

Abstract
Metastatic prostate cancer is treated with drugs that antagonize androgen action but most patients progress to a more aggressive form of the disease called castration-resistant prostate cancer, driven by elevated expression of the androgen receptor. Here we characterize the diarylthiohydantoins RD162 and MDV3100, two compounds optimized from a screen for non-steroidal antiandrogens that retain activity in the setting of increased androgen receptor expression. Both compounds bind to the androgen receptor with greater relative affinity than the clinically used antiandrogen bicalutamide, reduce the efficiency of its nuclear translocation and impair both DNA binding to androgen response elements and recruitment of coactivators. RD162 and MDV3100 are orally available and induce tumor regression in mouse models of castration-resistant human prostate cancer. Of the first 30 patients treated with MDV3100 in a phase I/II clinical trial, 13 of 30 (43 percent) showed sustained declines (by >50 percent) in serum levels of prostate specific antigen, a biomarker of prostate cancer. These compounds thus appear to be promising candidates for treatment of advanced prostate cancer.
Treatment of advanced prostate cancer is limited by the development of resistance to antiandrogen therapy. Castration-resistant prostate cancer (CRPC) is commonly associated with increased levels of androgen receptor (AR) gene expression, which can occur through AR gene amplification or other mechanisms (1, 2). Elevated AR levels are necessary and sufficient to confer resistance to antiandrogen therapy in mouse xenograft models (3). In addition, first generation AR antagonists such as bicalutamide (also called Casodex®) or flutamide demonstrate agonist properties in cells engineered to express higher AR levels. The partial agonism of these compounds is a potential liability, best illustrated clinically by the antiandrogen withdrawal response in which serum levels of prostate specific antigen (PSA) decline in patients after discontinuation of either of these AR antagonists (4). Collectively, these findings implicate increased AR levels as a molecular cause of drug resistance and suggest that second generation antiandrogens might be identified by their ability to retain antagonism in cells expressing excess AR.
Our earlier mutagenesis studies revealed that increased AR levels conferred resistance to antiandrogens in model systems only when the receptor contains a functional ligand binding domain (LBD) (3). Second generation antiandrogens could, in theory, be optimized to exploit this well characterized LBD. Co-crystal structures of wild-type AR bound to antagonists have not been solved, but a co-crystal of bicalutamide with mutant AR (in an agonist conformation), together with structural knowledge of estrogen receptor (ER)-α antagonists (5), suggests a steric clash mechanism in which the bulky phenyl ring on bicalutamide leads to a partial unfolding of AR (6). However, bicalutamide has relatively low affinity for AR (at least 30-fold reduced relative to the natural ligand dihydrotestosterone (DHT)) (7), suggesting that antagonism could be optimized by improved binding characteristics.
To search for improved antiandrogens, we selected the non-steroidal agonist RU59063 as a starting chemical scaffold on the basis of its relatively high affinity for AR (only 3-fold reduced compared to testosterone) and selectivity for AR over other nuclear hormone receptors (8, 9). Through an iterative process to be described in detail separately (see also US Patent Application 20070004753), we evaluated nearly 200 thiohydantoin derivatives of RU59063 for AR agonism and antagonism in human prostate cancer cells engineered to express increased levels of AR. Based on these structure activity relationships and further chemical modifications to improve serum half-life and oral bioavailability, the diarylthiohydantoins RD162 and MDV3100 were selected as the lead compounds for further biological studies (Fig. 1A). In a competition assay using 16β-[18F]fluoro-5α-DHT (18-FDHT) to measure relative AR binding affinity (10), both RD162 and MDV3100 bound AR in castration-resistant LNCaP/AR human prostate cancer cells (engineered to express higher levels of wild-type AR to mimic the clinical scenario) with 5-8 fold greater affinity than bicalutamide and only 2-3 fold reduced affinity relative to the derivative of the native ligand FDHT (Fig. 1B). RD162 binding to AR was specific, as there was little to no binding to the progesterone, estrogen or glucocorticoid receptors in an in vitro fluorescence polarization assay (table S1). We next compared the effects of RD162 and MDV3100 versus bicalutamide on androgen-dependent gene expression in LNCaP/AR cells. Expression of the AR _target genes PSA and transmembrane serine protease 2 (TMPRSS2) was induced by bicalutamide but not by RD162 or MDV3100 (Fig. 1C), indicating that RD162 and MDV3100 do not have agonist activity in a castration-resistant setting. Both RD162 and MDV3100 antagonized induction of PSA and TMPRSS2 by the synthetic androgen R1881 in parental LNCaP cells (fig. S1). In the human prostate cancer cell line VCaP which has endogenous AR gene amplification (11), RD162 and MDV3100 suppressed growth and induced apoptosis whereas bicalutamide did not (Fig. 1D, E). This growth suppression was reversed by co-treatment with the synthetic androgen R1881 which competes for AR binding (fig S2A) and was not observed in the AR-negative DU145 human prostate cancer cells (fig S2B). In addition, RD162 and MDV3100 inhibited the transcriptional activity of a mutant AR protein (W741C) isolated from a patient with acquired resistance to bicalutamide (fig. S3). The W741C substitution in the AR LBD causes bicalutamide to act as a pure agonist (12).
