

Abstract
PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a tumor suppressor that antagonizes signaling through the phosphatidylinositol-3-kinase–Akt pathway. We have demonstrated that subtle decreases in PTEN abundance can have critical consequences for tumorigenesis. Here, we used a computational approach to identify miR-22, miR-25, and miR-302 as three PTEN-_targeting microRNA (miRNA) families found within nine genomic loci. We showed that miR-22 and the miR-106b∼25 cluster are aberrantly overexpressed in human prostate cancer, correlate with abundance of the miRNA processing enzyme DICER, and potentiate cellular transformation both in vitro and in vivo. We demonstrated that the intronic miR-106b∼25 cluster cooperates with its host gene MCM7 in cellular transformation both in vitro and in vivo, so that the concomitant overexpression of MCM7 and the miRNA cluster triggers prostatic intraepithelial neoplasia in transgenic mice. Therefore, the MCM7 gene locus delivers two simultaneous oncogenic insults when amplified or overexpressed in human cancer. Thus, we have uncovered a proto-oncogenic miRNA-dependent network for PTEN regulation and defined the MCM7 locus as a critical factor in initiating prostate tumorigenesis.
Introduction
MicroRNAs (miRNAs) (1) are endogenous 18- to 24-nucleotide (nt) single-stranded RNA molecules that act as posttranscriptional regulators of gene expression. miRNAs are often organized as several clustered sequences that are transcribed by RNA polymerase II into long multicistronic precursors called pri-miRNAs. Mature, single-stranded miRNAs are generated from pri-miRNAs in a two-step process, with the first step taking place in the nucleus and the second in the cytoplasm (2). The second step, in which pre-miRNAs are processed to mature miRNAs, is catalyzed by the ribonuclease III (RNase III) DICER (1), which is variably expressed in cancer, suggesting a possible role for aberrant miRNA maturation in tumorigenesis (3, 4).
Of the ∼700 known mammalian miRNA genes, most are located within introns of protein-coding genes or within introns or exons of noncoding transcriptional units (TUs). The expression of intronic miRNAs largely coincides with that of the hosting TUs (5) because they are typically oriented in the same direction and are coordinately expressed with the pre–messenger RNA (pre-mRNA) in which they reside. Despite their large number, the function of only a few intronic miRNAs has been investigated (6), and even less is known about their functional relationship with their host proteins other than that they are cotranscribed in the same mRNA precursor.
miRNAs bind to sequences in the 3′ untranslated region (3′UTR) of _target genes; nucleotide pairing between nucleotides 2 to 7 of the miRNA (the miRNA seed sequence) and the corresponding sequence along the _target 3′UTR (seed match) is necessary for _target recognition (7). Once bound to a 3′UTR, miRNAs decrease protein translation or the stability of nascent mRNA strands, or both, to decrease production of the _target protein (8). miRNAs sharing the same seed sequence are grouped into families and are theorized to _target overlapping sets of genes (9). miRNAs play diverse roles in numerous cellular processes; in particular, miRNA abundance is altered during tumorigenesis, and miRNAs can act as onco-suppressors or oncogenes (10).
The tumor suppressor gene PTEN (phosphatase and tensin homolog deleted on chromosome 10) (1) encodes a phosphoinositide phosphatase that opposes the phosphatidylinositol 3-kinase (PI3K)–Akt pathway (11, 12). After stimulation of cells with growth factors, PI3K catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) into the second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3), which then recruits various proteins that contain a pleckstrin homology (PH) domain to the plasma membrane. Among these recruited proteins is the serine and threonine kinase Akt, which is activated through phosphorylation. In turn, active Akt phosphorylates various _target proteins to promote nutrient uptake, protein synthesis, cell survival, cell proliferation, cell motility, and angiogenesis (11). PTEN dephosphorylates PIP3 to PIP2, inhibiting Akt activation and thereby the PI3K-Akt signaling pathway.
Monoallelic loss or mutation of PTEN is detected in the early stages of many sporadic tumors, including prostate cancer (12). High degrees of Akt phosphorylation and hyperactivation of the Akt signaling pathway are hallmarks of tumors in which PTEN function is impaired. Small decreases in Pten protein have marked consequences on tumor initiation and progression, as demonstrated in vivo in a Pten hypomorphic allelic series in the mouse (13); thus, modulators of PTEN gene expression, such as miRNAs, may be predicted to have a profound effect on tumorigenesis. Here, we have identified nine loci that _target the tumor suppressor PTEN, thereby unraveling a complex proto-oncogenic miRNA network. Furthermore, we have defined a causal link between PTEN _targeting by the miR-106b∼25 cluster, the consequent decrease in PTEN abundance, and prostate tumorigenesis.
Results
PTEN abundance is regulated by multiple miRNA families
To identify PTEN-_targeting miRNAs, we filtered the output of four _target prediction algorithms [_targetScanS (14), PicTar (15), miRanda (16), and miRBase (17) (table S1)]. Our selection criteria considered miRNA families that (i) had oncogenic potential and (ii) were predicted to _target PTEN by multiple algorithms (see table S2 for details). The seven miRNA families that passed our dual-criteria approach are listed in Fig. 1A. The miR-193 and miR-198 families were not found to down-regulate PTEN; therefore, they were not investigated further. Two families, miR-17 and miR-19, had been previously identified as _targeting PTEN (18–21), confirming the reliability of our screen. The miR-17, miR-19, miR-22, miR-25, and miR-302 families were further characterized. The members of these families and the location of their seed matches in the PTEN 3′UTR are depicted in fig. S1A and Fig. 1C, respectively. The genomic organization indicates that different PTEN-_targeting miRNAs can be transcribed from the same multicistronic precursor (Fig. 1B).
Fig. 1.

PTEN-_targeting miR-17, 19, 22, 25, and 302 families. (A) List of the seven miRNA families that passed either of our selection criteria (CTR) (table S2). (B) Genomic organization of miRNAs belonging to miR-17 (red), miR-19 (yellow), miR-22 (brown), miR-25 (blue), and miR-302 (gray) families. miRNAs that do not _target PTEN are in white. Large boxes, pre-miRNAs; small black boxes, mature miRNAs. The miRNA clusters are named with the most upstream and the most downstream miRNA connected by a tilde (∼) (54). The chromosomes (chr.) in which the miRNA genes are located are indicated below the names. It is also specified whether the miRNA genes are independent TUs (intergenic) or are located in introns or exons of coding or noncoding TUs. (C) Schematic representation of the seed matches of miR-17, 19, 22, and 25 (_targetScan) and 302 (PicTar) families along the PTEN 3′UTR. The miR-17 and miR-302 families overlap in binding to the six-nucleotide oligomer Alu 5′-GCACTT-3′ motif (55). ORF, open reading frame.
To validate their ability to decrease PTEN abundance, we overexpressed a representative member of each of the five miRNA families as a synthetic short interfering–like molecule (si-miRNA) in the DU145 prostate cancer cell line [in which PTEN is wild type (22) and the expressed 3′UTR harbors all the predicted miRNA binding sites (fig. S1E)]. The abundance of both PTEN protein and transcript was reduced by all tested miRNAs (Fig. 2A and fig. S2). The miRNA-mediated decrease in PTEN abundance was confirmed functionally; miRNA overexpression led to increased abundance of the PTEN substrate PIP3 (Fig. 2B) and increased Akt phosphorylation (Fig. 2C). Direct interaction between the tested miRNAs and PTEN mRNA was verified with chimerical luciferase plasmids in which appropriate fragments of PTEN 3′UTR were cloned downstream of the luciferase reporter gene (Fig. 2D and fig.S1, B to D).
Fig. 2.
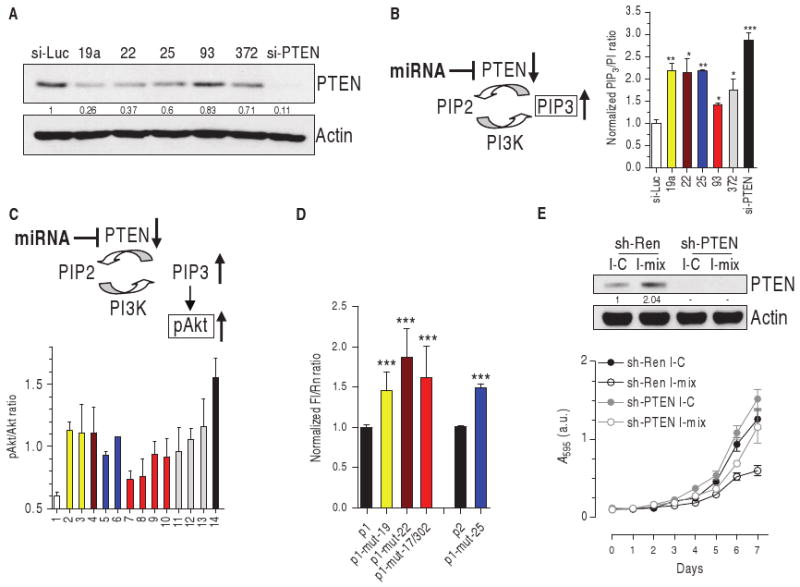
miR-17, 19, 22, 25, and 302 families decrease PTEN abundance and activate the Akt pathway. (A) Western blot of DU145 cells transiently transfected with control si-Luc, the indicated si-miRNAs, or the siRNA directed against human PTEN (si-PTEN). Quantification of PTEN protein is reported. (B) PIP3 detection in DU145 cells transiently transfected as above, serum-starved, and stimulated with 200 nM insulin for 5 min. (C) pAkt/Akt ratio in PWR-1E cells after the transient transfection of 1, si-Luc (white); 2 to 13, si-miRNAs: 19a, 19b (yellow, miR-19 family), 22 (brown), 25, 92a (blue, miR-25 family), 17, 20a, 93, 106b (red, miR-17 family), 302a, 372, 373 (gray, miR-302 family); 14, si-PTEN (black). (D) Wild-type or mutant p1 and p2 reporter plasmids (fig. S1, B and C) were transfected into DU145 cells. Twenty-four hours later, the luciferase activity of the mutant plasmids was higher than that of the corresponding wild-type plasmids, indicating that the introduced mutations in the seed matches impair miRNA binding to PTEN 3′UTR. ***P < 0.001. (E) DU145 cells were stably infected with a retroviral vector expressing a control shRNA directed against Renilla luciferase (sh-Ren) or an shRNA directed against human PTEN (sh-PTEN) and then transiently transfected with a control miRNA inhibitor (I-C) or a mix of the inhibitors against miR-19a/22/25/93/106b (I-mix). Top: Western blot showing PTEN 96 hours after transfection. Bottom: growth curve. See text for details.
