

Abstract
Histone deacetylase 2 (HDAC2) mediates the repression of pro-inflammatory genes by deacetylating core histones, RelA/p65 and the glucocorticoid receptor. Reduced level of HDAC2 is associated with steroid resistant inflammation caused by oxidants. However, the molecular mechanisms regulating HDAC2 in response to oxidants and unsaturated aldehydes is not known. Here, we report that oxidative stress imposed by cigarette smoke extract (CSE), and aldehyde acrolein induced phosphorylation of HDAC2 which was abolished by serine-alanine mutations at serine sites S394, S411, S422 and S424. HDAC2 phosphorylation required direct interaction with serine-phosphorylated protein kinase CK2α and involved reduced HDAC2 deacetylase activity. Furthermore, HDAC2 phosphorylation was required for HDAC2 interaction with transcription factors, co-repressor complex formation, CBP recruitment, acetylation on lysine residues and consequently increased transrepression activity. Thus, phospho-acetylation of HDAC2 negatively regulates its deacetylase activity which has implications in steroid resistance in chronic inflammatory conditions.
Keywords: HDAC2, acetylation, phosphorylation, inflammation, oxidants, lung
INTRODUCTION
Histone deacetylase 2 (HDAC2), a member of the class I family of histone deacetylases, catalyzes the removal of acetyl groups from ε-amino-terminal lysine tails of core histone proteins H3 and H4 and are generally associated with gene repression [1]. HDACs are prominently involved in cellular processes such as cell cycle regulation, inflammation, cellular proliferation and cancer and are thus tightly regulated by complex protein-protein interactions, sub-cellular localization, proteolysis and post-translational modifications [1–4]. HDAC2 is known to be a prominent member of three co-repressor complexes. HDAC1, HDAC2, RbAp46 and RbAp48, which is recruited to histone H4, form the core of the Sin3 and nucleosome remodeling and deacetylating complexes (NuRD). The complexes also contain other specific proteins such as Sin3, SAP18 and SAP30 for the Sin3 complex and Mi2, MTA2 and MBD3 for the NuRD/Mi2 complex [2] crucial for HDAC2 binding to DNA, recruitment to specific promoter sequences and modulating deacetylase activity [1, 5–7]. The regulation of HDAC2 activity and protein expression largely depends on several mechanisms such as protein-protein interaction, degradation and post-translational modifications [2]. Ours and several other labs have shown that HDAC2 activity is decreased in response to oxidative stress [8–11]. However, the signaling mechanism of HDAC2 reduction and the role of oxidative stress in regulating HDAC2 function by post-translational modifications, such as phosphorylation and/or acetylation have not been studied.
Glucocorticoids primarily exhibit potent anti-inflammatory properties through the repression of pro-inflammatory genes. Ligand-bound glucocorticoid receptors (GR) translocate from the cytoplasm to the nucleus, recruiting HDAC2 to promoters of pro-inflammatory genes thus shutting down gene transcription [12]. While low-dose inhaled corticosteroids are highly effective in asthma patients, inhaled or high-dose oral corticosteroids fail to markedly improve lung function or decrease abnormal inflammation in COPD patients [9, 13]. This suggests an important linkage between cigarette smoke-induced oxidative stress and the ability of steroids to suppress inflammation by the recruitment of co-repressor complexes to promoters of pro-inflammatory cytokines.
Cigarette smoke (CS) is a complex mixture of various chemical compounds. Both tar and gas phases of cigarette smoke contain/generate reactive oxygen species (ROS) and small unsaturated aldehydes, such as acrolein and 4-hydroxy-2-nonenal (4-HNE) which are involved in the pathogenesis of chronic obstructive pulmonary disease (COPD) [14, 15]. We had earlier observed a significant and exposure-dependent reduction in HDAC2 levels in epithelial cells and macrophages in-vitro and in the lungs of mice exposed to cigarette smoke [16]. Since HDAC2 had been shown to be uniquely phosphorylated in-vitro, and it possesses several protein kinase CK2 consensus sites at the C-terminal [17], we hypothesized that oxidants/CS and aldehyde-induced HDAC2 modulation involves a rapid induction of protein kinase CK2-mediated phosphorylation of HDAC2 on specific C-terminal serine residues and subsequent degradation. Based on this hypothesis, we sought to characterize HDAC2 phosphorylation in response to oxidants/aldehyde/CS extract (CSE) and its physiological role in regulating HDAC2 protein-protein interaction, enzymatic and gene repressive functions. We further determined the phosphorylation-dependent acetylation of HDAC2 by the recruitment of CREB binding protein (CBP) in response to CS.
MATERIALS AND METHODS
Materials
Trichostatin A (TSA) and acrolein were purchased from Sigma (St. Louis, MO). Okadaic acid was obtained from Alexis Biochemicals (Plymouth Meeting, PA). Lambda Phosphatase was obtained from Millipore (Burlington, MA). HDAC2, CK2α, CK2α′, CK2β, HDAC1, p53, p65, RbAp46/48, SAP30, CBP and Lamin B antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Mi2 Flag, monoclonal phospho-serine and β-actin antibodies were obtained from Sigma. Monoclonal anti-acetyl lysine and MBD3 antibodies were obtained from Cell Signaling (Danvers, MA).
Cell culture
Human bronchial epithelial cells (H292) were purchased from the American Type Tissue Culture Collection (Manassas, VA). 7 × 105 and 4 × 106 H292 cells were grown in 6-well and 100 mm dishes respectively in RPMI-1640 supplemented with 10% fetal bovine serum (HyClone Laboratories, Logan, UT), 2 mM L-glutamine, 100 μg/ml penicillin and 100 U/ml streptomycin in humidified atmosphere under 5% CO2 at 37°C.
Plasmid constructs
pME18S-HDAC2, pGal4-HDAC2 and flag-tagged HDAC2 mutant plasmids (S394A, S394/411/422/424A, aa1-400) were kind gifts from Dr. E. Seto (H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL). PathDetect pFR-Luc trans-reporter plasmid was purchased from Agilent Technologies (Cedar Creek, TX). CBP wild-type and CBP mutants (L1690K/C1691L) were kind gifts from Dr S. Ghosh (Columbia University Medical Center, New York, NY). pCMX-Gal4 plasmids were a kind gift from Dr. Chawnshang Chang (University of Rochester Medical Center, Rochester, NY).
Preparation of aqueous CSE
Research-grade cigarettes (3R4F) with a filter from the Kentucky Tobacco Research and Development Centre at the University of Kentucky (Lexington, KY) were used to prepare as described [11, 18]. Extract, defined as 10% CSE, was used for all experiments within 10 minutes of preparation. Air was bubbled into 10 ml of RPMI-1640 with 0.5% serum and pH adjusted to 7.4, filtered and used as control medium.
Transfections and luciferase reporter assay
For transfection assays, H292 cells were seeded at a density of 7.0 × 105 cells in 6 well culture plates or 4 × 106 cells in 100 mm dishes in a total volume of 2 or 7 ml RPMI-1640 containing 10% FBS without antibiotics overnight respectively. Cells were transiently transfected with control vector (Ctrl V) or flag-tagged wild-type HDAC2 (1–488), S394A, S394/422/424A, 1–400 aa HDAC2 mutants, CBP over-expression plasmid and CBP mutant plasmids (4–8 μg) lacking intrinsic HAT activity for 24 h with Lipofectamine 2000 according to manufacturer protocol. For co-transfections, EGFP or empty pcDNA3 vector plasmids were used to control for equal amount of DNA in all transfected wells. For siRNA assays, cells were seeded at a density of 3.0–5.0 × 105 cells in 6 well culture plates in a total volume of 2 ml RPMI-1640 containing 10% FBS without antibiotics overnight. 100 pmol of CK2α siRNA (Applied Biosystems, Austin, TX) or scrambled siRNA were transfected for 24 h with Lipofectamine 2000 according to manufacturer protocol. Cells were then washed once with 1X phosphate-buffered saline (PBS) and transfected with flag-tagged HDAC2 (1-488) for a further 24 h prior to treatment. Transfected cells were then washed in 1X PBS and treated with CSE (2.5 %) for 0.5 h. Cells were lysed for 30 min on ice in 50–100 μl lysis buffer (0.5 % NP-40, 25 mM Tris, 100 mM NaCl with protease and phosphatase inhibitors, pH 7.4).
