

Abstract
Previous Ussing chamber measurements of secretagogue-provoked changes in short circuit current in rectal suction biopsies of cystic fibrosis (CF) patients showed that in a minority of patients chloride secretion in response to cholinergic agonists is reduced but not completely absent. To assess a possible relationship between this phenomenon and both the genotype and the phenotype, we performed Ussing chamber experiments on rectal suction biopsies of 51 CF patients. The CF mutation was identified in 89 out of 102 CF alleles. No apparent chloride secretion was found in 30 CF patients (group I). Low residual chloride secretion was found in 11 CF patients (group II), while a relatively high residual secretion appeared in 10 CF patients (group III). Pancreatic function was preserved more frequently in CF patients displaying residual secretion: 0% in group I, 27% in group II, and 60% in group III (P < 0.001). The age at diagnosis (mean +/- SEM) in group III (18.4 +/- 6.6) was significantly different from group I (1.2 +/- 0.4, P < 0.01) and group II (3.5 +/- 1.4, P = 0.05). Residual chloride secretion was found in some of the 28 dF508 homozygous patients (three in group II, and one in group III), disclosing that other factors than the CF gene defect itself affect the transepithelial chloride transport. The age at diagnosis correlates significantly with the magnitude of the secretory response, even within the dF508 homozygous patients (r = 0.4, P < 0.05). We conclude that residual chloride secretion in CF is the pathophysiological basis of preserved pancreatic function and delayed presentation of the disease, which is not exclusively determined by the CF genotype.
Full text
PDF
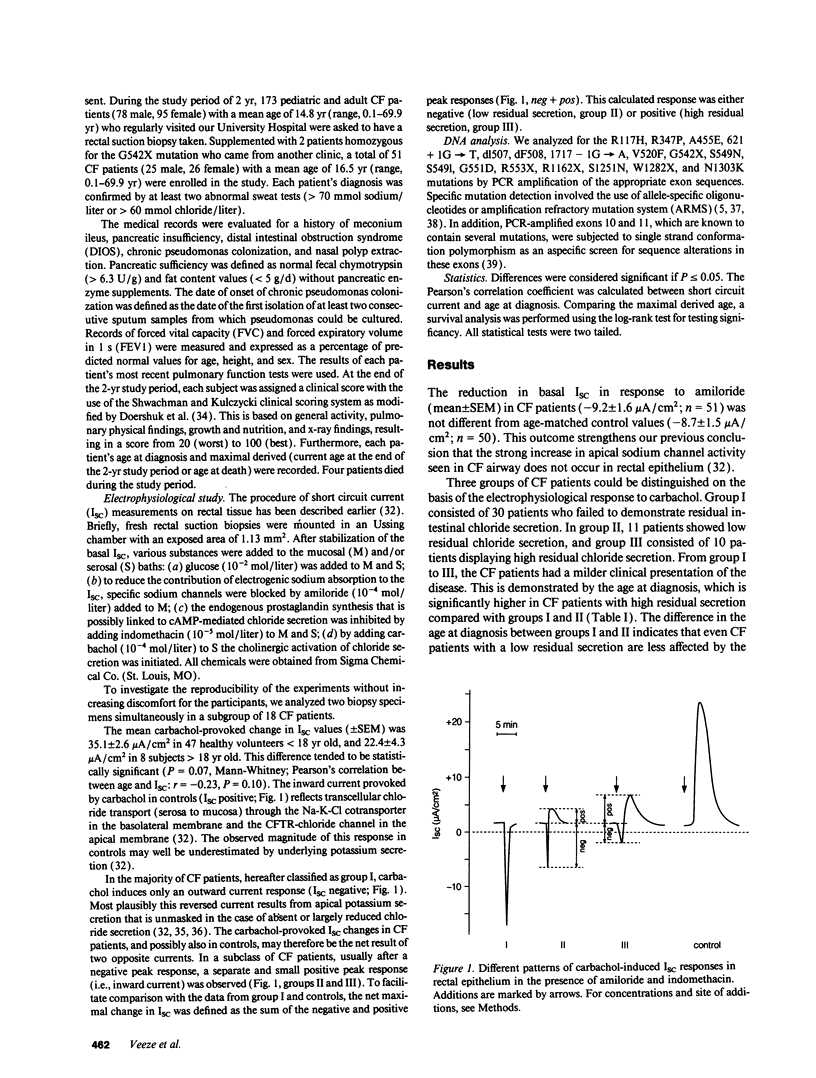
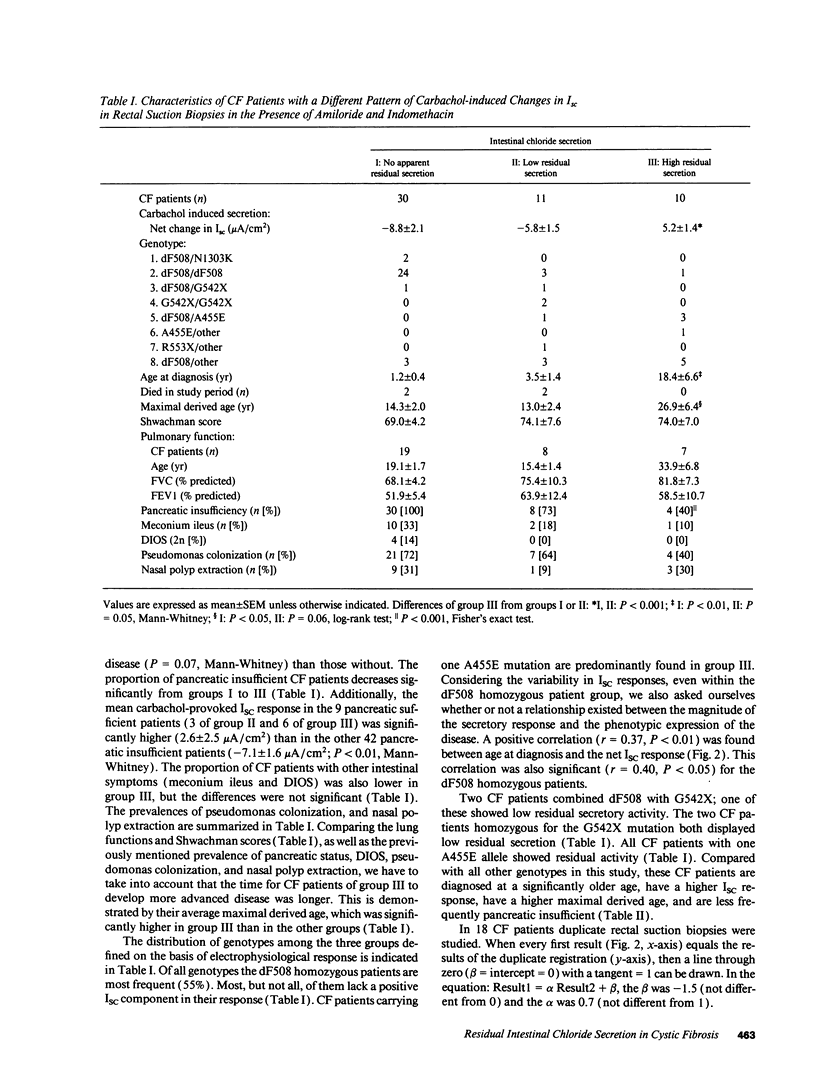
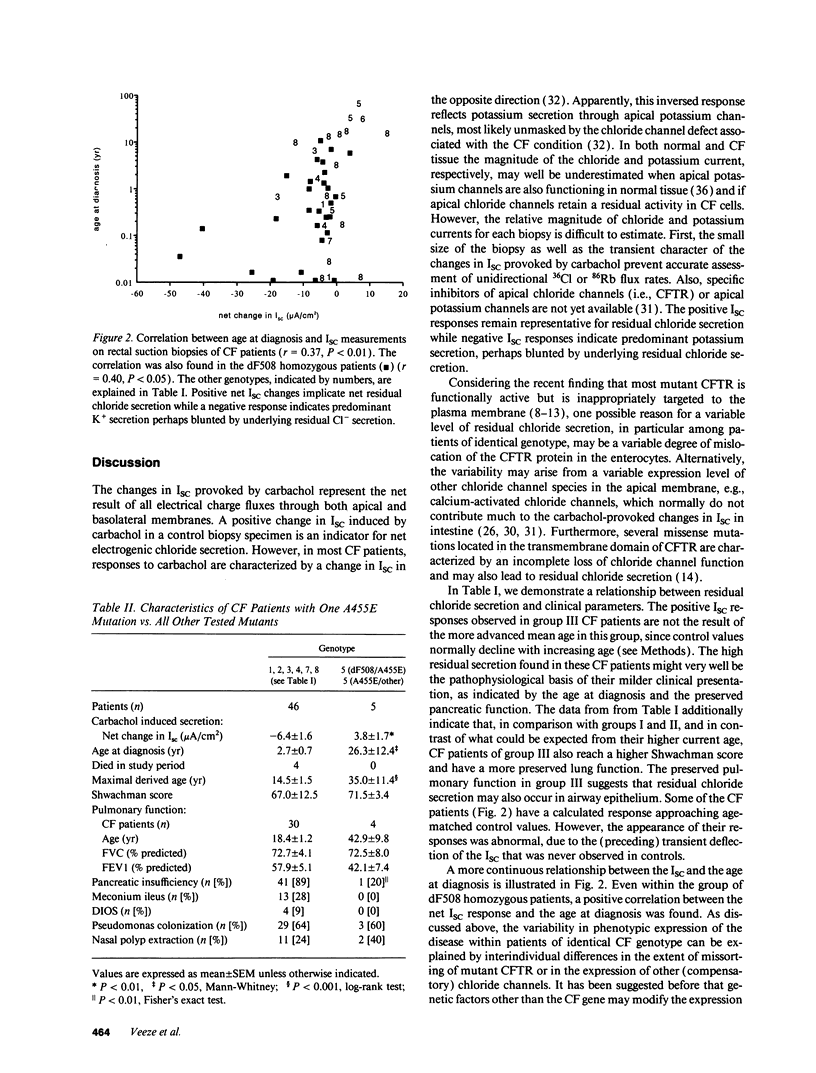


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., Welsh M. J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991 Jul 12;253(5016):202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Anderson M. P., Welsh M. J. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc Natl Acad Sci U S A. 1991 Jul 15;88(14):6003–6007. doi: 10.1073/pnas.88.14.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajnath R. B., Dekker K., Vaandrager A. B., de Jonge H. R., Groot J. A. Biphasic increase of apical Cl- conductance by muscarinic stimulation of HT-29cl.19A human colon carcinoma cell line: evidence for activation of different Cl- conductances by carbachol and forskolin. J Membr Biol. 1992 Apr;127(2):81–94. doi: 10.1007/BF00233281. [DOI] [PubMed] [Google Scholar]