Individual miRNAs binding to different seed matches on the same 3′UTR generally cooperate in translational repression (23). Indeed, we found that the combination of a representative miRNA for each of the families was more effective than any single miRNA at decreasing PTEN abundance (fig. S3A).
Finally, to validate that the PTEN-_targeting miRNAs we identified exerted their biological activity through their effects on PTEN, we generated DU145 cell lines stably transfected with short hairpin RNA (shRNA) _targeting a control gene Renilla luciferase (sh-Ren) or human PTEN (sh-PTEN). When sh-Ren–expressing cells were transiently transfected with a mix of antisense inhibitors of the identified PTEN-_targeting miRNAs (I-mix), PTEN abundance increased (Fig. 2E, top) and proliferation decreased (Fig. 2E, bottom) compared to cells transfected with a control miRNA inhibitor (I-C). In contrast, sh-PTEN–expressing cells, which had undetectable PTEN, showed no response to I-mix. These findings indicate that PTEN _targeting is necessary for the growth-promoting effects of members of the different miRNA families.
PTEN-_targeting miRNAs and DICER are abundant in human prostate cancer
Small differences in Pten abundance have marked consequences on prostate cancer initiation and progression (13); thus, we evaluated the possible involvement of the PTEN-_targeting miRNA families in the context of human prostate cancer cell lines and primary human prostate tumor samples. Based on our analysis, PTEN-_targeting miRNAs are organized into nine independent genomic clusters (Fig. 1B). We focused on four loci representative of the five PTEN-_targeting miRNA families: miR-22, miR-371∼373, and the paralogous miR-17∼92 and miR-106b∼25 clusters (Fig. 1B and table S2).
We performed real-time reverse transcription polymerase chain reaction (RT-PCR) on two prostate cell lines derived from normal epithelium, two derived from primary prostate carcinoma, and five derived from distant prostate carcinoma metastases. miR-22 was more abundant in both cell lines derived from primary carcinomas and in three of the five metastatic cell lines than in the lines derived from normal epithelium, suggesting that this miRNA might play a role in prostate cancer progression (Fig. 3A). A robust increase in the abundance of the three PTEN-_targeting components located in the miR-106b∼25 cluster (miR-25, miR-93, and miR-106b) was observed in all the prostate cancer cell lines compared to that in the normal cell lines (Fig. 3B). Of the PTEN-_targeting members of the miR-17∼92 cluster, abundance of only miR-19a was increased (Fig. 3C). This suggests that, although both the miR-106b∼25 and the miR-17∼92 clusters contain members of the same PTEN-_targeting miRNA families, only those in the first cluster are associated with prostate tumorigenesis. The PTEN-_targeting members of the miR-371∼373 cluster (miR-372 and miR-373) were not detectable in all the cell lines analyzed [see also (24)]. Therefore, this cluster was not investigated further.
Fig. 3.

Presence of miR-22, miR-106b∼25, and miR-17∼92 PTEN-_targeting miRNAs in human prostate cell lines. Mature miRNAs [miR-22 (A), miR-106b, miR-93, miR-25 (B), miR-17, miR-19a, miR-20a, miR-19b, and miR-92a (C)] were detected by real-time PCR analysis in the cell lines derived from the following (numbered 1 to 9 in order): RWPE-1 (normal prostate epithelium immortalized), PWR-1E (normal prostate epithelium immortalized), Ca-HpV-10 (primary prostate carcinoma), 22Rv1 (xenograft of a primary carcinoma), DU145, LnCaP, MDA-PCa-2b, PC3, and VCap (prostate carcinoma metastasized to distal organs). The ratio of the miRNA being tested to that of RNU24 (internal standard) in RWPE-1 was taken as 1. miR-302 family members belonging to the miR-371∼373 and miR-367∼302b clusters (Fig. 1B) were not detectable in all the cell lines analyzed.
The abundance of miR-22, miR-25, miR-93, and miR-106b was next examined by in situ hybridization (ISH) on a prostate tumor tissue microarray (TMA). This TMA contains 184 cases of tumor and matched nontumor tissues (table S3). miRNA abundance was scored as described in fig. S4. ISH showed that miR-22, miR-25, miR-93, and miR-106b were absent in most of the nontumor tissue samples, confirming the RT-PCR results in the cell lines derived from normal prostatic epithelium. Up to 53% of the tumor samples were, however, positive for these miRNAs (Fig. 4, A and B). TMA samples characterized as prostatic intraepithelial neoplasia (PIN, an early noninvasive malignant lesion of the prostate epithelium) consistently showed intermediate positivity (25 to 45% of positive cases for each miRNA). These results are in agreement with miRNA array data reported elsewhere (25, 26).
Fig. 4.
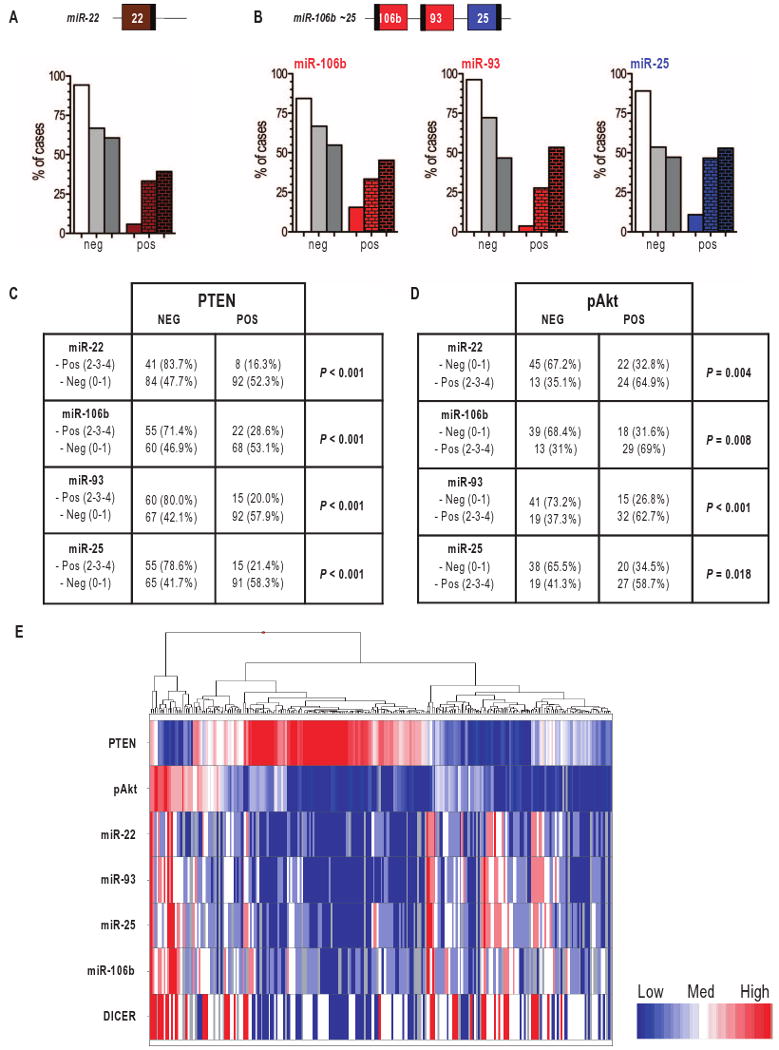
Presence of miR-22 and miR-106b∼25 miRNAs in human prostate tumor samples. (A and B) Score of the fractional presence of miR-22 (A) and miR-106b, miR-93 and miR-25 (B) in peritumoral tissue (left bars), PIN (middle bars), and prostate cancer (right bars), as detected by ISH. 0 to 1, negative (neg); 2 to 4: positive (pos). (C) Correlation between the lack of PTEN and the presence of miR-22 and miR-106b∼25 cluster miRNAs. (D) Correlation between the presence of pAkt and that of miR-22 and miR-106b∼25 cluster miRNAs. (E) Heat map of PTEN, pAkt, DICER, and miR-22/25/93/106b status in tumor samples. The heat map shows an inverse correlation between PTEN abundance and that of DICER, the miRNAs, and pAkt. The heat map was generated with TIBCO Spotfire Metrics (version 3.0) software.
To confirm that miR-22, miR-25, miR-93, and miR-106b are bona fide PTEN-_targeting miRNAs, we measured PTEN abundance by immunohistochemistry (IHC) in the same TMA. We identified a highly significant, inverse correlation between the abundance of PTEN and that of each of the four miRNAs (Figs. 4, C and E, and 5). Furthermore, we detected a direct correlation between each miRNA and phosphorylated Akt (pAkt) (Figs. 4, D and E, and 5). We also found that the concomitant presence of candidate miRNAs increased the probability of observing Akt phosphorylation in the TMA samples (fig. S5).