For luciferase assay, cells were transiently co-transfected with either pCMX-Gal4 or pGal4-HDAC2 with pFR-luc reporter. For CBP over-expression, cells were co-transfected with pCDNA3 empty vector, CBP wild-type or CBP (HAT-) with pGal4-HDAC2 and pFR-luciferase reporter. 24 h post transfection, cells were washed once with 1 × PBS, treated with CSE (2.5 %) for 0.5 h and then lysed in 80 μl/6-well plate 1X luciferase reporter lysis buffer (Promega, Madison, WI). 20 μg protein lysates was mixed with 100 μl of luciferase assay reagent and relative light units measured using a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA).
Immunoprecipitation and immunoblotting assays
Transfected H292 cells were lysed in 100 μl lysis buffer and 250 – 500 μg cell lysate was immunoprecipitated overnight with 40 μl EZview Red anti-flag M2 affinity gel (Sigma). Beads were washed with Tris-buffered Saline and immuno-complexes separated by SDS-polyacrylamide gel. For immunoprecipitation of CK2α, 500 μg cell lysates were incubated with 2 μg polyclonal CK2α antibodies overnight at 4°C and then immunoprecipitated with protein A/G Agarose beads (Santa Cruz Biotech) for 1 h at 4°C. Beads were washed with lysis buffer, boiled for 5 minutes in 2x sample buffer and immuno-complexes separated on SDS-polyacrylamide gels. Proteins were transferred overnight unto PVDF membranes (Millipore) and immunoblotted with respective primary antibodies. Primary antibodies were detected with respective secondary antibodies and membranes visualized with enhanced chemiluminescence (PerkinElmer, Waltham, MA).
Immunofluorescence
H292 cells were treated with CSE (2.5 %) for 0.5 h, washed and fixed with 4% PFA as described previously [19]. Slides were incubated with CK2α, CK2β goat polyclonal or α-tubulin mouse monoclonal antibody (1:100) overnight at 4 °C. Slides were mounted with ProLong Gold antifade reagent (Invitrogen) and images analyzed by a Leica confocal microscope or an Olympus inverted microscope.
Phosphatase activity assay
H292 cells, plated at 4.5 × 106 cells/well in 100 mm dishes overnight, were treated with CSE (2.5 %) for 0.5, 2 and 6 h or 100 nM okadaic acid for 1 h. Cells were washed once with cold 1X PBS and lysed in 300 μl phosphatase storage buffer (50 mM Tris HCl pH 8, 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% deoxycholate without phosphatase inhibitors) and PP2A activity measured using a Promega serine/threonine phosphatase assay kit (Promega, Madison, WI) according to manufacturer’s protocol. Briefly, samples were applied to a Sephadex G-25 resin column and centrifuged at 600 × g for 5 minutes at 4°C to remove endogenous phosphate. 5 μg of protein in 35 μl phosphatase storage buffer was mixed with 5 μl of 1 mM RRA(pT)VA phosphopeptide and 10 μl PPase-2A 5X reaction buffer (250 mM imidazole pH 7.2, 1 mM EGTA, 0.1 % β-mercaptoethanol and 0.5 mg/ml BSA) in a 96-well plate. Samples were incubated for 15 minutes and reactions stopped by the addition of 50 μl molybdate dye/additive mixture, incubated for a further 15 minutes and sample optical density (OD) determined with a microplate reader using a 630 nm filter. Phosphatase activity in pmol was determined using a standard curve obtained from free phosphate standards. All experiments were performed in quadruplicates.
HDAC2 activity assay
HDAC2 activity assay was determined as described [19]. Briefly, 500 –1000 μg HDAC2 was immunoprecipitated from whole cell lysates as described above. Beads were washed thoroughly and HDAC2 activity determined using an HDAC Color de Lys activity kit (Biomol, Plymouth Meeting, PA) according to manufacturer’s protocol.
Statistical analysis
Data expressed as mean±SE of triplicate experiments. Statistical analysis of significance was calculated using one-way Analysis of Variance (ANOVA) with STATVIEW software. ImageJ software was used for densitometry analysis. *P<0.05, **P<0.01, ### ***P<0.001.
RESULTS
CSE induces HDAC2 phosphorylation on serine sites 394, 422 and 424 in a phosphatase-independent manner
Previously, it had been shown that phosphorylation of HDAC2 modulates deacetylase activity [16, 17, 20] and protein kinase CK2 inhibition reverses cigarette smoke extract (CSE)-induced endogenous HDAC2 phosphorylation and degradation [19], however the mechanism and physiological relevance of CSE-induced HDAC2 phosphorylation remained largely unknown. To characterize CSE-induced HDAC2 phosphorylation, full length flag-tagged HDAC2 was first expressed in bronchial epithelial H292 cells (Fig 1A) which were then exposed to CSE (2.5%) for 0.5 h. Since we had previously shown CSE-induced covalent modification of HDAC2 at 4 h [11], it was important to choose a time point at which these modifications would be negligible and any functional modulation of HDAC2 activity would be due primarily to cellular signaling mechanisms such as phosphorylation. As expected, immunoprecipitated flag-tagged HDAC2 was significantly phosphorylated on serine residues which was de-phosphorylated by pre-treating immunoprecipitated beads with λ-phosphatase for 1 h at 30 °C prior to resolving samples on an SDS-polyacrylamide gel (Fig 1B, E).
Figure 1. HDAC2 phosphorylation requires serine sites S394, S422, S424 and is independent of phosphatase inhibition.

(A) Bronchial epithelial H292 cells were transfected with plasmid vector or flag-tagged WT HDAC2 (1-488) for 24 h using Lipofectamine 2000. 5 – 10 μg proteins were separated on a 7.5% SDS-PAGE gel and flag proteins detected with M2 monoclonal anti-flag antibodies. (B) H292 cells were transfected with flag-tagged WT HDAC2 for 24 h and then treated with CSE (2.5%) for 0.5 h, (C) acrolein (50 μM) for 1 h or (D) CSE (2.5%) for 0.5, 2 and 6 h. Immunoprecipitated flag proteins were probed with phosphoserine monoclonal antibodies by western blot. (E) Immunoprecipitated beads from H292 cells transfected with flag-tagged HDAC2 and treated with CSE (2.5%) were incubated with lambda PPase buffer or 400 units of lambda PPase for 30 minutes at 30 °C. Washed beads were then eluted with 2x Laemmli sample buffer and separated on a 7.5% SDS-PAGE gel. Blots were probed with phosphoserine antibodies. (F) Lysates from H292 cells treated with CSE (2.5 %), acrolein (50 μM) or exposed to hypoxia for 1 h were separated by SDS-PAGE gel and blots probed for HIF1A. (G) H292 cells were transfected with plasmid vector, flag-tagged WT HDAC2 (1-488) or S394A, S394/411/422/424A and 1-400 aa HDAC2 mutants for 24 h with Lipofectamine 2000. Cells were then treated with CSE (2.5%) for 0.5 h. Serine phosphorylation of immunoprecipitated flag proteins was determined by western blot. (H) H292 cells were treated with or without CSE (2.5%) for 0.5, 2 and 6 h with serine/threonine phosphatase (okadaic acid) used as a positive control. PP2A activity was determined by incubating 5 μg protein with PP2A specific-peptide substrate RRA(pT)VA and PP2A activity determined by reading optical density with microplate reader using a 630 nm filter (n = 4, ** p < 0.01).
