yu J, Gaffen SL. Interleukin-17: a novel inflammatory cytokine that bridges innate and adaptive immunity. Front Biosci. 2008;13:170–7. doi: 10.2741/2667. [DOI] [PubMed] [Google Scholar]
- 16.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 17.Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58:1152–67. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 18.Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonistic effects with IFN-γ and TNF-α. J Immunol. 1999;162:494–502. [PubMed] [Google Scholar]
- 19.Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–51. [PubMed] [Google Scholar]
- 20.Gaffen SL. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep. 2009;11:365–70. doi: 10.1007/s11926-009-0052-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–45. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lubberts E. IL-17/Th17 _targeting: on the road to prevent chronic destructive arthritis? Cytokine. 2008;41:84–91. doi: 10.1016/j.cyto.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–9. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 25.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–7. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 26.Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res. 2002;51:102–4. doi: 10.1007/BF02684010. [DOI] [PubMed] [Google Scholar]
- 27.Shen F, Gaffen S. Structure–function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine. 2008;41:92–104. doi: 10.1016/j.cyto.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogura H, Murakami M, Okuyama Y, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–36. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Shen F, Ruddy M, Plamondon P, Gaffen S. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol. 2005;77:388–99. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 30.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–80. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 31.Timmer TCG, Baltus B, Vondenhoff M, Huizinga TWJ, Tak PP, Verweij CL, Mebius RE, van der Pouw Kraan TCTM. Inflammation and ectopic lymphoid structures in rheumatoid arthritis synovial tissues dissected by genomics technology: identification of the interleukin-7 signaling pathway in tissues with lymphoid neogenesis. Arthritis Rheum. 2007;56:2492–502. doi: 10.1002/art.22748. [DOI] [PubMed] [Google Scholar]
- 32.Weyand CM, Goronzy JJ. Ectopic germinal center formation in rheumatoid synovitis. Ann N Y Acad Sci. 2003;987:140–9. doi: 10.1111/j.1749-6632.2003.tb06042.x. [DOI] [PubMed] [Google Scholar]
- 33.Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. 2000;12:1092–9. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- 34.Koenders MI, Kolls JK, Oppers-Walgreen B, et al. Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis. Arthritis Rheum. 2005;52:3239–47. doi: 10.1002/art.21342. [DOI] [PubMed] [Google Scholar]
- 35.Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167:141–9. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sylvester J, Liacini A, Li WQ, Zafarullah M. Interleukin-17 signal transduction pathways implicated in inducing matrix metalloproteinase-3, -13 and aggrecanase-1 genes in articular chondrocytes. Cell Signal. 2004;16:469–76. doi: 10.1016/j.cellsig.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Rifas L, Arackal S. T cells regulate the expression of matrix metalloproteinase in human osteoblasts via a dual mitogen-activated protein kinase mechanism. Arthritis Rheum. 2003;48:993–1001. doi: 10.1002/art.10872. [DOI] [PubMed] [Google Scholar]
- 38.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Reboul P, He Y, Jolicoeur FC, Pelletier JP. Modulation of TIMP-1 synthesis by antiinflammatory cytokines and prostaglandin E2 in interleukin 17 stimulated human monocytes/macrophages. J Rheumatol. 2001;28:712–8. [PubMed] [Google Scholar]
- 39.Jovanovic DV, Martel-Pelletier J, Di Battista JA, Mineau F, Jolicoeur F-C, Benderdour M, Pelletier J-P. Stimulation of 92-kd gelatinase (matrix metalloproteinase 9) production by interleukin-17 in human monocyte/macrophages. Arthritis Rheum. 2000;43:1134–44. doi: 10.1002/1529-0131(200005)43:5<1134::AID-ANR24>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 40.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. 1998;273:27467–73. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 41.Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moseley T, Haudenschild D, Rose L, Reddi A. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–74. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 43.Miossec P. Interleukin-17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum. 2003;48:594–601. doi: 10.1002/art.10816. [DOI] [PubMed] [Google Scholar]
- 44.Garrett-Sinha LA, John S, Gaffen SL. IL-17 and the Th17 lineage in systemic lupus erythematosus. Curr Opin Rheumatol. 2008;20:519–25. doi: 10.1097/BOR.0b013e328304b6b5. [DOI] [PubMed] [Google Scholar]
- 45.Hsu H, Yang P, Wang J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–75. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 46.Doreau A, Belot A, Bastid J, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–85. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 47.Hsu H, Wu Y, Yang P, et al. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J Immunol. 2007;178:5357–65. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- 48.Wang D. Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol. 2005;17:1179–91. doi: 10.1093/intimm/dxh295. [DOI] [PubMed] [Google Scholar]