Fig. 1.
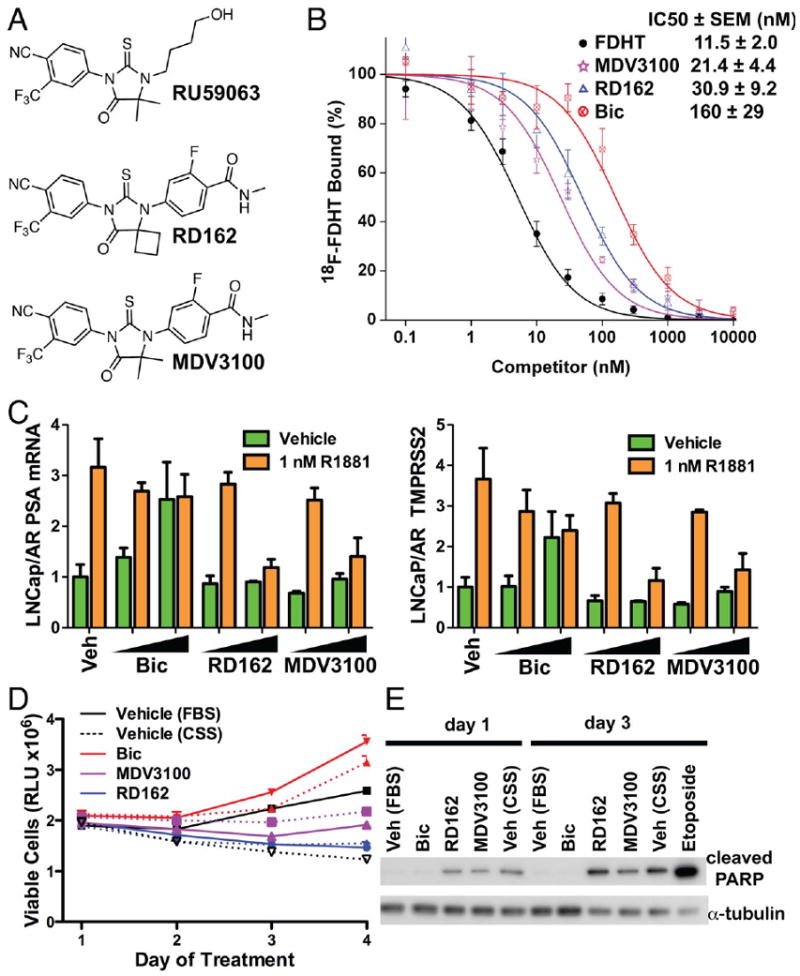
Effect of RD162 and MDV3100 in human prostate cancer cells in vitro. (A) Chemical structures of the parent arylthiohydantoin scaffold compound RU59063 and the novel AR antagonists RD162 and MDV3100. (B) Representative competition binding curve showing inhibition of 18F-FDHT equilibrium binding to AR by FDHT, RD162, MDV3100 and bicalutamide (Bic) in LNCaP/AR cells. The IC50 values from this experiment were: 5.1 nM (FDHT), 36 nM (MDV3100), 50 nM (RD162) and 159 nM (Bic) (error bars are reported as standard deviation of triplicate measurements). The inset shows the mean IC50 values (± SEM) from five replicate experiments. (C) qRT-PCR analysis of the AR-dependent genes PSA and TMPRSS2 in LNCaP/AR cells cultured in androgen-depleted media with 5% charcoal-stripped serum (CSS). Cells were treated for 8 hours with or without 1nM of the synthetic androgen R1881 combined with DMSO (Veh), bicalutamide (Bic, 1 and 10 μM), RD162 (1 and 10 μM) and MDV3100 (1 and 10 μM) (normalized to actin mRNA, Mean ± SD, n=3). (D) Effect of bicalutamide, RD162 or MDV3100 on cell proliferation. VCaP cells were treated with the indicated antiandrogen and concentration (dashed line = 1 μM, solid line = 10 μM) in media containing fetal bovine serum (FBS). Vehicle-treated cells were in media containing either FBS or CSS. The viable cell fraction was determined by CellTiter-GLO (n=3, error is SEM). (E) Effect of bicalutamide, RD162 or MDV3100 on cleavage of poly-ADP-ribose polymerase (PARP). VCaP cells were treated for 1 or 3 days with 10 μM antiandrogen in media containing FBS. Vehicle-treated cells were in media containing either FBS or CSS. Cells treated with 10 μM etoposide for 1 day served as a positive control for apoptosis. Whole cell lysates were analyzed by Western blot.