To characterize the stability of HDAC2 serine phosphorylation, we varied the time of CSE exposure from 0.5 h to time point at which HDAC2 protein degradation was first observed (6 h), HDAC2 serine phosphorylation was significant at 0.5 h, diminished by more than 50% at 2 h and was almost completely negligible at 6 h CSE treatment (Fig 1D). To determine if acrolein, a small unsaturated aldehyde and important constituent of CSE, would also induce HDAC2 phosphorylation, H292 cells were transfected with flag-tagged full length HDAC2 and then treated with acrolein (50 μM) for 1 h. Immunoprecipitated flag-tagged protein from treated cells were significantly phosphorylated on serine residues (Fig 1C).
To rule out any role for hypoxia in CSE or acrolein-induced HDAC2 phosphorylation, lysates from CSE and/or acrolein-treated cells were resolved on an SDS polyacrylamide gel and then assessed for the expression of hypoxia-inducible factor 1 alpha (HIF1α), an expression marker in response to reduced cellular oxygen concentrations. HIF1α expression was negligible in cells treated with CSE (2.5%) or acrolein (50 μM) compared to either cells exposed to media alone or 5 % oxygen for 1 h (Fig 1F).
Previous studies using radio-labeled wild-type and mutant HDAC2 constructs had identified serine sites S394, S422 and S424 as potential phospho-acceptor sites on HDAC2 [17], we therefore sought to determine if these sites were also critical for CSE-induced HDAC2 phosphorylation in-vivo. H292 cells were transfected with flag-tagged full length HDAC2 (1-488), a C-terminal 88-amino acid deletion mutant (1-400) and C-terminal serine to alanine mutants. Cells were then treated with CSE (2.5%) for 0.5 h, flag proteins immunoprecipitated using anti-flag affinity gel and resolved on an SDS-polyacrylamide gel. Both wild-type and S394A mutant HDAC2 were phosphorylated indicating the S394 site alone may not be crucial to CSE-induced HDAC2 phosphorylation. However, serine to alanine mutations on sites S394, S411, S422 and S424 significantly attenuated CSE-induced HDAC2 phosphorylation while the C-terminal 88-amino acid deletion mutant (1-400) construct was only modestly phosphorylated suggesting serine sites S422 and S424 as crucial for CSE-induced HDAC2 phosphorylation (Fig 1G).
To determine if phosphorylation of HDAC2 in response to CSE was dependent on a kinase-mediated mechanism or inhibition of phosphatase activity, PP2A serine/threonine phosphatase activity was measured in response to varying time points of exposure to CSE. Compared to non-treated controls, CSE significantly diminished PP2A activity at 6 h but had no effect at 0.5 h, time points at which CSE induced HDAC2 phosphorylation (Fig 1H).
Cigarette smoke induces CK2α phosphorylation
Since we observed that serine sites S394, S422 and S424 were required for CSE-induced HDAC2 phosphorylation and are conserved protein kinase CK2 phospho-acceptor sites on HDAC2, we speculated that CK2 would be involved in HDAC2 phosphorylation in response to CSE. Despite published reports indicating that the CK2 catalytic subunit is constitutively active [21, 22], we consistently observed a doublet band for CK2α in response to CSE (2.5%) for 0.5 h (Fig 2A) with no significant change in relative protein expression. To confirm if this doublet was due to CK2α phosphorylation, immunoprecipitated CK2α from bronchial epithelial (H292) cells was determined to be phosphorylated on serine residues in response to CSE (Fig 2B). Phosphatase treatment of CK2α immunoprecipitates completely abolished the doublet band in response to CSE (Fig 2C). Okadaic acid, a specific PP2A inhibitor which had earlier been shown to induce HDAC2 serine phosphorylation [19], also induced CK2α phosphorylation as shown by the presence of doublet bands (Fig 2D).
Figure 2. Cigarette Smoke Extract (CSE) induces CK2α serine phosphorylation.

(A) Bronchial epithelial H292 cells were treated with CSE (2.5 %) for 0.5 h. 10 μg protein was separated by SDS-PAGE and CK2α detected by western blot. (B) CK2α from control or CSE-treated H292 cells was immunoprecipitated and analyzed for serine phosphorylation. (C) Lysates from H292 cells treated with media alone or CSE were incubated with 400 units of lambda phosphatase for 30 minutes at 30 °C before separation by SDS-PAGE. CK2α expression was determined by western blot. (D) H292 cells were treated with indicated concentrations of okadaic acid for indicated time periods. CK2α expression was determined by western blot.
HDAC2 serine phosphorylation is associated with binding to CK2 catalytic but not regulatory subunits in response to CSE
The presence of conserved protein kinase CK2 sites that were highly phosphorylated on HDAC2 in response to CSE and acrolein, led us to hypothesize a potential direct interaction between HDAC2 and protein kinase CK2 catalytic subunits or the holoenzyme. Flag-tagged WT HDAC2 plasmids were transfected into H292 cells and then treated with CSE (2.5%) for 0.5 h. Both catalytic subunits of protein kinase CK2; CK2α, CK2α′ and the serine-phosphorylated form of CK2α were recruited to HDAC2 in response to CSE treatment (Fig 3A and 3B). To determine if this interaction was specific only to CSE, H292 cells were also treated with acrolein (25 and 50 μM) for 1 h. Acrolein-induced HDAC2 phosphorylation was associated with increased HDAC2 CK2α′ binding (Fig 3C) which, compared with CSE treatment, suggested that the extent of HDAC2 phosphorylation correlates with the level of CK2α binding. This finding further indicated a potential role for CK2α in HDAC2 phosphorylation.
Figure 3. Cigarette Smoke Extract (CSE) and acrolein induce HDAC2 interaction with catalytic subunits of CK2.

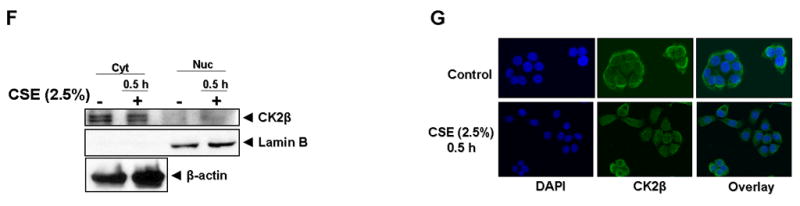
H292 cells were transfected with plasmid vector or flag-tagged WT HDAC2 (1-488) for 24 h using Lipofectamine 2000 and then treated with or without CSE (2.5 %) for 0.5 h. Flag proteins were immunoprecipitated and binding to (A) CK2α and (B) CK2α′ determined by western blot. (C) H292 cells transfected with flag-tagged WT HDAC2 were treated with CSE (2.5 %) for 0.5 h, and acrolein (25 μM and 50 μM) for 1 h. CK2α′ binding to immunoprecipitated flag-tagged HDAC2 was determined by western blot. (D) H292 cells were treated with or without CSE (2.5%) for 0.5 h. Nuclear and cytoplasmic extracts were probed for CK2α or (F) CK2β expression by western blot. (E) H292 cells were treated with CSE (2.5%) for 0.5 h, washed and fixed with 4% PFA. Slides were incubated with CK2α or (G) CK2β goat polyclonal antibody (1:100) overnight at 4 °C. Slides were mounted with ProLong Gold antifade reagent (Invitrogen) and images analyzed by a Leica confocal microscope.
HDAC2 is a predominantly nuclear protein while CK2β is mostly localized to the cytoplasm; hence we speculated CK2β must be translocated to the nucleus in response to CSE if it is required to _target CK2 catalytic subunits to HDAC2. Using both immunoblot and immunofluorescence techniques, we did not observe any significant translocation of CK2α to the nucleus (Fig 3D and E). However, CK2β, was not detected either in the nucleus or bound to HDAC2 both by immunoblotting and confocal microscopy in response to CSE (Figs 3F and G) suggesting that independent catalytic subunits of protein kinase CK2 but not the holoenzyme was sufficient for CSE and acrolein-induced HDAC2 phosphorylation and substrate specificity.