- 49.Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho I-C. Ets-1 is a negative regulator of Th17 differentiation. J Exp Med. 2007;204:2825–35. doi: 10.1084/jem.20070994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Groom J, Mackay F. B cells flying solo. Immunol Cell Biol. 2008;86:40–6. doi: 10.1038/sj.icb.7100142. [DOI] [PubMed] [Google Scholar]
- 51.Li X. Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine. 2008;41:105–13. doi: 10.1016/j.cyto.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 52.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–20. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 53.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 54.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 55.Duerr R, Taylor K, Brant S, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007;59:1073–83. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 57.Uhlig H, Powrie F. Mouse models of intestinal inflammation as tools to understand the pathogenesis of inflammatory bowel disease. Eur J Immunol. 2009;39:2021–6. doi: 10.1002/eji.200939602. [DOI] [PubMed] [Google Scholar]
- 58.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Camoglio L, Juffermans NP, Peppelenbosch M, te Velde AA, ten Kate FJ, van Deventer SJ, Kopf M. Contrasting roles of IL-12p40 and IL-12p35 in the development of hapten-induced colitis. Eur J Immunol. 2002;32:261–9. doi: 10.1002/1521-4141(200201)32:1<261::AID-IMMU261>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382–8. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 62.Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 63.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen T-E, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–52. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Connor W, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–9. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Awasthi A, Kuchroo VK. IL-17A directly inhibits TH1 cells and thereby suppresses development of intestinal inflammation. Nat Immunol. 2009;10:568–70. doi: 10.1038/ni0609-568. [DOI] [PubMed] [Google Scholar]
- 66.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 67.Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan J, Blumenschein W, Murphy E, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–87. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng Y, Danilenko D, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 70.Boniface K, Bernard F, Garcia M, Gurney A, Lecron J, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- 71.Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407–15. doi: 10.1111/j.1365-2249.2007.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. 2008;181:7420–7. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liang S, Tan X, Luxenberg D, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser L. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Broome A-M, Ryan D, Eckert RL. S100 protein subcellular localization during epidermal differentiation and psoriasis. J Histochem Cytochem. 2003;51:675–85. doi: 10.1177/002215540305100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peric M, Koglin S, Kim S-M, et al. IL-17A enhances vitamin D3-induced expression of cathelicidin antimicrobial peptide in human keratinocytes. J Immunol. 2008;181:8504–12. doi: 10.4049/jimmunol.181.12.8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–9. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 77.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 78.Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–60. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 79.Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002;110:493–7. doi: 10.1172/JCI15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langrish C, Chen Y, Blumenschein W, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Das J, Ren G, Zhang L, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206:2407–16. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 83.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 85.Yamazaki T, Yang XO, Chung Y, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol. 2008;181:8391–401. doi: 10.4049/jimmunol.181.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parsonage G, Filer A, Bik M, et al. Prolonged, granulocyte–macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha. Arthritis Res Ther. 2008;10:R47. doi: 10.1186/ar2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andoh A, Fujino S, Bamba S, Araki Y, Okuno T, Bamba T, Fujiyama Y. IL-17 selectively down-regulates TNF-alpha-induced RANTES gene expression in human colonic subepithelial myofibroblasts. J Immunol. 2002;169:1683–7. doi: 10.4049/jimmunol.169.4.1683. [DOI] [PubMed] [Google Scholar]
- 88.Yang D, Chen Q, Hoover D, Staley P, Tucker K, Lubkowski J, Oppenheim J. Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol. 2003;74:448–55. doi: 10.1189/jlb.0103024. [DOI] [PubMed] [Google Scholar]