To evaluate the activity of RD162 in vivo we first determined its pharmacokinetic properties in mice. RD162 was ∼50 percent bioavailable after oral delivery with a serum half-life of about 30 hours (Fig. 2A, table S2A). Trough levels observed 24 hours after a single 20 mg/kg oral dose (∼23 μM) exceeded concentrations expected to block AR activity based on the in vitro studies (∼1-10 μM). We evaluated the pharmacodynamic effects of RD162 on AR function in vivo by measuring luciferase activity of human LNCaP/AR xenografts grown in castrated male mice. These tumors coexpressed exogenous AR and the AR-dependent reporter construct ARR2-Pb-Luc (13). Luciferase activity was consistently reduced relative to vehicle control in mice treated for 5 days with 10 mg/kg RD162 daily by oral gavage (Fig. 2B), whereas lower doses of 0.1 and 1.0 mg/kg/day had minimal effect (fig. S4A). Commensurate with reduced AR transcriptional function, cellular proliferation of LNCaP/AR xenografts as measured by Ki-67 staining was substantially reduced after 5 days of RD162 treatment (fig. S4B).
Fig. 2.
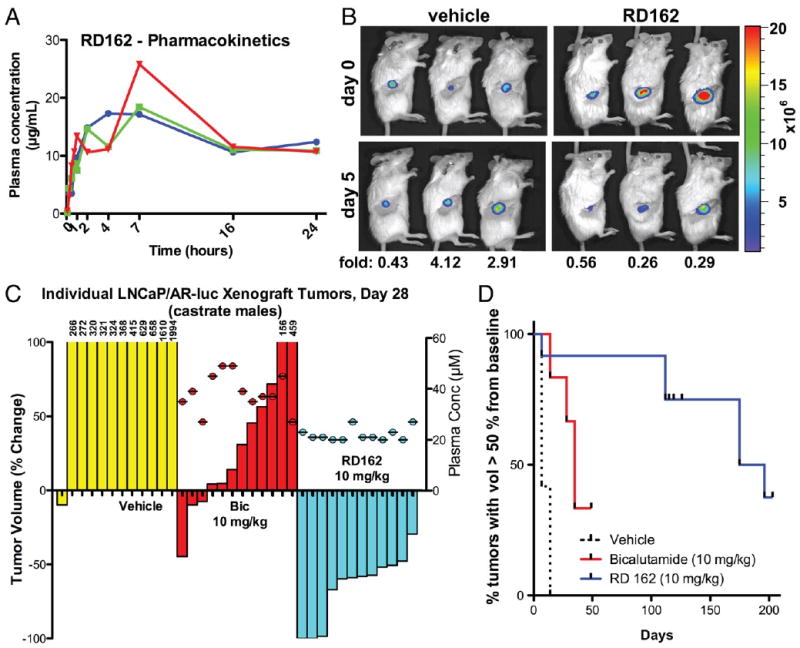
Activity of RD162 in mice. (A) Pharmacokinetic analysis of RD162 in male mice (n=3 per timepoint) dosed by oral gavage as a slurry of 20 mg/kg in 0.5% hydroxy-methyl-propyl-cellulose. (B) Pharmacodynamics of AR antagonism in castrate male mice after 5 days of treatment with daily oral RD162 (10 mg/kg) or vehicle control (n=3 mice per treatment group). Antagonism was measured by luciferase imaging of LNCaP/AR xenograft tumors expressing a luciferase reporter construct driven by the promoter of the probasin gene which is androgen regulated. Light emission at day 5 (2 hours after final dose) was quantified from a region of interest (ROI) drawn over the tumors and normalized for each animal to light emission at day 0. Luciferase activity was quantified at day 5 (normalized for each animal to day 0 luciferase activity) and expressed as fold-change as indicated for the individual mouse. Means ± SD: 2.5 ± 1.9 (Vehicle), 0.4 ± 0.2 (RD162). (C) Effects of RD162 and bicalutamide in a xenograft model of CRPC. Castrate male mice bearing LNCaP/AR tumors > 100 mm3 in size were treated by oral daily gavage with vehicle, bicalutamide (10 mg/kg) or RD162 (10 mg/kg). The percent change in volume for each tumor (12 tumors per treatment group) after 28 days is shown as a waterfall plot (y-axis, left). Plasma levels of bicalutamide and RD162 for each mouse were measured 20 hours after the final dose on day 28 (filled circles above the waterfall plot; y-axis, right). (D) Time to progression in mice continually treated as in (C). The percent change in volume for each tumor (12 tumors per treatment group) was assessed weekly. Events (tumor volume > 50% of baseline) were plotted on a Kaplan-Meier curve. Differences between all three groups were statistically significant: Bic vs R162 (p=0.04), Bic vs Veh (P<0.0001), RD162 vs Veh (P<0.0001) by log-rank (Mantel-Cox) test.