We also investigated the abundance of DICER, the enzyme that cleaves pre-miRNAs to release mature miRNAs, by IHC in the same TMA. We observed that increased DICER abundance was associated with tumor progression (Fig. 6, A and B). This finding is confirmed by analysis of an independent TMA (4) and is consistent with the observation that the oncogenic factor SOX4 increases DICER transcription (27). We also found a direct correlation between overexpression of candidate miRNAs and that of DICER (table S4 and Figs. 4E and 6C), suggesting that aberrant maturation contributes to the increase in abundance of these miRNAs.
Fig. 6.
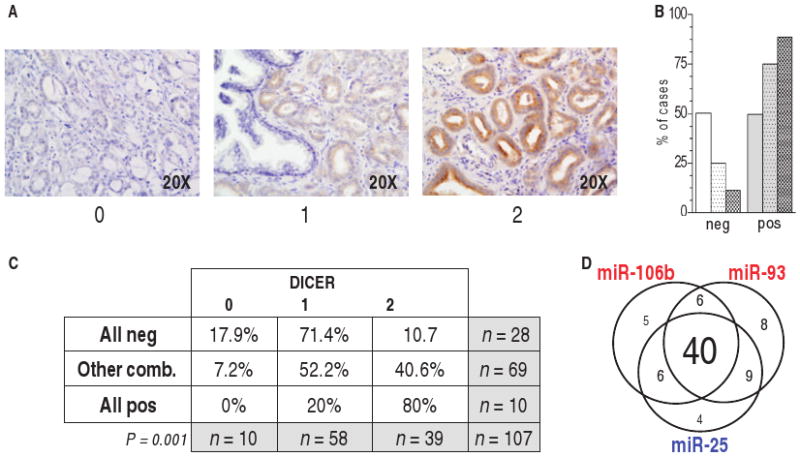
Correlation of DICER abundance in prostate tumor samples with that of miR-22 and miR-106b∼25 miRNAs. (A) TMA samples were considered negative (score, 0) or positive (score, 1 to 2) for DICER by IHC. (B) Presence of DICER in peritumoral tissues (left bars), PIN (middle bars), and prostate cancer (right bars). 0, negative; 1 to 2, positive. (C) Correlation between DICER presence and that of miR-22/miR-25/miR-93/miR-106b. (D) Venn diagram of tumor samples positive for one, two, or all three miRNAs in the miR-106b∼25 cluster. In most of the cases, the three miRNAs of the cluster were co-overexpressed.
miR-22 is a proto-oncogene
We performed transformation assays on mouse embryonic fibroblasts (MEFs) and found that miR-22 cooperated with the proto-oncogene c-MYC, proof of principle of its proto-oncogenic capacity (Fig. 7A). We also tested the ability of miR-22 to potentiate the proliferation of human prostate cancer cells. We generated DU145 cells that stably expressed a retroviral pri-miR-22 (PIG/22 cells), in which PTEN abundance and activity was decreased relative to that in control cells (Fig. 7B), and found that they formed greater numbers of colonies in soft agar compared to control cells (Fig. 7C). To determine whether the effects of miR-22 depended on decreased PTEN abundance, we retrovirally transduced DU145 cells with both pri-miR-22 and PTEN lacking its 3′UTR, so that it could not be _targeted by miRNAs. Exogenous 3′UTR-less PTEN overcame the proliferation-promoting effect of miR-22, indicating that this miRNA depends on PTEN down-regulation to exert its biological activity (Fig. 7D). When injected subcutaneously into the flank of nude mice, PIG/22 cells showed a proliferative advantage, as indicated by their generation of tumors that were twice the size of those generated by PIG-infected cells after 6 to 7 weeks of growth (Fig. 7E). miR-22 overexpression (Fig. 7, F, left, and G) and the concomitant decrease in PTEN mRNA abundance (Fig. 7F, right) were confirmed in PIG/22-derived tumors. These tumors demonstrated a proliferation rate higher than that of PIG-derived tumors, as shown by increased Ki67 staining, as well as a highly phosphorylated Akt as detected by IHC (Fig. 7G), both suggestive of hyperactivation of the Akt pathway.
Fig. 7.
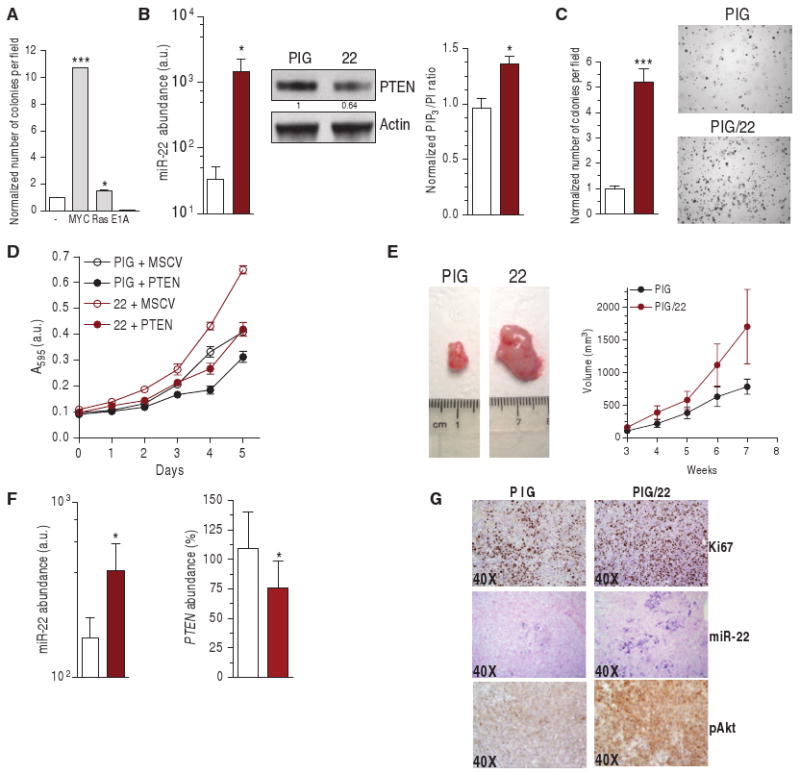
miR-22 proto-oncogenic activity. (A) Transformation assay performed in wild-type MEF by co-infecting MSCV/miR-22 and PIG, PIG/c-MYC, PIG/RasV12, or PIG/E1A. The number of colonies formed by MEFs double–infected with PIG and MSCV empty plasmids is taken as 1. (B) Abundance of mature miR-22 (left, real-time PCR), PTEN abundance (middle, Western blot) and PIP3 production (right) in stably infected DU145 cells. *P < 0.05. (C) Number of colonies formed in soft agar. ***P < 0.001. (D) Growth curve of DU145 cells stably infected with PIG/22 and MSCV-neo-Flag-PTEN plasmids. PTEN encoded by MSCV-neo-Flag-PTEN contains the entire PTEN open reading frame, but lacks the 3′UTR. (E) Representative tumor (left) and increase in tumor volume (right) formed after subcutaneous injection into nude mice (n = 6). (F and G) Analysis of the tumors: mature miR-22 (F, left) and PTEN mRNA (F, right) detection by real-time PCR. *P < 0.05. (G) IHC staining of the proliferation marker Ki67; ISH of miR-22; IHC of pAkt. (B, C, and F) White bars, PIG; brown bars, PIG/22.
Two potentially oncogenic elements—miR-106b∼25 and MCM7—are present in the same locus
All three components of the miR-106b∼25 cluster _targeted PTEN (Fig. 2A) and cooperated in decreasing PTEN abundance (fig. S3B). Thus, we analyzed this cluster for its oncogenic potential, with particular emphasis to its genomic context. miR-106b∼25 is located within intron 13 of the minichromosome maintenance protein 7 (MCM7) gene. MCM7 is one of six members of the highly conserved MCM2 to MCM7 family of DNA helicases that are essential for the initiation of DNA replication in eukaryotes (28). The abundance of MCM7 is commonly increased in human cancers and it is widely used as a diagnostic (29, 30) and prognostic (31) marker.
We first tested the proliferative consequences of overexpressing the miR-106b∼25 cluster by infecting DU145 prostate cancer cells with a retroviral vector expressing the ∼800–base pair (bp)-long intron that functions as pri-miRNA (PIG/106b∼25). miR-106b∼25–overexpressing cells showed decreased PTEN abundance and activity (fig. S6A) and an increased capacity to form colonies in soft agar (fig. S6B). To determine whether miR-106b∼25 functions through its effects on PTEN, we infected cells with the exogenous 3′UTR-less form of PTEN, which markedly decreased the number of colonies formed in soft agar by miR-106b∼25–overexpressing cells (fig. S6C). This indicates that the miR-106b∼25 cluster exerts its tumorigenic activity through a decrease in PTEN. The incomplete rescue by exogenous PTEN may be attributed to the ability of the three miRNAs in this cluster to _target genes other than PTEN (table S2). This notion is supported by the observation that an inefficient shRNA that decreased PTEN abundance to an extent similar to that elicited by the miRNAs was less effective than the miRNAs in promoting colony formation (compare fig. S7A with fig. S6B). miR-106b∼25–overexpressing cells were also injected subcutaneously into nude mice, where they formed bigger tumors compared to that in control cells (Fig. 8A; see fig. S6, D and E, for the analysis of the xenograft).
Fig. 8.