Pharmacological inhibition and silencing of protein kinase CK2 inhibit oxidative stress-induced HDAC2 phosphorylation
To determine if protein kinase CK2 was uniquely required for CSE-induced HDAC2 phosphorylation [17, 23], bronchial epithelial H292 cells were pre-treated with a specific CK2 inhibitor, 4, 5, 6, 7-tetrabromobenzotriazole (TBB) [24] for 2 h prior to exposure to CSE (2.5%) for 0.5 h. 50 μM TBB was sufficient at completely inhibiting CSE-induced HDAC2 phosphorylation (Fig 4A–B). TBB, at doses as low as 25 μM have also been observed to partially inhibit HDAC2 phosphorylation. Using a genetic approach, H292 cells were transfected with either scrambled or a 19-nucleotide siRNA sequence _targeted to human CK2α for 24 h. Cells were then subsequently transfected with flag-tagged wild-type HDAC2 for 24 h prior to treatment with CSE (2.5%) for 0.5 h. A 56 % knock-down in CK2α but not CK2α′ expression (Fig 4C) was sufficient to partially inhibit CSE-induced serine phosphorylation of HDAC2 (Fig 4D). Partial inhibition via CK2α knock-down compared to almost complete inhibition of phosphorylation with the specific CK2 inhibitor, TBB, suggested the possibility that CK2α′, the catalytic isoform of CK2α, is also involved in CSE-mediated HDAC2 phosphorylation and compensates for loss of CK2α.
Figure 4. CK2 is required for CSE-induced HDAC2 phosphorylation.

(A) H292 cells transfected with flag-tagged WT HDAC2 were pre-treated with TBB (50 μM) for 2 h prior to treatment with CSE (2.5%) for 0.5 h. Immunoprecipitated flag proteins were analyzed for serine phosphorylation by western blot. (B) Relative density of phospho-serine expression normalized to immunoprecipitated HDAC2. (C) H292 cells were transiently transfected with scrambled or 100 pmol CK2α siRNA for 24 h. Cells were washed and incubated for a further 24 h. Whole cell lysates were separated by SDS-PAGE and CK2α and CK2α′ expression analyzed by western blot. (D) H292 cells transfected with CK2α siRNA for 24 h were transfected for a further 24 h with flag-tagged WT HDAC2. Cells were then exposed to CSE (2.5 %) for 0.5 h and immunoprecipitated flag proteins analyzed for serine phosphorylation by western blot (n = 3, *** ### P<0.001).
CSE-induced HDAC2 phosphorylation is required for co-repressor binding and interaction with transcription factors but not HDAC2 subcellular localization
Based on earlier studies showing a disruption in HDAC2 binding to other proteins in the co-repressor complex in response to increased intracellular phosphorylation with okadaic acid [20], we initially hypothesized that CSE-mediated oxidative stress-induced HDAC2 phosphorylation would cause a disruption of the HDAC2 co-repressor complex. However, CSE (2.5 %) treatment of H292 cells transfected with wild-type flag-tagged HDAC2 for 0.5 h showed a significant increase in HDAC2 associated with HDAC1 (Fig 5A) compared with media-alone treatments. As earlier observed with immunoprecipitation reactions showing increased p53 interaction with HDAC1/2 via mSin3A in response to doxycycline-induced DNA damage [25], we hypothesized that CSE-induced HDAC2 phosphorylation causes significantly increased HDAC2 interaction with p53. As expected, only immunoprecipitated flag-tagged wild-type HDAC2 from H292 cells exposed to CSE (2.5%) showed interaction with the p53 transcription factor (Fig 5B). To further determine if the increased interaction was dependent on phosphorylation, H292 cells were transfected with either flag-tagged HDAC2 serine to alanine mutants on sites S394, S411, S422 and S424 or an HDAC2 C-terminal 88-amino acid deletion mutant and then exposed to CSE (2.5%) for 0.5 h. Consistent with earlier results showing abrogation of serine phosphorylation with loss of serine site mutations S394, S411, S422 and S424 but not S411, S422 and S424 alone, HDAC1 interaction was significantly lower in the S394, S411, S422 and S424 mutant but not the C-terminal deletion mutant (Fig 5C) suggesting phosphorylation is strongly required for HDAC2 co-repressor interactions. Inhibiting HDAC2 phosphorylation by pre-treating cells with N-acetyl-L-cysteine (NAC), a cysteine source for the synthesis of the antioxidant GSH, for 2 h prior to exposure to CSE significantly attenuated HDAC2 interaction with SAP30, a Sin3-specific complex protein, MBD3, a Mi-2/nucleosome remodeling and deacetylase (NURD)-specific co-repressor complex protein, RBAp46/48, a component of both Sin3 and NURD complexes and p65, a transcription factor involved in inflammation and known to be actively deacetylated by HDAC1/HDAC2 [26] (Fig 5D). To further confirm the role of CK2-mediated HDAC2 phosphorylation in increased HDAC2 interaction with co-repressor proteins and transcription factors, H292 cells transfected with flag-tagged wild-type HDAC2 were pre-treated with either 40 or 90 μM TBB for 2 and 0.5 h respectively and then exposed to CSE (2.5%) for 0.5 h. TBB significantly inhibited CSE-induced HDAC2 interaction with HDAC1 and p65 in a dose-dependent manner (Fig 5E).
Figure 5. Cigarette Smoke Extract (CSE)-induced HDAC2 phosphorylation regulates co-repressor formation, interaction with transcription factors and deacetylase activity but not cellular localization.


(A) H292 cells were transfected with plasmid vector or flag-tagged WT HDAC2 (1-488) for 24 h using Lipofectamine 2000 and then treated with or without CSE (2.5 %) for 0.5 h. Flag proteins were immunoprecipitated and binding to HDAC1 and (B) p53 determined by western blot. (C) H292 cells were transfected with plasmid vector, flag-tagged WT HDAC2 (1-488) or S394/411/422/424A and 1-400 aa HDAC2 mutants for 24 h with Lipofectamine 2000. Cells were then treated with CSE (2.5%) for 0.5 h. HDAC1 interaction with immunoprecipitated flag proteins was determined by western blot. (D) Flag-tagged HDAC2-transfected H292 cells were pre-treated with NAC (2 mM) for 2 h prior to 0.5 h treatment with CSE (2.5 %). Immunoprecipitated flag proteins were separated by SDS-PAGE and binding to SAP30, RbAp46/48, MBD3 and RelA/p65 analyzed by western blot. (E) Flag-tagged HDAC2-transfected H292 cells were pre-treated with TBB 40 or 90 μM for 2 h or 0.5 h respectively prior to treatment with CSE (2.5 %) for 0.5 h. Immunoprecipitated flag proteins were analyzed for HDAC1 and p65 binding by western blot. (F) Immunoprecipitated flag proteins from NAC pre-treatment experiment were analyzed for HDAC deacetylase activity using a deacetylase activity kit. (G) H292 cells were transfected with siRNA _targeting CK2α for 24 h. Cells were then washed and transfected with flag-tagged WT HDAC2 for a further 24 h prior to treating with CSE (2.5 %) for 0.5 h. Immunoprecipitated flag proteins were analyzed for deacetylase activity. (H) H292 cells were grown on coverslips and treated with CSE (2.5%) or acrolein (50 μM) for 0.5 and 1 h respectively. Cells were fixed with 4 % PFA then incubated with HDAC2 rabbit polyclonal antibody overnight at 4 °C. Images from slides, mounted with ProLong Gold antifade reagent, were then analyzed with an Olympus inverted microscope (n = 3, * P<0.05, ** P<0.01).