- Bear C. E., Li C. H., Kartner N., Bridges R. J., Jensen T. J., Ramjeesingh M., Riordan J. R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell. 1992 Feb 21;68(4):809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Berger H. A., Travis S. M., Welsh M. J. Regulation of the cystic fibrosis transmembrane conductance regulator Cl- channel by specific protein kinases and protein phosphatases. J Biol Chem. 1993 Jan 25;268(3):2037–2047. [PubMed] [Google Scholar]
- Berschneider H. M., Knowles M. R., Azizkhan R. G., Boucher R. C., Tobey N. A., Orlando R. C., Powell D. W. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988 Jul;2(10):2625–2629. doi: 10.1096/fasebj.2.10.2838365. [DOI] [PubMed] [Google Scholar]
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990 Nov 16;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- DOERSHUK C. F., MATTHEWS L. W., TUCKER A. S., NUDLEMAN H., EDDY G., WISE M., SPECTOR S. A 5 YEAR CLINICAL EVALUATION OF A THERAPEUTIC PROGRAM FOR PATIENTS WITH CYSTIC FIBROSIS. J Pediatr. 1964 Nov;65:677–693. doi: 10.1016/s0022-3476(64)80152-9. [DOI] [PubMed] [Google Scholar]
- Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992 Aug 27;358(6389):761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Dharmsathaphorn K., Pandol S. J. Mechanism of chloride secretion induced by carbachol in a colonic epithelial cell line. J Clin Invest. 1986 Feb;77(2):348–354. doi: 10.1172/JCI112311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drumm M. L., Wilkinson D. J., Smit L. S., Worrell R. T., Strong T. V., Frizzell R. A., Dawson D. C., Collins F. S. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991 Dec 20;254(5039):1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Ferrie R. M., Schwarz M. J., Robertson N. H., Vaudin S., Super M., Malone G., Little S. Development, multiplexing, and application of ARMS tests for common mutations in the CFTR gene. Am J Hum Genet. 1992 Aug;51(2):251–262. [PMC free article] [PubMed] [Google Scholar]