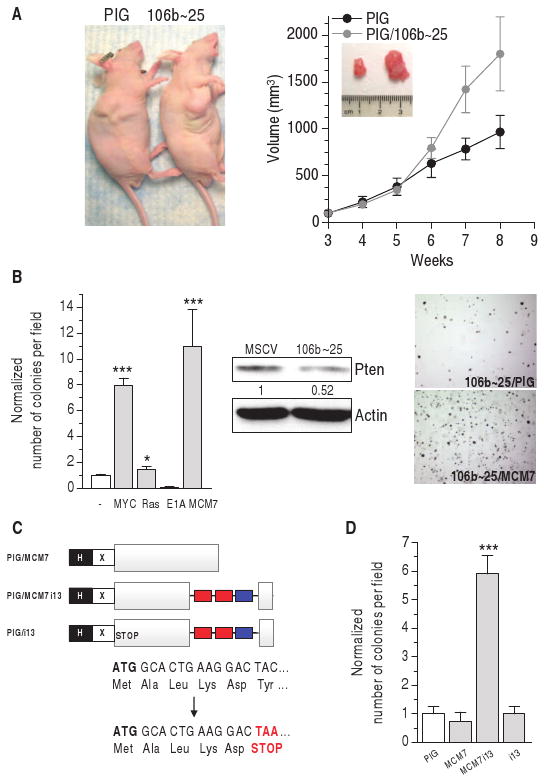
miR-106b∼25 cluster proto-oncogenic activity. (A) Representative tumor (left) and increase in tumor volume (right) after subcutaneous injection into nude mice of DU145 stably infected with the miR-106b∼25 cluster (n = 6). (B) Left: transformation assay performed in wt MEF by co-infecting MSCV/miR-106b∼25 and PIG empty, PIG/c-MYC, PIG/RasV12, PIG/E1A, or PIG/MCM7 plasmids. The number of colonies formed by MEFs double-infected with PIG and MSCV empty plasmids is taken as 1. Center: decrease in Pten abundance caused by the cluster as measured by Western blot. Right: representative fields of miR-106b∼25/PIG and miR-106b∼25/MCM7 colonies. ***P < 0.001. (C) Schematic representation of the PIG/MCM7, PIG/MCM7i13, and PIG/i13 plasmids. PIG/MCM7 plasmid expresses only MCM7; PIG/MCM7i13 expresses both MCM7 and the three miRNAs belonging to the miR-106b∼25 cluster, as a result of the splicing and maturation of the recombinant intron 13; PIG/i13 is derived from PIG/MCM7i13 by insertion of a point mutation that transforms the sixth amino acid of MCM7 (TAC, Tyr) into a stop codon (TAA, STOP). Therefore, this plasmid expresses only the intronic miRNAs. H, HA tag; X, Xpress tag; gray box, MCM7 coding sequence; red/blue boxes, miR-106b/93 and miR-25 genes. (D) Transformation assay performed in wt MEFs with the plasmids shown in (C). ***P < 0.001.
To further investigate the oncogenic capabilities of miR-106b∼25, we performed transformation assays in MEFs. Although miR-106b∼25 expresses miRNAs belonging to different families (miR-25 and miR-93/106b), it behaved as a single functional unit that cannot efficiently transform MEF on its own (Fig. 8B). This is consistent with the fact that, in most of the cases, the three miRNAs share an expression pattern (Fig. 6D) and confirms that lowering Pten alone is not sufficient to confer fullblown transformation in MEFs (32). However, the miR-106b∼25 cluster transformed MEFs when combined with c-MYC or MCM7 (Fig. 8B). A specific decrease in Pten abundance by an shRNA shows the same pattern of transformation (sh-Pten + c-MYC and sh-Pten + MCM7), although with lower efficiency, suggesting that Pten _targeting is required by the intronic miRNAs to transform MEFs (fig. S7B).
Cooperation of miR-106b∼25 and MCM7 in cellular transformation indicates that the MCM7 locus contains not one, but two oncogenic units. To validate this notion, we generated a construct that drives the expression of both the miR-106b∼25 cluster and MCM7 protein from the same TU (PIG/MCM7i13), mimicking the organization of the MCM7 locus itself (Fig. 8C; see fig. S8 for the characterization of the chimerical construct). Only the PIG/MCM7i13 and not control constructs transformed MEFs (Fig. 8D), providing further evidence that this locus contains two oncogenic elements that promote cellular transformation.
The MCM7 locus can initiate prostate tumorigenesis
To characterize the role of the bi-oncogenic MCM7 locus in prostate tumorigenesis in vivo, we generated a transgenic mouse model in which human MCM7 protein and the PTEN-_targeting miR-106b∼25 intronic cluster were selectively overexpressed in prostate epithelium. We placed the MCM7i13 construct described above (Fig. 8C) under the control of the prostate-specific rat probasin promoter ARR2PB (subsequently referred to as Pb) (33, 34). Pronuclear injection of the Pb/MCM7i13 transgenic construct (Fig. 9A) resulted in the generation of nine independent lines of mice, as indicated by Southern blot (Fig. 9B, top) and PCR (Fig. 9B, bottom) analyses. Prostate-specific expression of the transgenic mRNA was determined by real-time PCR in these nine lines (1, 2, 12, 18, 21, 37, 46, 54, and 62). Line 21 did not show any expression of the transgene; the other transgenic lines were characterized as low expressors (LE; lines 12, 37, and 46), medium expressors (MEs; lines 1, 2, and 62), and high expressors (HEs; lines 18 and 54) (Fig. 9C and fig. S9A). In these mice, overexpressed MCM7i13 mRNA abundance was mirrored by an increase in the abundance of mature miR-106b, 93, and 25 (fig. S9B). Real-time PCR data were confirmed by MCM7 and miR-93 detection in preneoplastic prostates by IHC and ISH, respectively (fig. S9C).
Fig. 9.
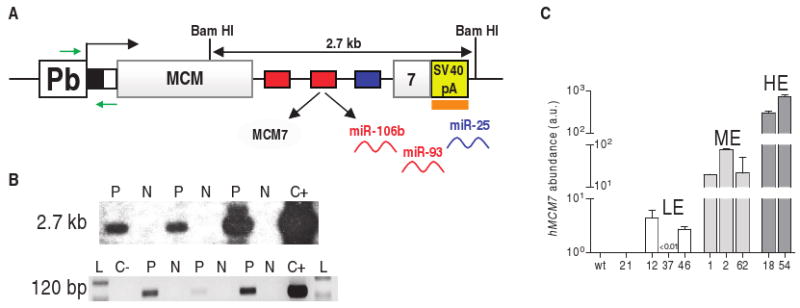
Pb/MCM7i13 transgenic construct. (A) Pb, prostate-specific rat probasin promoter ARR2PB; black box, HA tag; white box, Xpress tag; gray boxes, human MCM7 coding sequence; red and blue boxes, miR-93/106b and miR-25 genes; yellow box, SV40 poly(A) (SV40 pA); orange box, probe used for Southern analysis of transgenic lines; green arrows, primers used for PCR analysis of transgenic lines and for genotyping. (B) Top: Southern blot analysis. Genomic DNA extracted from tails was digested with Bam HI and hybridized with a probe complementary to the SV40 poly(A) region as shown in (A). The Bam HI–digested Pb/MCM7i13 plasmid was used as positive control (C+). Bottom: PCR analysis. A forward primer complementary to the probasin promoter and a reverse primer complementary to the tag were used, as shown in (A). Empty Pb and Pb/MCM7i13 plasmids were used as negative (C−) and positive (C+) controls. L, 100-bp ladder. Three representative positive (P) and negative (N) mice are shown. (C) Abundance of the transgenic construct measured by real-time PCR in nine different lines. A primer pair specific for human MCM7 coding sequence was used. LE, ME, HE: low, medium, and high expressors, respectively. Human MCM7 was not detectable in wild-type (wt) mice, confirming the specificity of the primers. See also fig. S9A.
Next, we examined the incidence of prostate-specific lesions in a cohort of 1-year-old transgenic mice from the various lines. Although the anterior (AP) and ventral (VP) prostates appeared normal, the dorsolateral prostates (DLPs) displayed multifocal proliferative lesions, the severity of which was directly correlated with expression of the transgene (Fig. 10A). LE lines manifested signs of hyperplasia, whereas ME and, to a greater extent, HE lines developed the distinctive architectural growth features that characterize PIN, including hyperplastic growth in disorganized multicellular layers, loss of epithelial cell polarity, nuclear atypia (large and irregular nuclei and prominent nucleoli), aberrant mitotic activity (Fig. 10B, left panels, dotted arrow), and cribriform growth patterns with intraepithelial lumen formation (Fig. 10B, left panels, straight arrows). Hyperproliferation was confirmed by an increase in the proliferation-specific marker Ki67 (Fig. 10B, right panels), and hyperactivation of the Akt pathway as a result of Pten down-regulation (Fig. 10C) was reflected by an increase in phosphorylation of Akt (Fig. 10C) and of its downstream effector S6 (36) (Fig. 10B, middle panels). Thus, the combined overexpression of MCM7 and its host miRNA cluster can initiate prostate tumorigenesis.
Fig. 10.