To determine whether phosphorylation was associated with HDAC2 enzymatic activity, flag-tagged wild-type HDAC2 was immunoprecipitated from H292 cells pre-treated with NAC for 2 h prior to exposure to CSE (2.5%) for 0.5 h. NAC pre-treatment significantly attenuated CSE-induced decrease in HDAC2 deacetylase activity (Fig 5F) suggesting serine phosphorylation is a possible negative regulator of HDAC2 activity by CSE-mediated oxidative stress. Since NAC is a thiol antioxidant, it was important to see whether direct modulation of the CK2-HDAC2 phosphorylation pathway would affect HDAC2 activity. H292 cells, transfected with either a scrambled sequence or 100 pmol of a siRNA sequence _targeted to CK2α, were exposed to either media alone or CSE (2.5%) for 0.5 h. Cells were then harvested and specific HDAC2 activity measured. Consistent with the pattern observed with NAC pre-treatment, partial CK2α knock-down rescued CSE-induced HDAC2 enzymatic activity almost to control levels (Fig 5G). Based on earlier findings showing reduced nuclear expression of HDAC2 following long-term exposure to CSE [19] and that cellular localization of class II HDACs such as HDAC4 is actively regulated by phosphorylation [27], we hypothesized that CSE-induced phosphorylation of HDAC2 would induce nucleo-cytoplasmic shuttling. H292 cells were exposed to CSE (2.5 %) and acrolein (50 μM) for 0.5 and 1 h respectively at time points where induction of serine phosphorylation was observed. However, HDAC2 remained exclusively localized to the nucleus (Fig 5H) suggesting localization of class I HDACs is unaffected by phosphorylation.
CBP-mediated HDAC2 acetylation is dependent on phosphorylation
Despite the presence of at least 12 conserved lysine residues on the C-terminal domain of HDAC2 and earlier findings suggesting HDAC1 is also acetylated in response to dexamethasone [28], it remained largely unknown as to whether HDAC2 was also modified by acetylation. To determine this, H292 cells were transfected with flag-tagged full length HDAC2 and then treated with or without CSE (2.5%) for 0.5 h. Consistent with our hypothesis, full length HDAC2 was strongly acetylated in response to CSE (Fig 6A). To further validate this, flag-tagged wild-type HDAC2 was transfected into H292 cells treated with acrolein (50 μM) for 1 h, using CSE as a positive control. Consistent with earlier results showing acrolein-induced HDAC2 phosphorylation, immunoprecipitated flag-tagged HDAC2 showed significant acetylation of lysine residues compared to untreated controls (Fig 6B).
Figure 6. HDAC2 acetylation requires phosphorylation-induced CBP recruitment.

(A) H292 cells, transfected with flag-tagged WT HDAC2 were treated with CSE (2.5%) for 0.5 h and (B) Acrolein (50 μM) for 1 h. Immunoprecipitated flag proteins were analyzed for global acetylation by western blot. (C) Immunoprecipitated flag proteins from H292 cells treated with CSE (2.5%) for 0.5 h were analyzed for CBP binding by western blot. (D) H292 cells were transiently co-transfected with CBP WT or (E) CBP HAT mutant for 24 h prior to CSE treatment. Immunoprecipitated flag-tagged proteins were analyzed for lysine residue acetylation or (F) serine phosphorylation by western blot. G) H292 cells were transfected with plasmid vector, flag-tagged WT HDAC2 (1-488) or S394/411/422/424A mutant for 24 h with Lipofectamine 2000. Cells were treated with CSE (2.5%) for 0.5 h. Acetylation of immunoprecipitated flag proteins was determined by western blot.
Since both CREB binding protein (CBP) and HDAC2 had been recently mapped to be actively recruited to promoters and intergenic regions of active genes [29], we speculated that CBP would be involved in HDAC2 acetylation most possibly via a direct protein-protein interaction. To determine a possible interaction between HDAC2 and CBP, flag-tagged wild-type HDAC2 was transfected into H292 cells and then treated with CSE (2.5%) for 0.5 h. Immunoprecipitated flag-tagged HDAC2 showed significant interaction with CBP only in CSE-treated cells compared with media-alone controls (Fig 6C).
To further establish a role for CBP in HDAC2 interaction, we obtained both wild-type (CBP+) and HAT activity-deficient CBP mutants (CBP- L1690K and C1691L) [30] and co-transfected with flag-tagged wild-type HDAC2 in H292 cells. Cells were then exposed to CSE (2.5%) for 0.5 h and flag-tagged proteins immunoprecipitated and immunoblotted for acetylation of lysine residues. However, over-expression of wild-type CBP failed to enhance CSE-induced HDAC2 acetylation (Fig 6D) suggesting the possibility of specific CBP binding sites on HDAC2. Consistent with a prominent role for CBP in HDAC2 acetylation, over-expression of a dominant negative HAT-deficient CBP mutant completely abrogated CSE-induced HDAC2 acetylation of lysine residues (Fig 6E). The abrogation of HDAC2 acetylation, however, had no effect on HDAC2 phosphorylation in the presence of over-expressed dominant negative CBP (Fig 6F) suggesting that acetylation was more likely a consequence of CSE-induced HDAC2 phosphorylation.
To further determine whether acetylation was dependent on phosphorylation, we speculated that an HDAC2 mutant deficient in critical phosphorylation sites would not be acetylated in response to CSE. H292 cells were transfected with flag-tagged wild-type or S394, S411, S422 and S424 mutant HDAC2 and then treated with CSE (2.5%) for 0.5 h. Consistent with our hypothesis, mutant HDAC2 was only minimally acetylated compared to the full length wild-type HDAC2 (Fig 6G).
Acetylation modulates HDAC2 transrepression
HDAC2 plays two distinct cellular roles; deacetylation of substrate proteins or histones (enzymatic activity) and repression of gene transcription (functional activity) [2]. Since CK2α knock-down and NAC-mediated inhibition of HDAC2 phosphorylation in response to CSE increased HDAC2 enzymatic activity, it was important to determine whether phosphorylation would affect HDAC2 functional activity. To determine this, H292 cells were co-transfected with plasmids encoding both Gal4-HDAC2 [17] or Gal4-DBD and a luciferase reporter plasmid containing five tandem yeast Gal4 binding site repeats (Fig 7A). Transfected cells were then treated with CSE (2.5%) or acrolein (50 μM) for 0.5 and 1 h respectively and then assayed for luciferase expression. Consistent with other reports [17], HDAC2 significantly repressed luciferase transcription in response to CSE treatment compared to untreated controls alone (Fig 7B). Acrolein, however, did not show a significant effect on luciferase expression compared to CSE-treated cells. This may be explained by data showing acrolein induces far less HDAC2 serine phosphorylation compared to CSE (data not shown) which is associated with a significant difference in CK2α′ binding to HDAC2 (Fig 3C). To determine the specific role of HDAC2 acetylation in gene transrepression, H292 cells were co-transfected with Gal4-HDAC2, a pFR-luciferase reporter plasmid and either a CBP wild-type or mutant lacking HAT activity. Cells were then treated with or without CSE (2.5%) for 0.5 h, lysed and then assayed for luciferase activity. Surprisingly, over-expression of wild-type CBP significantly increased HDAC2 transrepression activity which was attenuated by over-expression of mutant CBP with no HAT activity (Fig 7C), suggesting that acetylation is the more dominant regulator of HDAC2 transrepression activity.
Figure 7. HDAC2 acetylation is critical for HDAC2 functional activity.

(A) Schematic for yeast Gal4 2-hybrid assay. HDAC2-Gal4 plasmids were co-transfected with a pFR-luciferase reporter tagged to a promoter with 5 tandem yeast Gal 4 repeats. (B) H292 cells were co-transfected with either pCMX-Gal4 or Gal4-HDAC2 plasmids with a pFR-luciferase reporter plasmid. Cells were treated for 0.5 or 1 h with CSE (2.5%) or acrolein (50 μM) respectively. 20 μg cell lysates were analyzed for luciferase expression (n=3). (C) H292 cells were co-transfected with pcDNA3 vector, CBP wild-type or CBP (HAT-) plasmids with HDAC2-Gal4 and a pFR-luciferase reporter. Cells were exposed to either media alone or CSE (2.5%) for 0.5 h. 20 μg cell lysates were analyzed for luciferase expression (n=3).