- Frizzell R. A., Rechkemmer G., Shoemaker R. L. Altered regulation of airway epithelial cell chloride channels in cystic fibrosis. Science. 1986 Aug 1;233(4763):558–560. doi: 10.1126/science.2425436. [DOI] [PubMed] [Google Scholar]
- Goldstein J. L., Shapiro A. B., Rao M. C., Layden T. J. In vivo evidence of altered chloride but not potassium secretion in cystic fibrosis rectal mucosa. Gastroenterology. 1991 Oct;101(4):1012–1019. doi: 10.1016/0016-5085(91)90728-4. [DOI] [PubMed] [Google Scholar]
- Greenwald L., Biagi B. A. Interaction between carbachol and vasoactive intestinal peptide in cells of isolated colonic crypts. Am J Physiol. 1992 May;262(5 Pt 1):G940–G944. doi: 10.1152/ajpgi.1992.262.5.G940. [DOI] [PubMed] [Google Scholar]
- Halley D. J., Veeze H. J., Sandkuyl L. A., Wesby-van Swaay E., van Damme N. H., Deelen W. H., Witte J. E., Niermeijer M. F. The mutation delta F508 on Dutch cystic fibrosis chromosomes: frequency and relation to patients age at diagnosis. Hum Genet. 1990 Sep;85(4):407–408. doi: 10.1007/BF02428281. [DOI] [PubMed] [Google Scholar]
- Halley D. J., van Damme N. H., Deelen W. H., Oostra B. A., Jahoda M. G., Sachs E. S., Los F. J., Niermeijer M. F. Prenatal detection of major cystic fibrosis mutation. Lancet. 1989 Oct 21;2(8669):972–972. doi: 10.1016/s0140-6736(89)90973-2. [DOI] [PubMed] [Google Scholar]
- Halm D. R., Frizzell R. A. Active K transport across rabbit distal colon: relation to Na absorption and Cl secretion. Am J Physiol. 1986 Aug;251(2 Pt 1):C252–C267. doi: 10.1152/ajpcell.1986.251.2.C252. [DOI] [PubMed] [Google Scholar]
- Hamosh A., Trapnell B. C., Zeitlin P. L., Montrose-Rafizadeh C., Rosenstein B. J., Crystal R. G., Cutting G. R. Severe deficiency of cystic fibrosis transmembrane conductance regulator messenger RNA carrying nonsense mutations R553X and W1316X in respiratory epithelial cells of patients with cystic fibrosis. J Clin Invest. 1991 Dec;88(6):1880–1885. doi: 10.1172/JCI115510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen H. K., Nir M., Høiby N., Koch C., Schwartz M. Severity of cystic fibrosis in patients homozygous and heterozygous for delta F508 mutation. Lancet. 1991 Mar 16;337(8742):631–634. doi: 10.1016/0140-6736(91)92449-c. [DOI] [PubMed] [Google Scholar]
- Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat Genet. 1992 Aug;1(5):321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kerem E., Corey M., Kerem B. S., Rommens J., Markiewicz D., Levison H., Tsui L. C., Durie P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). N Engl J Med. 1990 Nov 29;323(22):1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- Knowles M. R., Stutts M. J., Spock A., Fischer N., Gatzy J. T., Boucher R. C. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science. 1983 Sep 9;221(4615):1067–1070. doi: 10.1126/science.6308769. [DOI] [PubMed] [Google Scholar]
- Kristidis P., Bozon D., Corey M., Markiewicz D., Rommens J., Tsui L. C., Durie P. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet. 1992 Jun;50(6):1178–1184. [PMC free article] [PubMed] [Google Scholar]
- Li C., Ramjeesingh M., Reyes E., Jensen T., Chang X., Rommens J. M., Bear C. E. The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet. 1993 Apr;3(4):311–316. doi: 10.1038/ng0493-311. [DOI] [PubMed] [Google Scholar]
- Orita M., Suzuki Y., Sekiya T., Hayashi K. Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics. 1989 Nov;5(4):874–879. doi: 10.1016/0888-7543(89)90129-8. [DOI] [PubMed] [Google Scholar]
- Osborne L., Santis G., Schwarz M., Klinger K., Dörk T., McIntosh I., Schwartz M., Nunes V., Macek M., Jr, Reiss J. Incidence and expression of the N1303K mutation of the cystic fibrosis (CFTR) gene. Hum Genet. 1992 Aug;89(6):653–658. doi: 10.1007/BF00221957. [DOI] [PubMed] [Google Scholar]
- Puchelle E., Gaillard D., Ploton D., Hinnrasky J., Fuchey C., Boutterin M. C., Jacquot J., Dreyer D., Pavirani A., Dalemans W. Differential localization of the cystic fibrosis transmembrane conductance regulator in normal and cystic fibrosis airway epithelium. Am J Respir Cell Mol Biol. 1992 Nov;7(5):485–491. doi: 10.1165/ajrcmb/7.5.485. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 Sep 8;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Santis G., Osborne L., Knight R. A., Hodson M. E. Independent genetic determinants of pancreatic and pulmonary status in cystic fibrosis. Lancet. 1990 Nov 3;336(8723):1081–1084. doi: 10.1016/0140-6736(90)92566-z. [DOI] [PubMed] [Google Scholar]