To assess the therapeutic activity of RD162 in CRPC, we measured the effect of daily 10 mg/kg oral RD162 treatment on established LNCaP/AR tumors growing in castrate male mice. After 28 days of treatment, 11 of 12 tumors in vehicle-treated mice increased in size by ∼2-20 fold (Fig. 2C, yellow bars). Bicalutamide retains some activity in this model as one tumor regressed by 50 percent and four tumors did not change substantially in size (stable disease), but the remaining seven tumors progressed on treatment with an increase in tumor volume up to 5-fold (Fig. 2C, red bars). Plasma levels of bicalutamide in all mice exceeded 25 μM (Fig. 2C, see y-axis on right), which is well above concentrations typically achieved in patients (14). In contrast, all twelve tumors in the RD162-treated mice regressed: nine tumors by greater than 50 percent and three tumors that were no longer palpable (Fig. 2C, cyan bars). Median time to tumor progression in the RD162-treated group was 186 days versus 35 days in bicalutamide-treated mice (Fig. 2D). The superiority of RD162 over bicalutamide in this model is unlikely explained by pharmacokinetic properties as plasma levels of RD162 were somewhat lower (mean 24 μM) than those of bicalutamide (mean 40 μM) (Fig. 2C, see y-axis on right). Furthermore, in vitro protein-binding studies revealed a modest increase in protein-bound RD162 and MDV3100 relative to bicalutamide (table S2B). Tumor responses were also observed with MDV3100 (fig. S5) and in castration-resistant LNCaP/HR xenografts derived by serial passage in castrate male mice (rather than by forced AR overexpression) as well as in a distinct castration-resistant xenograft model LAPC4/AR (fig. S6).
Two lines of evidence suggest that the activity of RD162 in these mice is mediated through AR inhibition rather than through off-_target effects. First, antitumor activity in the LNCaP/AR model is dose-dependent, with some slowing of tumor growth at 0.1 mg/kg RD162 and a few tumor regressions at 1 mg/kg (fig. S7), correlating closely with the effect of these same doses on AR transcriptional activity in the luciferase imaging experiment (fig. S4A). Second, neither bicalutamide nor RD162 impaired the growth of AR-negative DU145 prostate cancer xenografts (fig. S8).