Dexamethasone blocks pro-inflammatory gene expression induced by LPS but not CSE
Steroid resistance is a major hallmark of moderate to severe COPD and has been associated with loss of HDAC2 [31, 32]. As we had earlier shown that HDAC2 serine phosphorylation in response to CSE is directly associated with HDAC2 degradation [19], it was important to compare the ability of corticosteroids to inhibit IL-8 release in response to CSE or LPS which did not induce HDAC2 phosphorylation in our system (data not shown). Cells were pre-treated with low dose dexamethasone (0.1 μM) for 2 h prior to CSE or LPS exposure for 1 h. There was no significant difference in IL-8 release between samples treated with media alone or dexamethasone alone (<150±9 pg/ml). However, while dexamethasone failed to inhibit IL-8 release in response to CSE treatment (CSE 1554±54 pg/ml vs. CSE+DEX 1665±65, n=4), cells treated with dexamethasone + LPS showed a 40% decrease in IL-8 release (809±32 pg/ml, p<0.01, n=4) compared to LPS-alone treatments (1354±26 pg/ml). These results suggest that HDAC2 serine phosphorylation is a key mechanism of steroid resistance in COPD and is consistent with earlier observations showing increased HDAC2 serine phosphorylation in mouse lungs with sub-chronic CS exposure which subsequently leads to loss of nuclear HDAC2 protein [19].
DISCUSSION
Ligand-bound corticosteroid receptors translocate from the cytoplasm to the nucleus, recruiting HDAC2 to promoters of pro-inflammatory genes thus shutting down gene transcription [12]. Corticosteroids are ineffective at attenuating pro-inflammatory gene expression in COPD [9, 33–35], and it has been suggested that the inability of corticosteroids to recruit HDAC2 or the presence of defective or post-translationally modified HDAC2 may explain the abnormal inflammatory response and steroid insensitivity observed which is a hallmark of COPD [31, 32, 36–39]. We had earlier shown increased serine phosphorylation of endogenous HDAC2 in macrophages and lung epithelial cells in response to CSE which was sensitive to CK2 inhibitor treatment [19]. Endogenous HDAC2 in lungs of mice exposed to cigarette smoke was also phosphorylated both on serine and threonine residues and correlated with HDAC2 degradation [19]. As oxidant-mediated HDAC2 degradation was attenuated with CK2α inhibition [19], we speculated that early serine phosphorylation of HDAC2 in response to either CS or acrolein would play a prominent role in regulating HDAC2 enzymatic or functional activity. We consistently observed a rapid but transient increase in HDAC2 serine phosphorylation that was sensitive to phosphatase treatment. HDAC2 phosphorylation peaked as early as 0.5 h and was lost by 6 h after exposure to CSE. No change in relative HDAC2 protein expression and covalent protein-protein modifications was observed at the early time point selected [11], indicating that observed CSE modulation of HDAC2 activity was solely due to phosphorylation. Since hypoxia had been implicated in phosphorylating HDAC2 [16], we had to overcome the potential problem that the observed CSE effect was due primarily to the induction of a hypoxic environment. However, neither CSE nor acrolein induced any significant level of hypoxia inducible factor alpha (HIF1α) compared to cells exposed to hypoxia.
Furthermore, it was plausible that the CSE-mediated effect observed was primarily due to increased intracellular phosphorylation status in response to inhibition of serine phosphatases, we therefore measured PP2A activity. We chose PP2A for several reasons: okadaic acid, which had been shown to phosphorylate HDAC2 [19, 20] is a potent inhibitor of PP2A; PP2A, unlike PP2B and C, does not possess a metal ion requirement and PP1 mostly dephosphorylates proteins phosphorylated by PKA. Since PKA does not phosphorylate HDAC2 at least in-vitro [17], we did not expect PP1 to be wholly important if at all phosphatase activity is instrumental to HDAC2 phosphorylation at early time points. However, CSE had no effect on PP2A activity at time points of peak HDAC2 phosphorylation, ruling out a potential role for phosphatase inhibition mediating HDAC2 phosphorylation in response to CSE. Elegant work by Seto’s group determined three possible phosphorylation sites on HDAC2 that could possibly be involved in CSE-induced HDAC2 phosphorylation [17]. However, we did not observe any significant change in HDAC2 phosphorylation when the S394 site was mutated. The most significant loss of phosphorylation occurred when all three sites, S394/422/424 were mutated or sites S422/424 were lost with truncation of the HDAC2 C-terminal domain. However, both mutants still retained some HDAC2 phosphorylation, indicating the presence of other possible phosphorylation sites yet to be identified. Interestingly, the possibility of HDAC2 phosphorylation on other residues such as threonine or tyrosine remains unknown. We had earlier shown that CSE induces HDAC2 phosphorylation on threonine residues both in MonoMac6 cells and in mouse lungs following subchronic CS exposure [19] and closely follows a similar pattern as serine phosphorylation. Kinase prediction software such as NetPhos 2.0, which accurately predicts serine protein kinase CK2-mediated phosphorylation of HDAC2 serine sites Ser394, Ser422 and Ser424, suggests threonine 278 may be potentially phosphorylated by PKC. However, previous studies using 32P-labeled HDAC2 did not reveal any threonine phosphorylation [17]. We speculate that threonine phosphorylation may be functionally relevant to HDAC2 similar to serine phosphorylation since it is also induced in response to oxidative stress. The functional relevance of HDAC2 threonine phosphorylation in-vivo, if any, remains unknown and will however require future studies.
Although CK2 is generally regarded to be a constitutively active enzyme with CK2α undergoing autophosphorylation on Tyr182 and Tyr188 [40], we were surprised to consistently observe a CK2α phosphatase-sensitive gel retardation band with cells exposed to both CSE and okadaic acid. Immunoprecipitation of CK2α showed it was highly phosphorylated on serine residues and suggested a form of CSE-induced CK2α activation involving serine phosphorylation. Interestingly, multiple CK2α gel retardation bands were observed following prolonged treatment with or increased concentration of okadaic acid. We speculate that these bands may represent CK2α hyperphosphorylation as prolonged okadaic acid treatment may activate other kinases which then induce phosphorylation of CK2α at multiple phosphorylation sites. HDAC2 phosphorylation in-vitro was uniquely dependent on protein kinase CK2, a constitutively active pleiotropic serine/threonine kinase found as a tetrameric holoenzyme with catalytic subunit made up of dimers of αα, αα′ or α′α′ while 2 β subunits constitute the regulatory subunit [41]. Consistent with our hypothesis that CSE-induced HDAC2 phosphorylation in-vivo was largely mediated by CK2, we observed a strong correlation between HDAC2 phosphorylation and binding to both catalytic subunits of the protein kinase CK enzyme (CK2α and CK2α′) either with CSE or acrolein treatment with no significant nuclear translocation contrary to earlier reports [16]. The nuclear pool of CK2α was more highly phosphorylated as evidenced by the presence of the gel retardation band, which was consistent with HDAC2 being a nuclear protein. CK2β, the regulatory subunit of the protein kinase CK2 holoenzyme, plays two prominent roles; determining substrate specificity for the holoenzyme and increasing activity of both catalytic subunits [42]. However, we failed to detect any significant level of CK2β in the nucleus and consequently no interaction with HDAC2. Unlike CK2 inhibition, knock-down of CK2α was less efficient at attenuating CSE-induced HDAC2 phosphorylation. We speculated that this was due to a compensatory role for CK2α′ in the absence of CK2α.