- 89.Kolls J, McCray P, Chan Y. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 2008;8:829–35. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Happel K, Dubin P, Zheng M, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aujla S, Chan Y, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berger T, Togawa A, Duncan G, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:1834–9. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flo T, Smith K, Sato S, Rodriguez D, Holmes M, Strong R, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–21. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 95.Shen F, Hu Z, Goswami J, Gaffen S. Identification of common transcriptional regulatory elements in interleukin-17 _target genes. J Biol Chem. 2006;281:24138–48. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 96.Chan YR, Liu JS, Pociask DA, et al. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol. 2009;182:4947–56. doi: 10.4049/jimmunol.0803282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zheng Y, Valdez P, Danilenko D, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 98.Ishigame H, Kakuta S, Nagai T, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 99.Yu JJ, Ruddy MJ, Conti HR, Boonanantanasarn K, Gaffen SL. The interleukin-17 receptor plays a gender-dependent role in host protection against Porphyromonas gingivalis-induced periodontal bone loss. Infect Immun. 2008;76:4206–13. doi: 10.1128/IAI.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu J, Ruddy M, Wong G, Sfintescu C, Baker P, Smith J, Evans R, Gaffen S. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood. 2007;109:3794–802. doi: 10.1182/blood-2005-09-010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hamada S, Umemura M, Shiono T, et al. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against Listeria monocytogenes infection in the liver. J Immunol. 2008;181:3456–63. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 103.Khader SA, Pearl JE, Sakamoto K, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–95. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 104.Khader S, Bell G, Pearl J, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 105.Lin Y, Ritchea S, Logar A, et al. IL-17 is required for Th1 immunity and host resistance to the intracellular pathogen Francisella tularensis LVS. Immunity. 2009;5:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang X, Gao L, Lei L, et al. A MyD88-dependent early IL-17 production protects mice against airway infection with the obligate intracellular pathogen Chlamydia muridarum. J Immunol. 2009;183:1291–300. doi: 10.4049/jimmunol.0803075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dongari-Bagtoglou A, Fidel P. The host cytokine responses and protective immunity in oropharyngeal candidiasis. J Dent Res. 2005;84:966–77. doi: 10.1177/154405910508401101. [DOI] [PubMed] [Google Scholar]
- 108.Huang W, Na L, Fidel P, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–31. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 109.Conti H, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. 2009;5:487–97. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol. 2008;6:67–78. doi: 10.1038/nrmicro1815. [DOI] [PubMed] [Google Scholar]
- 112.Tangye SG, Cook MC, Fulcher DA. Insights into the role of STAT3 in human lymphocyte differentiation as revealed by the hyper-IgE syndrome. J Immunol. 2009;182:21–8. doi: 10.4049/jimmunol.182.1.21. [DOI] [PubMed] [Google Scholar]
- 113.Zelante T, De Luca A, Bonifazi P, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol. 2007;37:2695–706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- 114.Yao Z, Fanslow WC, Seldin MF, Rousseau A-M, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–21. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 115.Kohyama S, Ohno S, Isoda A, et al. IL-23 enhances host defense against vaccinia virus infection via a mechanism partly involving IL-17. J Immunol. 2007;179:3917–25. doi: 10.4049/jimmunol.179.6.3917. [DOI] [PubMed] [Google Scholar]
- 116.Patera AC, Pesnicak L, Bertin J, Cohen JI. Interleukin 17 modulates the immune response to vaccinia virus infection. Virology. 2002;299:56–63. doi: 10.1006/viro.2002.1400. [DOI] [PubMed] [Google Scholar]
- 117.Crowe CR, Chen K, Pociask DA, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. 2009;183:5301–10. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hou W, Kang HS, Kim BS. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J Exp Med. 2009;206:313–28. doi: 10.1084/jem.20082030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–9. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 120.Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–9. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- 121.Qian Y, Liu C, Hartupee J, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–56. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 122.Chang S, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–7. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 123.Liu C, Qian W, Qian Y, et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. 2009;2:ra63. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ruddy M, Wong G, Liu X, Yamamoto H, Kasayama S, Kirkwood K, Gaffen S. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–67. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 125.Tohyama M, Hanakawa Y, Shirakata Y, et al. IL-17 and IL-22 mediate IL-20 subfamily cytokine production in cultured keratinocytes via increased IL-22 receptor expression. Eur J Immunol. 2009;39:2779–88. doi: 10.1002/eji.200939473. [DOI] [PubMed] [Google Scholar]
- 126.Woltman AM, de Haij S, Boonstra JG, Gobin SJ, Daha MR, van Kooten C. Interleukin-17 and CD40-ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. J Am Soc Nephrol. 2000;11:2044–55. doi: 10.1681/ASN.V11112044. [DOI] [PubMed] [Google Scholar]
- 127.Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–41. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 128.Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis M-A, Miossec P. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112–20. doi: 10.4049/jimmunol.0801967. [DOI] [PubMed] [Google Scholar]