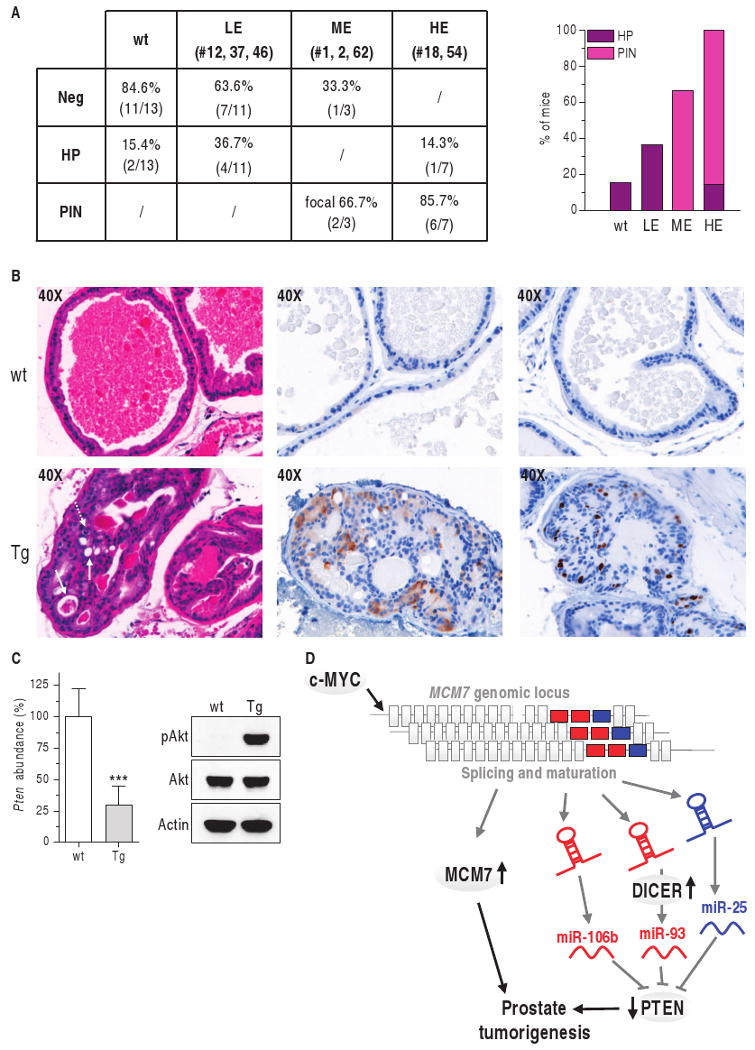
The MCM7 locus can initiate prostate tumorigenesis. (A) Incidence of hyperplasia (HP) and PIN in the DLP of 1-year-old transgenic mice. (B) Analysis of 1-year-old transgenic mice belonging to HE line 18. H&E staining (left) shows PIN in the DLP, as indicated by loss of cell polarity, abnormal proliferation, presence of mitotic figures (dotted arrow), and intraepithelial lumen formation (straight arrows). PIN glands are characterized by aberrant pS6 phosphorylation (middle, brown staining) and by an increased number of Ki67-positive cells (right, brown staining). (C) Decreased Pten mRNA (real-time PCR) is associated with increased Akt phosphorylation (Western blot). ***P < 0.001. (D) Model of MCM7 locus oncogenicity in prostate cancer. The MCM7 locus contains two oncogenic and cooperating elements: MCM7 (gray) and the PTEN-_targeting miR-106b∼25 miRNAs in intron 13. This locus undergoes genomic amplification in prostate cancer. c-MYC increases transcription of both MCM7 and the miR-106b∼25 miRNAs and increased DICER abundance leads to increased miRNA maturation.
We also generated a transgenic mouse model in which human MCM7 protein alone was selectively _targeted to prostatic epithelium (fig. S10A). Two independent lines with varying MCM7 expression were studied further. Pb/MCM7 line 88 expressed the transgenic construct in amounts comparable to those of Pb/MCM7i13 line 18 (fig. S10B). Nonetheless, none of the mice from these two lines developed cancer or an overt phenotype by 1 year of age (fig. S10, C and D). This indicates that MCM7 alone cannot initiate tumorigenesis in the prostate.
Discussion
Our analysis points to the existence of a complex network of miRNAs that control PTEN expression. Given the critical role of PTEN as a prostate cancer tumor suppressor, we have been able to identify a number of genetic elements that are implicated in the development of prostate tumors. Our identification of these elements has, in turn, led to a number of unexpected conclusions.
Using an approach that integrates four published prediction algorithms and a meta-analysis of the published literature, we have identified and validated three PTEN-_targeting miRNA families—miR-22, miR-25, and miR-302—encoded by up to nine proto-oncogenic loci. We found that different miRNA families, located at the same or distinct loci, can cooperatively modulate PTEN abundance. In particular, miR-93 and miR-106b, which have mild effects on PTEN individually, cooperate with miR-25 to substantially decrease PTEN abundance. This fine-tuning of PTEN abundance is relevant in tumorigenesis, because even slight variations in the abundance of this potent oncosuppressor can have marked effects on tumor initiation and progression (13). The importance of such a network of cooperating regulators is underlined by the observation that complete inactivation of PTEN can trigger fail-safe mechanisms such as cellular senescence (32). Therefore, it is tempting to speculate that PTEN heterozygosity, when accompanied by miRNA-mediated down-regulation, might be more effective at promoting tumorigenesis than complete homozygous loss of PTEN, a conclusion supported by analysis of tumorigenesis in a Pten hypomorphic allelic series in the mouse (13).
We found that these PTEN-_targeting miRNA families were aberrantly expressed in human cancers in which their ability to decrease PTEN may contribute to their oncogenic activity. For example, the miR-106b∼25 locus on chromosome 7, which is entirely composed of PTEN-_targeting miRNAs (miR-106b, miR-93, and miR-25), is markedly overexpressed and genetically amplified in prostate cancer [Figs. 3B and 4B; (37)]. Furthermore, miR-106b∼25 abundance is inversely correlated with PTEN abundance in human prostate cancer (Fig. 4, C and E). This locus is distinct from the paralogous miR-17∼92 cluster, which is composed of PTEN- and non–PTEN-_targeting miRNAs and is not consistently overexpressed in prostate cancer (Fig. 3C and fig. S11). On the other hand, the miR-302 family, which we did not find expressed in prostate cancer, is highly expressed in breast cancer (38), where decreases in PTEN abundance are frequently observed as well (35). Collectively, these findings suggest that different combinations of miRNA families and genomic loci may decrease PTEN abundance in different tumor types.
We identified miRNA processing as a regulatory control critical in cancer. The abundance of DICER, the enzyme that catalyzes the final step of miRNA maturation, correlated with that of candidate miRNAs and was directly associated with tumor progression (Fig. 6), suggesting that aberrant regulation of miRNA maturation contributes to prostate cancer.
We found that miR-22 and the miR-106b∼25 cluster acted as proto-oncogenes. miR-22 transformed MEFs when combined with c-MYC, as did the miR-106b∼25 cluster when combined with either c-MYC or MCM7. The cooperative oncogenic ability of the miR-106b∼25 cluster with c-MYC or MCM7 is pertinent to human prostate cancer because c-MYC is genetically amplified in up to 30% of prostate cancers (39) and is overexpressed in most (40). MCM7 transcription is enhanced by N-MYC (41, 42) and c-MYC (41, 43, 44), an observation corroborated by in vivo data in c-MYC transgenic mice (34). Our findings support the notion that the oncogenic activity of c-MYC may also involve its transcriptional activation of PTEN-_targeting miRNAs (45).
Notably, the cooperation of miR-106b∼25 and MCM7 in cellular transformation represents a functional interaction between an intronic miRNA cluster and its host protein in tumorigenesis, and suggests the existence of a single gene locus that harbors two independent and cooperating oncogenic elements. To confirm this hypothesis in vivo, we designed a transgenic model in which the overexpression of MCM7 protein and the PTEN-_targeting intronic miR-106b∼25 cluster is selectively driven in the mouse prostate. MCM7 and the miRNA cluster were co-overexpressed to mimic the physiologic condition because they share the same primary transcript (46). Transgenic mice developed PIN at 1 year of age with high penetrance (>80%) (Fig. 10). The onset and the features of Pb/MCM7i13-driven PIN were similar to those in Pten+/− mice (47) and are consistent with the ∼50% decrease in Pten abundance caused by the miR-106b∼25 cluster. Furthermore, Pb/MCM7i13 mice show a hyperactivation of the Akt pathway, similar to that observed in Pten+/− mice (33). Another similarity between the Pten+/− and Pb/MCM7i13 models is that PIN lesions occur mainly in the DLP. This finding is remarkable because this lobe corresponds to the peripheral zone of the human gland, which undergoes transformation in most human prostate cancer cases.
MCM7 overexpression alone is not sufficient to drive the oncogenic process in vitro or in vivo (fig. S10). MCM7 has been previously shown to potentiate tumorigenesis in the skin (48) and to increase the growth of prostate cell lines both in vitro and in a xenograft model (37). Furthermore, MCM7 abundance increases with tumor progression in both human samples and mouse models (30). MCM7 is widely used as a diagnostic and prognostic marker for cancer, and it is believed to be a more sensitive readout than Ki67 for the detection of proliferating cells (31, 49). Nevertheless, the Pb/MCM7 model we describe here indicates that MCM7 alone cannot be considered an initiation factor in prostate tumorigenesis. In contrast, the transgenic mouse overexpressing both MCM7 and intron-13 miRNAs develops PIN with high penetrance, and MCM7 cooperates with its intronic miRNAs in in vitro transformation assays. Thus, MCM7 and the intron-13 miRNAs cooperate in oncogenesis. This is pertinent to human tumorigenesis, in which the entire MCM7 locus is amplified (37).
Although the number of miRNAs associated with human malignancies is constantly increasing (10), there is limited evidence causally linking specific miRNAs to specific forms of cancer. miR-155 can induce leukemia when ectopically overexpressed in B cells (50). The prostate-specific transgenic mouse models presented here indicate that the aberrant expression of miRNAs is an initiating event for solid tumorigenesis as well (Fig. 10D).