Previous studies showed that LY294002, a specific Akt inhibitor can significantly attenuate CK2α activation [43] suggesting a role for other kinases, such as Akt in CSE-mediated HDAC2 phosphorylation. Theophylline, a methylxanthine derivative and PI-3K inhibitor [44], has been used in combination with corticosteroids to improve clinical outcome in patients with asthma at doses too low to inhibit phosphodiesterases (PDE4) [45]. Consistent with our earlier work showing that inhibition of CK2 significantly attenuates CSE-induced HDAC2 degradation associated with decreased HDAC2 serine phosphorylation [19], theophylline-mediated inhibition of PI3K and subsequent attenuation of HDAC2 serine phosphorylation improves HDAC2 activity and steroid response in COPD macrophages [37, 46]. Interestingly, PI-3Kδ null mice are more responsive to steroid treatment following CS-induced airway inflammation [47]. These studies, along with a report showing CK2 can modulate Akt activity by direct interaction [48], suggest a potential interplay between CK2 and Akt in HDAC2 serine phosphorylation. Since inhibition of HDAC2 phosphorylation has also been shown to restore steroid function in macrophages via increased HDAC2 activity [49], inhibitors of CK2 may equally be effective therapies for improving steroid response in COPD and chronic inflammatory diseases.
HDAC2 enzymatic activity is dependent on the formation of a co-repressor complex, we therefore speculated that HDAC2 phosphorylation in response to CSE would disrupt HDAC2 co-repressor complexes, consistent with reports showing a similar trend with okadaic acid treatment [20]. However, HDAC2 phosphorylation was associated with a significant increase in HDAC2 interaction with SAP30, RbAp46/48, MBD3 and HDAC1 which is attenuated by mutation of serine sites S394/422/424A, NAC and TBB inhibition of HDAC2 phosphorylation in response to CSE. We also observed an increase in interaction with the transcription factors p65 and p53 which are known substrates for HDAC2 [26, 50]. This finding was consistent with previous reports showing that phosphorylated HDAC2 preferentially interacts with both transcriptional activators and repressors such as YY1, p53 and Sp3 [23, 51]. CSE consistently decreased HDAC2 enzymatic activity which was rescued by pre-treatment with NAC or CK2α knockdown. Since we speculated that 0.5 h exposure to CSE was an early response insufficient to induce HDAC2 covalent modifications, the decrease in HDAC2 deacetylase activity had to be a consequence of phosphorylation by a mechanism yet unknown. While cellular localization of class II HDACs such as HDAC4, is actively regulated by serine phosphorylation [27, 52, 53], HDAC2 localization remained almost exclusively nuclear in response to both CSE and acrolein treatments.
The presence of several conserved lysine residues especially clustered around the C-terminal domain of HDAC2 and the recent finding that HDAC1 is acetylated in response to dexamethasone [28, 54] prompted us to consider the possibility of HDAC2 acetylation. Both CSE and acrolein induced significant HDAC2 acetylation associated with the recruitment of CBP. This is consistent with a recent report showing both HATs such as CBP and HDAC2 are enriched on actively transcribed genes [29]. Over-expression of wild-type CBP did not affect levels of HDAC2 acetylation, indicating the possibility of saturation of specific CBP binding sites on HDAC2. Over-expression of dominant negative CBP lacking constitutive HAT activity (CBP-) attenuated CSE-induced HDAC2 acetylation. The loss of acetylation on HDAC2 serine mutants indicated phosphorylation was an absolute requirement for acetylation and not vice versa.
HDAC2 was first discovered as a negative regulator of gene transcription [55], it was thus important to determine whether HDAC2 phosphorylation modulated HDAC2 functional activity. HDAC2 functional activity, as measured by repression of luciferase gene expression, was slightly higher in response to oxidant-derived CSE. This was consistent with earlier findings showing serine site HDAC2 mutants efficiently repressed gene function as well as wild-type HDAC2 [17]. The increased HDAC2 transrepression activity with wild-type CBP over-expression suggests acetylation and not phosphorylation is the dominant regulator of HDAC2 functional activity. We speculate that HDAC2 phosphorylation leads to the recruitment of co-repressor proteins and transcription factors with intrinsic HAT activity such as p300/CBP leading to HDAC2 acetylation with the goal of shutting down transcription of active pro-inflammatory genes turned on in response to oxidative stress. This is supported by reports indicating co-repressor proteins are actively involved in recruiting HDAC2 to promoters [56].
There is sufficient evidence implicating CK2 as a potential pro-inflammatory kinase. Apart from being induced by stress signals such as UV radiation [57], CK2 induces phosphorylation and rapid turnover of IκBα leading to nuclear accumulation of RelA/p65 [58]. Increased RelA/p65 transcriptional activity in response to TNFα has also been shown to involve CK2-mediated interaction and phosphorylation of RelA/p65 [59]. Since we had earlier shown that CK2-mediated phosphorylation of HDAC2 induces subsequent proteasomal degradation [19] and our current data suggests decreased HDAC2 activity with increased serine phosphorylation, we speculate that inhibition of CK2 may be a crucial step in reversing steroid resistance associated with a decline in HDAC2 protein expression and aberrant RelA/p65 activation. However, determining whether inhibition of CK2 could reverse cigarette smoke-mediated steroid resistance in our model is beyond the scope of this work and is currently being investigated.
In summary, we describe a novel mechanism of oxidant-mediated regulation of HDAC2 via protein kinase CK2-mediated phosphorylation (Fig 8). CS-derived oxidants and aldehydes induce phosphatase-independent HDAC2 phosphorylation on serine sites S394, S422, S424 which requires interaction with CK2 catalytic but not regulatory subunits that are almost entirely exclusive to the cytoplasm. CK2α phosphorylation of HDAC2 is associated with serine phosphorylation of CK2α both in response to oxidants and increasing intracellular phosphorylation with okadaic acid treatment. Pharmacological inhibition and CK2α silencing suggests this is uniquely mediated by catalytic but not the regulatory subunits of the protein kinase CK2 holoenzyme which are almost exclusively localized in the cytoplasm. HDAC2 phosphorylation is absolutely required for co-repressor complex formation, interaction with transcription factors and decreased deacetylase activity. We also describe a novel acetylation of HDAC2 which requires phosphorylation at specific HDAC2 serine residues, which is mediated by the recruitment of CBP and is involved in modulating HDAC2 transrepression. These findings may have implications in understanding steroid resistance in response to oxidant/aldehyde-induced stress and devising pharmacological therapies for patients with COPD and severe asthmatics.
Figure 8. Schematic for oxidant/aldehyde regulation of HDAC2.

Schematic for cigarette smoke/oxidant/aldehyde-mediated regulation of HDAC2. CK2α-mediated phosphorylation of HDAC2 induces increased co-repressor complex formation and transcription factor recruitment. Phospho-acetylation of HDAC2 leads to decreased in its deacetylase activity and hence steroid resistance.