In considering mechanisms for the superiority of RD162 and MDV3100 over bicalutamide in these models, we examined potential effects on AR protein expression in vitro and found no obvious changes (fig. S9). Bicalutamide impairs AR transcriptional activity by promoting the assembly of transcription complexes that incorporate corepressors such as NCoR and SMRT rather than coactivators such as SRC1 at the promoters of AR _target genes (15, 16). The partial agonism of bicalutamide revealed in the setting of increased AR expression is associated with AR recruitment to enhancer regions and aberrant recruitment of coactivators to these transcription complexes, leading to _target gene activation rather than repression (3). Since RD162 and MDV3100 do not display agonism in AR-overexpressing cells (Fig. 1C), we explored whether they exert different effects on AR transcription complex assembly. In chromatin immunoprecipitation experiments both R1881 and bicalutamide promoted AR recruitment to the PSA enhancer and TMPRSS2 enhancer in LNCaP/AR cells, whereas only R1881 promoted recruitment in parental LNCaP cells. Neither RD162 nor MDV3100 recruited AR to enhancer regions in either cell line (Fig. 3A, fig S10). To test whether altered AR localization in RD162 or MDV3100-treated cells might explain this result, we visualized transfected AR-EYFP by confocal microscopy in live LNCaP cells. AR-EYFP was predominantly cytoplasmic in the absence of androgen but largely nuclear after treatment with R1881 or bicalutamide (Fig. 3B). The ratio of nuclear versus cytoplasmic AR in RD162- or MDV3100-treated cells was ∼5-fold reduced relative to bicalutamide. To determine whether the nuclear AR in RD162 or MDV3100-treated cells is competent for DNA binding, we used a VP16-AR fusion protein that activates an AR-dependent luciferase reporter. Due to the strong transactivation and nuclear localization properties provided by the VP16 domain, this AR construct is not dependent on ligand induced conformational changes for nuclear localization or coactivator recruitment (17). As expected, wild-type AR was activated only by R1881 and not by the antiandrogens, whereas both R1881 and bicalutamide activated VP16-AR (Fig. 3C). In contrast, neither RD162 nor MDV3100 activated VP16-AR (Fig. 3C), providing further evidence that these compounds impair AR DNA binding. First generation nonsteroidal antiandrogens induce a conformational change in AR that, while distinct from that conferred by the natural ligands testosterone and DHT, remains competent to bind certain LxxLL or FxxLF motif-containing coactivator proteins. The interaction induced by this conformational change can be modeled in vitro and quantified by fluorescence resonance energy transfer (FRET) (18). DHT and bicalutamide both promoted dose-dependent interaction of the AR LBD with an FxxLF containing peptide, whereas RD162 and MDV3100 did not (Fig. 3D). Based on the AR localization, DNA binding and FxxLF peptide interaction studies, we hypothesize that RD162 and MDV3100 induce a conformational change in AR distinct from that induced by bicalutamide. Future crystallographic studies could provide further insight into its mechanism of action and guide efforts to develop additional antiandrogens.