Materials And Methods
Reagents
The following antibodies and reagents were used: antibodies against Hsp-90 #61041 (Becton Dickinson); PTEN #9559 (WB), pAkt (Ser473) #9271 (WB), #3787 (IHC), Akt #9272, pS6 #2211 (Cell Signaling); DICER #CG006 (Clonegene); MSCV-neo (Clontech); siGENOME non-_targeting small interfering RNA (siRNA) #2 (si-Luc), si-miR-19a, si-miR-19b, si-miR-22, si-miR-25, si-miR-92a, si-miR-17, si-miR-20a, si-miR-93, si-miR-106b, si-miR-302a, si-miR-372, si-miR-373, si-PTEN, siGLO RISC-free control siRNA miRNA antisense inhibitor negative control #1, miR-19a antisense inhibitor, miR-22 antisense inhibitor, miR-25 antisense inhibitor, miR-93 antisense inhibitor, miR-106b antisense inhibitor, Dharmafect 1 (Dharmacon); miR-17, miR-19a, miR-20a, miR-22, miR-25, miR-93, and miR-106b 3′ digoxigenin (DIG)–labeled LNA ISH probes (Exiqon); lipofectamine 2000, Trizol reagent, deoxyribonuclease I (DNase I) amplification grade, SuperScript II reverse transcriptase, Dulbecco's modified Eagle's medium (DMEM), RPMI 1640, fetal bovine serum (FBS), keratinocyte serum-free medium, epidermal growth factor (EGF), bovine pituitary extract (BPE), antibody against Xpress Tag, pcDNA3.1/His C, geneticin (G418), hygromycin, antibody against PTEN (IHC) #51-2400 (Invitrogen); human MCM7 complementary DNA (cDNA) #MHS1011-75176 (Open Biosystems); pGL3-Control (Firefly luciferase), pRL-TK (Renilla luciferase), Dual-Luciferase reporter assay (Promega); fraction V bovine serum albumin (BSA), polybrene, insulin, antibody against actin #A3853, anti-FLAG antibody #F3165, puromycin (Sigma); QuantiTect Sybr Green PCRkit (Qiagen); antibody against MCM7 #9966 (Santa Cruz Biotechnology); QuikChange II XL Site-Directed Mutagenesis Kit, Herculase Taq polymerase (Stratagene); Ki67 antibody #VP-K451 (Vector Laboratories).
Plasmids
To construct the p1 and p2 chimeric luciferase plasmids, we removed the multicloning site of pGL3-control plasmid from its original position and inserted it into the Xba I site located downstream of the luciferase STOP codon. In this way, pGLU plasmid was obtained. An ∼1.7- (p1) or 0.5-kilo–base pair (kbp) (p2) fragment of PTEN 3′UTR was then amplified from genomic DNA and cloned into the Kpn I and Xho I sites of pGLU. The QuikChange II XL Site-Directed Mutagenesis Kit was used to generate the mutated forms of these plasmids (fig. S1C).
PIG/22 retroviral vector was obtained by cloning a ∼0.55 genomic fragment encompassing human pri-miR-22 into Bgl II and Xho I sites of MSCV-PIG (51) vector (puromycin resistance). The ∼0.8–kbp-long intron 13 of human MCM7 was cloned analogously and PIG/106b∼25 vector was obtained. The corresponding MSCV plasmids (hygromycin resistance) were obtained by subcloning in Bgl II and Xho I sites.
MSCV-neo-Flag-PTEN was generated by cloning the open reading frame of human PTEN (NM_000314.4) in the Eco RI and Xho I sites of MSCV-neo. The Flag epitope (AspTyrLysAspAspAspAspLys) was then inserted upstream of the ATG in Eco RI.
pSUPER-shEGFP and pSUPER-sh-Pten plasmids were previously generated in the laboratory (32). sh-Pten is complementary to nucleotides 274 to 291 in the open reading frame of both mouse and human PTEN.
Cells and culture conditions
Wild-type MEFs were isolated from 13.5-day mouse embryos. After the legs, tail, liver, and head were removed, each embryo was mechanically fragmented and then incubated with trypsin–phosphate-buffered saline (PBS) 1:3 (10 ml total) at 37°C for 5 min. Five milliliters was removed and collected in 20 ml of DMEM + 10% FBS. Fresh trypsin (5 ml) was then added and the above-mentioned procedure was repeated three to four times. The collected cells were then spun down and seeded in a 6-cm dish. Cells were trypsinized at confluency (passage 1, P1). The propagation protocol 3T9 (9 × 105 cells per 100–mm-diameter dish transferred every 3 days) was followed. MEFs at P1 to P3 were used.
U2OS, Phoenix A and E cells were grown in DMEM +10% FBS. RWPE-1, PWR-1E, and Ca-HpV-10 were grown in keratinocyte medium + EGF + BPE. 22Rv1, DU145, LnCap, MDA-PCa-2b, and PC3 were grown in RPMI 1640 + 10% FBS. All cell lines were obtained from American Type Culture Collection and grown in penicillin-streptomycin and glutamine-containing medium at 37°C in a humidified atmosphere with 6% CO2.
Dual-luciferase reporter assay
DU145 cells were seeded at a density of 6 × 104 cells per 24-well plate. Twenty-four hours later, 400 ng of p1 or p2 plasmid per well were cotransfected with 80 ng of pRL-TK. Lipofectamine 2000 was used as transfectant according to the manufacturer's recommendations. Twenty-four hours after transfection, luciferase activity was measured and normalized as in (20). In the case of plasmid-miRNA inhibitor cotransfection, 200 ng of p1/p1-mut-17/302 and 10 nmol of I-C/I-93 were transfected. Cells were analyzed as described above.
Transient transfection
DU145 (1.5 × 105) and PWR-1E (0.8 × 105) were seeded in 12-well dishes, respectively. The following day they were transfected with 100 nM siRNAs/si-miRNAs or 400 nM miRNA inhibitors with Dharmafect 1 according to the manufacturer's recommendations. With this protocol, more than 90% of cells were positive for the fluorescent siGLO RISC-free control siRNA. Six hours after transfection, cells were seeded for growth curves (see below). Otherwise, the day after transfection, cells were trypsinized and reseeded in 12-well plates for subsequent collection and analysis.
DU145 and MEF retroviral infection
For retrovirus-mediated gene transfer of DU145, Phoenix A cells (3 × 106) were plated in a 10-cm poly-d-lysine–coated dish and, 16 hours later, were transfected with PIG retroviral plasmids using Lipofectamine 2000. Forty-eight hours later, the virus-containing medium (10 ml) was filtered and mixed with 5 ml of freshly prepared medium supplemented with polybrene (4 μg/ml). Cells (5 × 105) were plated in a 10-cm dish. Sixteen hours later, the medium was replaced with viral supernatant. Puromycin (2 μg/ml) was administered 48 hours after infection. The cells were selected for 2 days and then used for the various assays. For double infection, Phoenix A cells were transfected with empty PIG or the miRNA-expressing plasmids (PIG/22, PIG/106b∼25), which carry puromycin resistance, and MSCV-neo empty or MSCV-neo-Flag-PTEN plasmids, which carry G418 resistance. The medium for infection contained 5 ml of freshly prepared medium, 5 ml of puro-resistant viral supernatant, and 5 ml of G418-resistant viral supernatant. Selection with puromycin (2 μg/ml) + G418 (500 μg/ml) was started 48 hours after infection and continued for 1 week. The selection medium was changed daily.
Double infection of MEF was performed similarly, except that Phoenix E cells instead of A cells were used. Phoenix E cells were transfected with MCSV/22 or MCSV/106b∼25 plasmids, which carry hygromycin resistance (51), and PIG/c-MYC, PIG/RasV12, PIG/E1A (51), or PIG/MCM7 puromycin-resistant plasmids. For sh-Pten experiments, Phoenix E cells were transfected with pSUPER-shPten, which carries puromycin resistance, and MSCV/c-MYC, MSCV/RasV12, MSCV/E1A, or MSCV/MCM7 hygromycin-resistant plasmids. The medium used to infect MEFs contained 5 ml of freshly prepared medium, 5 ml of hygromycin-resistant viral supernatant, and 5 ml of puromycin-resistant viral supernatant. Forty-eight hours after infection, selection was started and carried on for 4 days [the first two with puromycin (2 μg/ml) + hygromycin (75 μg/ml), the second two with only hygromycin (75 μg/ml)]. The selection medium was changed daily.
Phosphoinositide analysis
DU145 cells were seeded at 3 × 105 cells per six-well dish. The following day, they were transfected with the different si-miRNAs as described. Six hours after transfection, cells of one six-well plate were trypsinized and replated in two 10-cm plates. The following day, they were labeled for 24 hours with [3H]inositol (10 mCi/ml) for 24 hours in inositol-free DMEM supplemented with 10% FBS and 0.5% BSA. They were then serum-starved for 24 hours in inositol-free DMEM with [3H]inositol (10 mCi/ml) and 0.5% BSA, but without FBS. After 5 min of stimulation with 200 nM insulin, cells were lysed in 1 M HCl. Lipids were extracted in chloroform-methanol (1:1, vol/vol) and deacylated as described (52). Phosphatidylinositides were separated by anion-exchange high-performance liquid chromatography (Beckman), detected by a flow scintillation analyzer (Perkin-Elmer), and quantified with ProFSA software (Perkin-Elmer). The 3H-labeled PI3P (phosphatidylinositol 3-phosphate), PIP2, and PIP3 peaks were identified by 32P-labeled in vitro synthesized internal lipid standards, prepared with baculovirus-expressed PI3K. For the [3H]inositol labeling, the counts in each peak were normalized against the counts found in the phosphatidylinositol peak.
PCR analysis
Total RNA was extracted with Trizol reagent according to the manufacturer's instructions.
Mature miRNAs were detected with TaqMan miRNA assays (Applied Biosystems) at the Biopolymers Facility (Harvard Medical School). RNU24 (human) or sno202 (mouse) were used as internal standards.
To determine the length of PTEN 3′UTR, total RNA was subjected to DNase treatment and retrotranscription (1 μg of RNA per vial). PCR was then performed with Herculase Taq polymerase.