Acknowledgments
This study was supported by the NIH-NHLBI Grant R01-HL-085613, NIEHS Toxicology training grant ES-07026 and NIEHS Environmental Health Sciences Center Grant ES-01247. We thank Dr. E. Seto for the HDAC2 flag-tagged wild-type and mutant plasmids; Dr. S. Ghosh for the CBP plasmids; Dr. Chawnshang Chang for the pCMX-Gal4 plasmids and Drs. G. Pryhuber, J. Finkelstein and S. Maggirwar for their advice during the course of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sengupta N, Seto E. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 3.Villagra A, Sotomayor EM. E. Seto Oncogene. 29:157–173. doi: 10.1038/onc.2009.334. [DOI] [PubMed] [Google Scholar]
- 4.Kramer OH. Trends Pharmacol Sci. 2009;30:647–655. doi: 10.1016/j.tips.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- 6.Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, Howard BH. J Biol Chem. 2001;276:6817–6824. doi: 10.1074/jbc.M007372200. [DOI] [PubMed] [Google Scholar]
- 7.Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- 8.Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Biochem Biophys Res Commun. 2004;315:240–245. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 9.Keatings VM, Jatakanon A, Worsdell YM, Barnes PJ. Am J Respir Crit Care Med. 1997;155:542–548. doi: 10.1164/ajrccm.155.2.9032192. [DOI] [PubMed] [Google Scholar]
- 10.Osoata GO, Yamamura S, Ito M, Vuppusetty C, Adcock IM, Barnes PJ, Ito K. Biochem Biophys Res Commun. 2009;384:366–371. doi: 10.1016/j.bbrc.2009.04.128. [DOI] [PubMed] [Google Scholar]
- 11.Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–L57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 12.Rhen T, Cidlowski JA. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 13.Adcock IM, Ito K. Curr Opin Pharmacol. 2004;4:257–262. doi: 10.1016/j.coph.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Pryor WA, Stone K. Ann N Y Acad Sci. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. [DOI] [PubMed] [Google Scholar]
- 15.Rahman I, Adcock IM. Eur Respir J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 16.Pluemsampant S, Safronova OS, Nakahama K, Morita I. Int J Cancer. 2008;122:333–341. doi: 10.1002/ijc.23094. [DOI] [PubMed] [Google Scholar]
- 17.Tsai SC, Seto E. J Biol Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- 18.Kode A, Yang SR, Rahman I. Respir Res. 2006;7:132. doi: 10.1186/1465-9921-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Am J Respir Cell Mol Biol. 2009;40:464–473. doi: 10.1165/rcmb.2008-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galasinski SC, Resing KA, Goodrich JA, Ahn NG. J Biol Chem. 2002;277:19618–19626. doi: 10.1074/jbc.M201174200. [DOI] [PubMed] [Google Scholar]
- 21.Filhol O, Cochet C. Cell Mol Life Sci. 2009;66:1830–1839. doi: 10.1007/s00018-009-9151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinna LA. Int J Biochem Cell Biol. 1997;29:551–554. doi: 10.1016/s1357-2725(96)00142-2. [DOI] [PubMed] [Google Scholar]
- 23.Sun JM, Chen HY, Moniwa M, Litchfield DW, Seto E, Davie JR. J Biol Chem. 2002;277:35783–35786. doi: 10.1074/jbc.C200378200. [DOI] [PubMed] [Google Scholar]
- 24.Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA. FEBS Lett. 2001;496:44–48. doi: 10.1016/s0014-5793(01)02404-8. [DOI] [PubMed] [Google Scholar]
- 25.Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ, George DL. Genes Dev. 1999;13:2490–2501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashburner BP, Westerheide SD, Baldwin AS., Jr Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X, Ito A, Kane CD, Liao TS, Bolger TA, Lemrow SM, Means AR, Yao TP. J Biol Chem. 2001;276:35042–35048. doi: 10.1074/jbc.M105086200. [DOI] [PubMed] [Google Scholar]
- 28.Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, Hendarwanto A, Martinez ED, Chen Y, Lu H, Adkins NL, Stavreva DA, Wiench M, Georgel PT, Schiltz RL, Hager GL. Mol Cell. 2006;22:669–679. doi: 10.1016/j.molcel.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McInerney EM, Mullen TM, Glass CK, Rosenfeld MG. Science. 1998;279:703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- 31.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 32.Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alsaeedi A, Sin DD, McAlister FA. Am J Med. 2002;113:59–65. doi: 10.1016/s0002-9343(02)01143-9. [DOI] [PubMed] [Google Scholar]
- 34.Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthews JL, Barnes PJ. Am J Respir Crit Care Med. 1999;160:1635–1639. doi: 10.1164/ajrccm.160.5.9811058. [DOI] [PubMed] [Google Scholar]
- 35.Hattotuwa KL, Gizycki MJ, Ansari TW, Jeffery PK, Barnes NC. Am J Respir Crit Care Med. 2002;165:1592–1596. doi: 10.1164/rccm.2105025. [DOI] [PubMed] [Google Scholar]
- 36.Cosio BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF, Adcock IM. Am J Respir Crit Care Med. 2004;170:141–147. doi: 10.1164/rccm.200305-659OC. [DOI] [PubMed] [Google Scholar]
- 37.Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. J Exp Med. 2004;200:689–695. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes PJ. Annu Rev Physiol. 2009;71:451–464. doi: 10.1146/annurev.physiol.010908.163257. [DOI] [PubMed] [Google Scholar]
- 39.Barnes PJ, Adcock IM. Lancet. 2009;373:1905–1917. doi: 10.1016/S0140-6736(09)60326-3. [DOI] [PubMed] [Google Scholar]
- 40.Vilk G, Weber JE, Turowec JP, Duncan JS, Wu C, Derksen DR, Zien P, Sarno S, Donella-Deana A, Lajoie G, Pinna LA, Li SS, Litchfield DW. Cell Signal. 2008;20:1942–1951. doi: 10.1016/j.cellsig.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Litchfield DW. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinna LA. J Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 43.Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, Waterfield MD. Biochem J. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foukas LC, Daniele N, Ktori C, Anderson KE, Jensen J, Shepherd PR. J Biol Chem. 2002;277:37124–37130. doi: 10.1074/jbc.M202101200. [DOI] [PubMed] [Google Scholar]
- 45.Ukena D, Harnest U, Sakalauskas R, Magyar P, Vetter N, Steffen H, Leichtl S, Rathgeb F, Keller A, Steinijans VW. Eur Respir J. 1997;10:2754–2760. doi: 10.1183/09031936.97.10122754. [DOI] [PubMed] [Google Scholar]
- 46.Ito K, Lim S, Caramori G, Cosio B, Chung KF, Adcock IM, Barnes PJ. Proc Natl Acad Sci U S A. 2002;99:8921–8926. doi: 10.1073/pnas.132556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marwick JA, Caramori G, Stevenson CS, Casolari P, Jazrawi E, Barnes PJ, Ito K, Adcock IM, Kirkham PA, Papi A. Am J Respir Crit Care Med. 2009;179:542–548. doi: 10.1164/rccm.200810-1570OC. [DOI] [PubMed] [Google Scholar]
- 48.Guerra B. Int J Oncol. 2006;28:685–693. [PubMed] [Google Scholar]
- 49.Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, Marwick JA, Chakravarty P, Fletcher D, Whittaker P, Megson IL, Kirkham PA, Rahman I. Am J Respir Cell Mol Biol. 2008;39:312–323. doi: 10.1165/rcmb.2008-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Insinga A, Monestiroli S, Ronzoni S, Carbone R, Pearson M, Pruneri G, Viale G, Appella E, Pelicci P, Minucci S. EMBO J. 2004;23:1144–1154. doi: 10.1038/sj.emboj.7600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun JM, Chen HY, Davie JR. J Biol Chem. 2007;282:33227–33236. doi: 10.1074/jbc.M703549200. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Randall WR, Schneider MF. J Cell Biol. 2005;168:887–897. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paroni G, Cernotta N, Dello Russo C, Gallinari P, Pallaoro M, Foti C, Talamo F, Orsatti L, Steinkuhler C, Brancolini C. Mol Biol Cell. 2008;19:655–667. doi: 10.1091/mbc.E07-06-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo Y, Jian W, Stavreva D, Fu X, Hager GL, Bungert J, Huang S, Qiu Y. J Biol Chem. 2009;284:901–910. doi: 10.1074/jbc.M109.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang WM, Inouye C, Zeng Y, Bearss D, Seto E. Proc Natl Acad Sci U S A. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 57.Kato T, Jr, Delhase M, Hoffmann A, Karin M. Mol Cell. 2003;12:829–839. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- 58.McElhinny JA, Trushin SA, Bren GD, Chester N, Paya CV. Mol Cell Biol. 1996;16:899–906. doi: 10.1128/mcb.16.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr J Biol Chem. 2000;275:32592–32597. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]