Fig. 3.
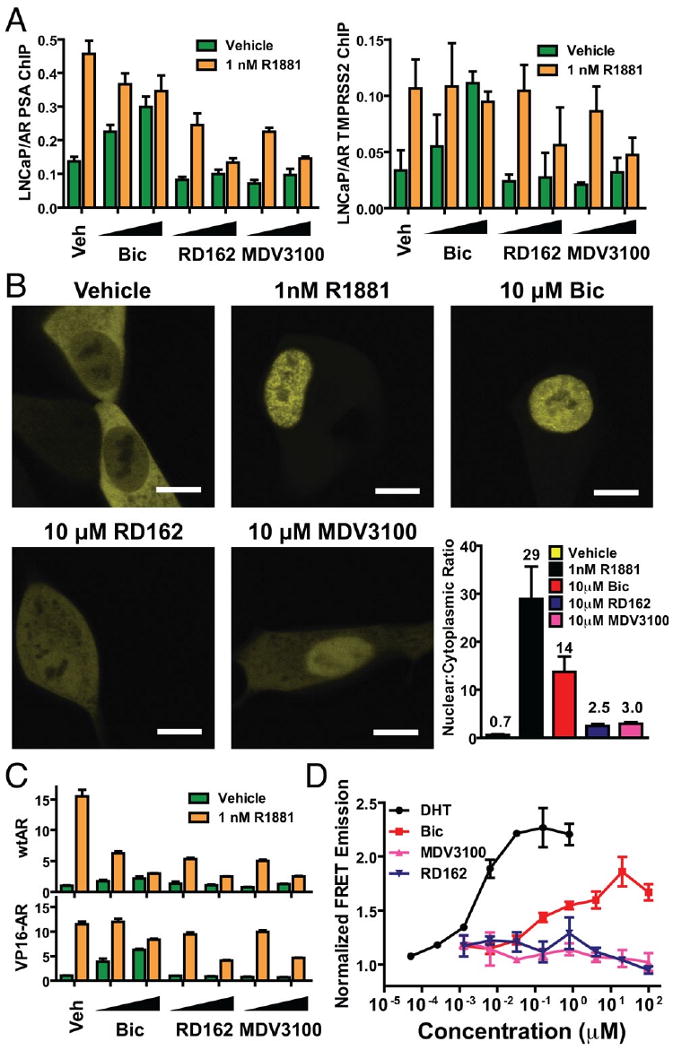
RD162 and MDV3100 impair AR nuclear translocation, DNA binding and coactivator peptide recruitment. (A) Chromatin immunoprecipitation analysis of AR in LNCaP cells. The cells were cultured in androgen-depleted media with 5% CSS and treated for 8 hours with or without 1nM R1881 combined with DMSO (Veh), bicalutamide (Bic, 1 and 10 μM), RD162 (1 and 10 μM) and MDV3100 (1 and 10 μM). Real-time PCR quantification of immunoprecipitated PSA enhancer and TMPRSS2 enhancer is shown (% input mean ± SD, n=3). (B) Representative confocal microscopic images (scale bar = 10 μm) of LNCaP cells transfected with AR-EYFP in androgen-depleted media with 5% CSS and treated with DMSO, 1nM R1881, 10 μM bicalutamide, 10 μM RD162 or 10 μM MDV3100. The ratio of nuclear to cytoplasmic fluorescence intensity of individual cells were calculated (n=3, mean ± SEM). (C) Activation of an androgen-regulated reporter gene by VP16-AR. Cos-7 cells were cotransfected with ARE(4x)-luciferase plasmid and either wild-type AR or VP16-AR fusion protein. Cells were treated for 24 hours with or without 1nM R1881 combined with DMSO (Veh), bicalutamide (Bic, 1 and 10 μM), RD162 (1 and 10 μM) and MDV3100 (1 and 10 μM) for 24 hours. A luciferase assay was conducted using cell lysates and relative light units shown (n=3, mean ± SEM). (D) In vitro fluorescence resonance energy transfer (FRET) analysis of the interaction between AR LBD and FxxLF coactivator peptide. Increasing concentrations of DHT, bicalutamide, RD162 or MDV3100 were incubated with purified AR LBD, terbium-labeled anti-AR antibody and fluorescein-labeled AR FxxLF coactivator peptide (4 replicates). FRET between terbium and fluorescein, indicative of binding of FxxLF peptide to AR-LBD, was measured by ratio of fluorescence emission at 525 nm (fluorescein emission) to 488 nm (terbium emission) after excitation at 322 nm (terbium excitation). Normalized ratio is plotted (mean of 2 experiments with SEM).
Because of its activity in CRPC xenograft models and its favorable drug-like properties, MDV3100 was selected for clinical development. In an ongoing phase I/II clinical trial, 30 men with CRPC who have progressed on first-line antiandrogens, 12 of whom had also failed taxane-based chemotherapy (table S3A and B), received either 30 mg (n=3) or 60 mg (n=27) of daily oral MDV3100. Twenty-two patients had a sustained decline in serum PSA levels for at least 12 weeks, which in 13 of these patients represented a PSA decrease of more than 50 percent (Fig. 4, table S3C). MDV3100 was well tolerated, with 11 patients remaining on study longer than 25 weeks. All discontinuations at these doses were due to disease progression. Results from 110 additional patients who received MDV3100 at higher doses will be reported separately. While preliminary, these clinical data appear promising and validate the persistent role of AR in driving castration-resistant disease.
Fig. 4.