To evaluate hPTEN, mPten, hMCM7, and intron 13 mRNA concentrations, real-time PCR was carried out with Sybr Green fluorescence. Two microliters of cDNA was used in a 20-μl reaction. ACTIN (human) or GAPDH (mouse) was used as an internal standard.
Relative quantification of gene expression was performed with the comparative CT method (53).
Western blot
Transfected cells grown for specified time points were collected and lysed [50 mM tris (pH 8.0), 1 mM EDTA, 1 mM MgCl2, 1% NP-40, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM NaF, and protease inhibitors]. Proteins (30 μg per lane) were separated on 10% SDS–polyacrylamide gel and transferred to nitrocellulose membranes. Immunoblotting of the membranes was performed with the following primary antibodies: antibodies against PTEN (1:1000), Flag (1:1000), pAkt (Ser473) (1:1000), Akt (1:1000), MCM7 (1:2000), XPress (1:1000), Actin (1:10000), and Hsp90 (1:1000). Signals were revealed after incubation with the recommended secondary antibody coupled to peroxidase by enhanced chemiluminescence. Scanned images were quantified with ImageJ software.
In situ hybridization
ISH on TMAs was performed on 5-μm paraffin sections with 3′ DIG– labeled miRNA LNA (locked nucleic acid) probes with an automatic stainer (Discovery XT, Ventana Medical Systems Inc.). Cells were baked overnight at 60°C, dewaxed, postfixed in 4% paraformaldehyde (PFA) for 12 min, and then digested in proteinase K solution (15 μg/ml) for 4 min. Hybridization was performed overnight at 22°C below the melting temperature (Tm) of each probe with a commercial hybridization buffer (RiboHybe, Ventana Medical Systems). Two 16-min posthybridization washes in 2× SCC were performed at 4°C above the hybridization temperature. Then, sections were incubated for 40 min with a biotinylated antibody against DIG (InnoGenex). Detection with streptavidin–alkaline phosphatase and BCIP/NBT (bromochloroindolyl phosphate–nitro blue tetrazolium) substrates was performed for 10 hours with the BlueMap kit (Ventana Medical Systems). A Nikon Eclipse 50i microscope was used.
Immunohistochemistry
IHC was performed on 5-mm paraffin sections with the avidin-biotin-peroxidase method. The following primary antibodies were used: Ki67 (1:1000), pAkt (Ser473) (1:30), PTEN (1:100), pS6 (1:500), and DICER (1:250). Antigen retrieval was performed with citrate buffer (Ki67, DICER, pS6, PTEN) or 1 mM EDTA solution (pAkt). pAkt staining was considered specific only when cytoplasmic, and tumor cases were considered positive when staining in neoplastic cells was stronger than in normal prostate epithelium. PTEN abundance was scored semi-quantitatively with an intensity × quantity product (intensity: 0 = negative, 1 = weak, 2 = moderate, 3 = strong; quantity: 0 = negative, 1 = 1 to 9% of examined cells positive, 2 = 0 to 39% of cells positive, 3 = 40 to 69% of cells positive, 4 = 70 to 100% of cells positive). Scores ranging from 0 to 12 were averaged across replicate cores. For DICER, a classic three-category–based score was used, as shown in Fig. 6A. A Nikon Eclipse 50i microscope was used.
Cell proliferation
Six hours after transfection, the cells of one 12-well dish were trypsinized, resuspended in 50 ml of medium, and plated in eight sets of three wells of a 12-well plate. Starting from the following day (day 0), 1 set of wells per day was washed once with PBS, fixed in 10% formalin solution for 10 min at room temperature, and then kept in PBS at 4°C. At day 7, all the wells were stained with crystal violet. After lysis with 10% acetic acid, the absorbance was read at 595 nm.
Growth in semisolid medium
The bottom layer was obtained by covering six-well dishes with 3 ml of 0.6% agar in DMEM. The following day, 5 × 104 infected cells were plated on this bottom layer in triplicate in 2 ml of 0.3% agar in DMEM + 10% FBS. Colonies were counted after 3 to 4 (DU145) or 4 to 6 (MEF) weeks at 40× and 100× magnification, respectively. Five fields for each well were counted. A Nikon Eclipse TE300 microscope was used. Images were acquired with IPLab software.
Injection into nude mice
NCR nude mice strain (4 to 6 weeks old; Taconic) were subcutaneously injected into the right flank with 4 × 106 infected cells. Tumor size was assessed weekly by caliper measurement. Tumor volume was calculated with the formula D × d2 × π/6, where D and d are the long and the short sides of the tumor.
Patient material and TMAs
Four TMAs were constructed from archival formalin-fixed, paraffin-embedded (FFPE) radical prostatectomy specimens from 184 cases of prostate cancer from the Department of Pathology of Brigham and Women's Hospital. Representative regions of each tumor were selected for coring on the basis of the corresponding hematoxylin and eosin (H&E)–stained full section. Three 0.6-mm-diameter cores of each tumor were included in the TMAs along with three cores of normal prostate tissue from the same patient (for 177 cases). Three cores with foci of PIN were available for 24 cases. Informed written consent was obtained from all patients, and the use of patient material in this study was approved by our internal institutional review board. Clinicopathological characteristics of the cohort of patients included in the study are summarized in table S3.
Generation of MCM7 and MCM7i13 transgenic mice
Inserts from pcDNA/MCM7 and pcDNA/MCM7i13 plasmids (fig. S8A) were excised by Hind III/Eco RI digestion and ligated to the Pst I/Eco RI– restricted ARR2PB-containing plasmid (33), after blunting one extremity to obtain the Pb/MCM7 and the Pb/MCM7i13 plasmids. The fragments containing Pb-MCM7-SV40polyA and Pb-MCM7i13-SV40polyAwere released by Not I/Pvu I digestion and injected into the pronuclei of fertilized oocytes in a B6/CBA F1 hybrid background. Southern blot analysis was performed according to standard procedures. The PCR primers used for genotyping are the following: forward primer, 5′ GCTAGAACTAGTGGATCCCCC 3′, and reverse primer, 5′ CATCGTCGTACAGATCCCGAC 3′
Mouse histopathology
Prostates were extracted from euthanized mice and fixed in 4% PFA overnight at 4°C. They were then transferred into 70% ethanol, embedded in paraffin, sectioned and stained with H&E in accordance with standard procedures. IHC and ISH on prostate sections were performed as described above.
Statistical analysis
In vitro and in vivo data were analyzed with unpaired t test (GraphPad Prism, GraphPad Software Inc.). Values of P < 0.05 were considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001). The mean ± SD of three independent experiments is reported. Chi-squared test was used for the analysis of TMA samples (http://www.quantpsy.org).
Supplementary Material
Fig. S1. Direct interaction of miR-17, 19, 22, 25, and 302 with PTEN 3′UTR.
Fig. S2. miR-17, 19, 22, 25, and 302 families cause a decrease in the abundance of PTEN mRNA.
Fig. S3. Cooperation between different miRNAs in decreasing PTEN abundance.
Fig. S4. Score categories for in situ hybridization of miRNAs.
Fig. S5. The concomitant presence of miR-22, miR-25, miR-93, and miR-106b increases the probability of Akt phosphorylation.
Fig. S6. Analysis of DU145 stably infected with members of the miR-106b∼25 cluster.
Fig. S7. sh-Pten partially mimics miR-22 and miR-106b∼25 cluster.
Fig. S8. Constructs for expressing human MCM7 and the intronic miR-106b∼25 cluster.
Fig. S9. Transgene expression in the Pb/MCM7i13 lines.
Fig. S10. MCM7 cannot initiate prostate tumorigenesis.
Fig. S11. Detection of miR-17∼92 miRNAs by ISH in prostate tumor samples.
Table S1. Criteria used by four different prediction algorithms.
Table S2. Criterion 1 and/or Criterion 2 are fulfilled by seven miRNA families.
Table S3. Clinical and pathological characteristics of the patients.
Table S4. Correlation between miRNA up-regulation and DICER overexpression in prostate tumor samples.
Table S5. Primers used to generate chimeric luciferase plasmids.
Table S6. Primers used to generate PIG/22 and PIG/106b∼25 plasmids.
Table S7. Primers used to detect human PTEN 3′UTR by PCR.
Table S8. Primers used to detect human PTEN, MCM7 and intron 13 mRNA by real-time PCR.
Table S9. Primers used to detect mouse Pten by real-time PCR.
Fig. 5.
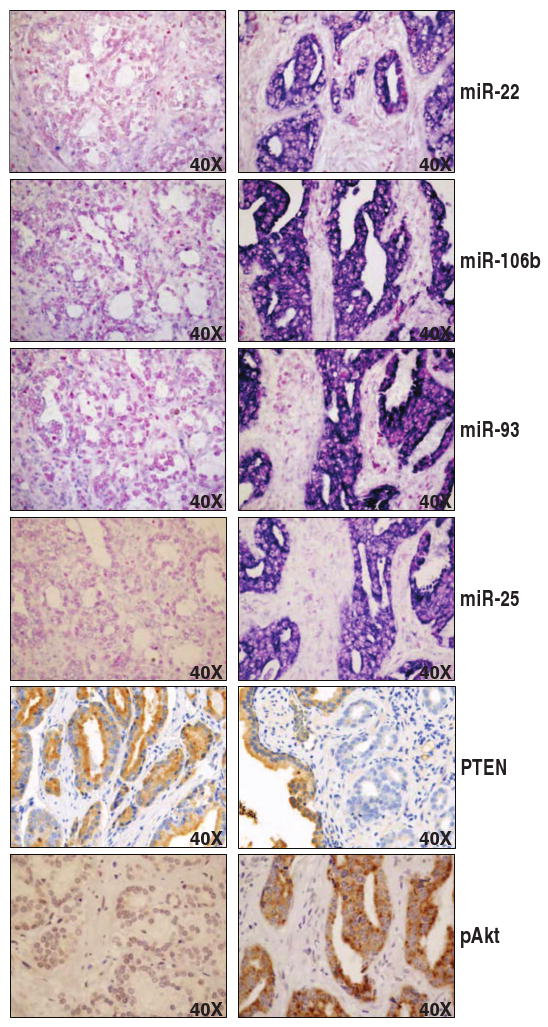
Representative pictures of the prostate TMA. A tumor specimen negative for miRNAs and pAkt and positive for PTEN (left) and a tumor specimen positive for miRNAs and pAkt and negative for PTEN (right) are shown.