- Santis G., Osborne L., Knight R. A., Hodson M. E. Linked marker haplotypes and the delta F508 mutation in adults with mild pulmonary disease and cystic fibrosis. Lancet. 1990 Jun 16;335(8703):1426–1429. doi: 10.1016/0140-6736(90)91448-j. [DOI] [PubMed] [Google Scholar]
- Sheppard D. N., Rich D. P., Ostedgaard L. S., Gregory R. J., Smith A. E., Welsh M. J. Mutations in CFTR associated with mild-disease-form Cl- channels with altered pore properties. Nature. 1993 Mar 11;362(6416):160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- Tabcharani J. A., Chang X. B., Riordan J. R., Hanrahan J. W. Phosphorylation-regulated Cl- channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991 Aug 15;352(6336):628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- Taylor C. J., Baxter P. S., Hardcastle J., Hardcastle P. T. Failure to induce secretion in jejunal biopsies from children with cystic fibrosis. Gut. 1988 Jul;29(7):957–962. doi: 10.1136/gut.29.7.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. J., Hughes H., Hardcastle P. T., Hardcastle J. Genotype and secretory response in cystic fibrosis. Lancet. 1992 Jan 4;339(8784):67–68. doi: 10.1016/0140-6736(92)90201-d. [DOI] [PubMed] [Google Scholar]
- Tsui L. C. The spectrum of cystic fibrosis mutations. Trends Genet. 1992 Nov;8(11):392–398. doi: 10.1016/0168-9525(92)90301-j. [DOI] [PubMed] [Google Scholar]
- Vaandrager A. B., Bajnath R., Groot J. A., Bot A. G., De Jonge H. R. Ca2+ and cAMP activate different chloride efflux pathways in HT-29.cl19A colonic epithelial cell line. Am J Physiol. 1991 Dec;261(6 Pt 1):G958–G965. doi: 10.1152/ajpgi.1991.261.6.G958. [DOI] [PubMed] [Google Scholar]
- Vaandrager A. B., van den Berghe N., Bot A. G., de Jonge H. R. Phorbol esters stimulate and inhibit Cl- secretion by different mechanisms in a colonic cell line. Am J Physiol. 1992 Feb;262(2 Pt 1):G249–G256. doi: 10.1152/ajpgi.1992.262.2.G249. [DOI] [PubMed] [Google Scholar]
- Veeze H. J., Sinaasappel M., Bijman J., Bouquet J., de Jonge H. R. Ion transport abnormalities in rectal suction biopsies from children with cystic fibrosis. Gastroenterology. 1991 Aug;101(2):398–403. doi: 10.1016/0016-5085(91)90017-f. [DOI] [PubMed] [Google Scholar]
- Welsh M. J., Liedtke C. M. Chloride and potassium channels in cystic fibrosis airway epithelia. 1986 Jul 31-Aug 6Nature. 322(6078):467–470. doi: 10.1038/322467a0. [DOI] [PubMed] [Google Scholar]