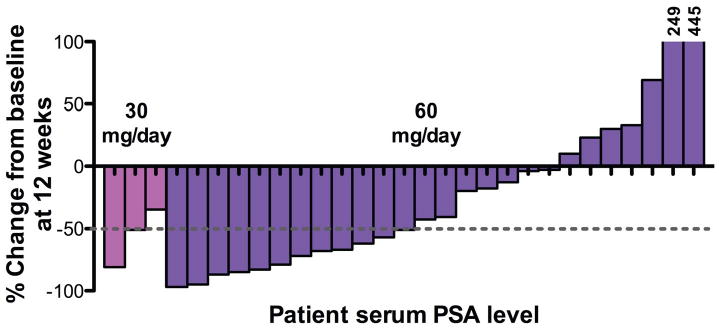
PSA response data in the first 30 patients receiving MDV3100 in Phase I/II trial. Thirty men with CRPC were treated with oral daily MDV3100 at doses of 30 mg/day (n=3) or 60 mg/day (n=27). The percent change in serum PSA for each patient after 12 weeks is shown as a waterfall plot. Patient 30 came off study prior to 12 weeks due to disease progression and is therefore not shown.
Supplementary Material
Acknowledgments
We thank S. Balk and M. Diamond for plasmids, E. De Stanchina, N. Wu, Q. Weige and C. Wa for assistance with pharmacokinetic and serum protein binding studies; O. Ouerfelli, A. Dilhas, H. Zhao and G. Yang for chemistry support, the MSKCC molecular cytology core for immunohistochemistry, S. Cai, E. Burnazi and the MSKCC Radiochemistry Core for 18F-FDHT production; the MSKCC Small-Animal Imaging Core Facility, D. Liang for technical support and D. Danila for helpful discussions. Funded in part by the Prostate Cancer Foundation, the National Cancer Institute and DOD PC051382 Prostate Cancer Research Program Clinical Consortium Award. N.J.C. is supported by the Charles H. Revson Foundation. Y.C. is supported by an ASCO Young Investigator Award and AACR BMS Fellowship. C.L.S. is a Doris Duke Distinguished Clinical Scientist. C.T., S.O., J.W., D.W., C.C., M.J. and C.L.S. are co-inventors on patent applications covering RD162, MDV3100 and related compounds. C.L.S. and M.J. have been paid consultants for and have stock options in Medivation, Inc. D.T. H. is an employee of Medivation, Inc.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/1168175/DC1
Materials and Methods
Figs. S1 to S10
Tables S1 to S3
References
References
- 1.Scher HI, Sawyers CL. J Clin Oncol. 2005;23:8253. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 2.Taplin ME, Balk SP. J Cell Biochem. 2004;91:483. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 3.Chen CD, et al. Nat Med. 2004;10:33. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 4.Kelly WK, Slovin S, Scher HI. Urol Clin North Am. 1997;24:421. doi: 10.1016/s0094-0143(05)70389-x. [DOI] [PubMed] [Google Scholar]
- 5.Shiau AK, et al. Cell. 1998;95:927. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 6.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Proc Natl Acad Sci U S A. 2005;102:6201. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolvenbag GJ, Furr BJ, Blackledge GR. Prostate Cancer Prostatic Dis. 1998;1:307. doi: 10.1038/sj.pcan.4500262. [DOI] [PubMed] [Google Scholar]
- 8.Teutsch G, et al. J Steroid Biochem Mol Biol. 1994;48:111. doi: 10.1016/0960-0760(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 9.Van Dort ME, Robins DM, Wayburn B. J Med Chem. 2000;43:3344. doi: 10.1021/jm000163y. [DOI] [PubMed] [Google Scholar]
- 10.Larson SM, et al. J Nucl Med. 2004;45:366. [PubMed] [Google Scholar]
- 11.Liu W, et al. Neoplasia. 2008;10:897. doi: 10.1593/neo.08428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida T, et al. Cancer Res. 2005;65:9611. doi: 10.1158/0008-5472.CAN-05-0817. [DOI] [PubMed] [Google Scholar]
- 13.Ellwood-Yen K, Wongvipat J, Sawyers C. Cancer Res. 2006;66:10513. doi: 10.1158/0008-5472.CAN-06-1397. [DOI] [PubMed] [Google Scholar]
- 14.Cockshott ID, Cooper KJ, Sweetmore DS, Blacklock NJ, Denis L. Eur Urol. 1990;18 3:10. doi: 10.1159/000463972. [DOI] [PubMed] [Google Scholar]
- 15.Hodgson MC, et al. J Biol Chem. 2005;280:6511. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- 16.Baek SH, et al. Proc Natl Acad Sci U S A. 2006;103:3100. doi: 10.1073/pnas.0510842103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. J Biol Chem. 2002;277:26321. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 18.Ozers MS, et al. Biochemistry. 2007;46:683. doi: 10.1021/bi061321b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.