Acknowledgments
We thank C. Gewinner for providing sh-Ren and sh-PTEN retroviral vectors and for assistance with lipid extraction and growth in semisolid medium; A. Tuccoli for advice in the analysis of the _target prediction algorithms; and A. Papa for helpful discussion.
Funding: L.P. is supported by a fellowship from the American Italian Cancer Foundation. L.S. is supported by a fellowship from the Canadian Institute of Health Sciences. P.S. is supported in part by the Associazione Umbra Leucemie e Linfomi (AULL). P.P.P. is supported by NIH grant R01 CA-82328-0.
Footnotes
Author contributions: L.P. and P.P.P. designed the project. L.P., L.S., L.R., A.F., M.S.S., R.M.H., P.S., S.V., A. E., G.F., and L.Ra. performed the experiments. L.P., L.S., L.R., R.M.H., and P.P.P. wrote the paper. M.L., and P.P.P. supervised the project. All authors critically discussed the results and the manuscript.
Competing interests: The authors declare that they do not have any competing financial, personal, or professional interest.
References and Notes
- 1.Nomenclature: PTEN, human protein; PTEN, human mRNA and gene; Pten, mouse protein; Pten, mouse mRNA and gene; DICER, human protein; DICER, human mRNA or gene; MCM7, human protein; MCM7, human mRNA and gene; miR-X, mature miRNA; miR-X, miRNA gene.
- 2.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 3.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 4.Chiosea S, Jelezcova E, Chandran U, Acquafondata M, McHale T, Sobol RW, Dhir R. Up-regulation of Dicer, a component of the microRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–1820. doi: 10.2353/ajpath.2006.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 7.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA _targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 9.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foà R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ventura A, Jacks T. MicroRNAs and cancer: Short RNAs go a long way. Cell. 2009;136:586–591. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA _targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 15.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA _target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 16.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA _targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, _targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, Rajewsky K. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132:860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA _targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 21.Takakura S, Mitsutake N, Nakashima M, Namba H, Saenko VA, Rogounovitch TI, Nakazawa Y, Hayashi T, Ohtsuru A, Yamashita S. Oncogenic role of miR-17-92 cluster in anaplastic thyroid cancer cells. Cancer Sci. 2008;99:1147–1154. doi: 10.1111/j.1349-7006.2008.00800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doench JG, Sharp PA. Specificity of microRNA _target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, Nojima H, Looijenga LH, Agami R. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 25.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, Yfantis HG, Stephens RM, Croce CM. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene _targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scharer CD, McCabe CD, Ali-Seyed M, Berger MF, Bulyk ML, Moreno CS. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res. 2009;69:709–717. doi: 10.1158/0008-5472.CAN-08-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa A, Onesti S. The MCM complex: (Just) a replicative helicase? Biochem Soc Trans. 2008;36:136–140. doi: 10.1042/BST0360136. [DOI] [PubMed] [Google Scholar]
- 29.Levesque MH, El-Alfy M, Berger L, Labrie F, Labrie C. Evaluation of AIbZIP and Cdc47 as markers for human prostatic diseases. Urology. 2007;69:196–201. doi: 10.1016/j.urology.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 30.McCarthy S, Caporali A, Enkemann S, Scaltriti M, Eschrich S, Davalli P, Corti A, Lee A, Sung J, Yeatman TJ, Bettuzzi S. Green tea catechins suppress the DNA synthesis marker MCM7 in the TRAMP model of prostate cancer. Mol Oncol. 2007;1:196–204. doi: 10.1016/j.molonc.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laitinen S, Martikainen PM, Tolonen T, Isola J, Tammela TL, Visakorpi T. EZH2, Ki-67 and MCM7 are prognostic markers in prostatectomy treated patients. Int J Cancer. 2008;122:595–602. doi: 10.1002/ijc.23145. [DOI] [PubMed] [Google Scholar]
- 32.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A, Carver B, Gerald W, Cordon-Cardo C, Pandolfi PP. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 2008;22:2172–2177. doi: 10.1101/gad.1699608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 35.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 36.Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, Bikoff R, Ma H, Kantoff PW, Golub TR, Loda M, Sellers WR. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: The MPAKT model. Proc Natl Acad Sci USA. 2003;100:7841–7846. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, Michalopoulos G, Yu YP, Luo JH. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25:1090–1098. doi: 10.1038/sj.onc.1209134. [DOI] [PubMed] [Google Scholar]
- 38.Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R, Nair S, Egan DA, Li A, Huang G, Klein-Szanto AJ, Gimotty PA, Katsaros D, Coukos G, Zhang L, Puré E, Agami R. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10:202–210. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- 39.DeMarzo AM, Nelson WG, Isaacs WB, Epstein JI. Pathological and molecular aspects of prostate cancer. Lancet. 2003;361:955–964. doi: 10.1016/S0140-6736(03)12779-1. [DOI] [PubMed] [Google Scholar]
- 40.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, Isaacs WB, Luo J, De Marzo AM. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, Schramm A, Christiansen H, Eilers M, Eggert A, Berwanger B. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 2008;122:699–704. doi: 10.1002/ijc.23153. [DOI] [PubMed] [Google Scholar]
- 42.Shohet JM, Hicks MJ, Plon SE, Burlingame SM, Stuart S, Chen SY, Brenner MK, Nuchtern JG. Minichromosome maintenance protein MCM7 is a direct _target of the MYCN transcription factor in neuroblastoma. Cancer Res. 2002;62:1123–1128. [PubMed] [Google Scholar]
- 43.Koppen A, Ait-Aissa R, Koster J, van Sluis PG, Ora I, Caron HN, Volckmann R, Versteeg R, Valentijn LJ. Direct regulation of the minichromosome maintenance complex by MYCN in neuroblastoma. Eur J Cancer. 2007;43:2413–2422. doi: 10.1016/j.ejca.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic _targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 46.Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 48.Honeycutt KA, Chen Z, Koster MI, Miers M, Nuchtern J, Hicks J, Roop DR, Shohet JM. Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis. Oncogene. 2006;25:4027–4032. doi: 10.1038/sj.onc.1209435. [DOI] [PubMed] [Google Scholar]
- 49.Padmanabhan V, Callas P, Philips G, Trainer TD, Beatty BG. DNA replication regulation protein Mcm7 as a marker of proliferation in prostate cancer. J Clin Pathol. 2004;57:1057–1062. doi: 10.1136/jcp.2004.016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in Eμ-miR155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maeda T, Hobbs RM, Merghoub T, Guernah I, Zelent A, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP. Role of the proto-oncogene Pokemon in cellular transformation and ARF repression. Nature. 2005;433:278–285. doi: 10.1038/nature03203. [DOI] [PubMed] [Google Scholar]
- 52.Serunian LA, Auger KR, Cantley LC. Identification and quantification of polyphosphoinositides produced in response to platelet-derived growth factor stimulation. Methods Enzymol. 1991;198:78–87. doi: 10.1016/0076-6879(91)98010-4. [DOI] [PubMed] [Google Scholar]
- 53.Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, Zeng C, Baron A, Hunger SP, Varella-Garcia M, Gemmill R, Brizard F, Brizard A, Roche J. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16:186–195. doi: 10.1038/sj.leu.2402354. [DOI] [PubMed] [Google Scholar]
- 54.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. _targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smalheiser NR, Torvik VI. Alu elements within human mRNAs are probable microRNA _targets. Trends Genet. 2006;22:532–536. doi: 10.1016/j.tig.2006.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Direct interaction of miR-17, 19, 22, 25, and 302 with PTEN 3′UTR.
Fig. S2. miR-17, 19, 22, 25, and 302 families cause a decrease in the abundance of PTEN mRNA.
Fig. S3. Cooperation between different miRNAs in decreasing PTEN abundance.
Fig. S4. Score categories for in situ hybridization of miRNAs.
Fig. S5. The concomitant presence of miR-22, miR-25, miR-93, and miR-106b increases the probability of Akt phosphorylation.
Fig. S6. Analysis of DU145 stably infected with members of the miR-106b∼25 cluster.
Fig. S7. sh-Pten partially mimics miR-22 and miR-106b∼25 cluster.
Fig. S8. Constructs for expressing human MCM7 and the intronic miR-106b∼25 cluster.
Fig. S9. Transgene expression in the Pb/MCM7i13 lines.
Fig. S10. MCM7 cannot initiate prostate tumorigenesis.
Fig. S11. Detection of miR-17∼92 miRNAs by ISH in prostate tumor samples.
Table S1. Criteria used by four different prediction algorithms.
Table S2. Criterion 1 and/or Criterion 2 are fulfilled by seven miRNA families.
Table S3. Clinical and pathological characteristics of the patients.
Table S4. Correlation between miRNA up-regulation and DICER overexpression in prostate tumor samples.
Table S5. Primers used to generate chimeric luciferase plasmids.
Table S6. Primers used to generate PIG/22 and PIG/106b∼25 plasmids.
Table S7. Primers used to detect human PTEN 3′UTR by PCR.
Table S8. Primers used to detect human PTEN, MCM7 and intron 13 mRNA by real-time PCR.
Table S9. Primers used to detect mouse Pten by real-time PCR.