

Abstract
With the advent of treatments and diseases such as AIDS resulting in increasing numbers of patients with suppressed immune systems, fungal diseases are an escalating problem. Candida albicans is the most common of these fungal pathogens, causing infections in many of these patients. It is therefore important to understand how immunity to this fungus is regulated and how it might be manipulated. Although work has been done to identify the receptors, fungal moieties, and responses involved in anti-Candida immunity, most studies have investigated interactions with myeloid or lymphoid cells. Given that the first site of contact of C. albicans with its host is the mucosal epithelial surface, recent studies have begun to focus on interactions of C. albicans with this site. The results are startling yet in retrospect obvious, indicating that epithelial cells play an important role in these interactions, initiating responses and even providing a level of protection. These findings have obvious implications, not just for fungal pathogens, but also for identifying how host organisms can distinguish between commensal and pathogenic microbes. This review highlights some of these recent findings and discusses their importance in the wider context of infection and immunity.
Keywords: HIV/AIDS, mycology, innate immunity, oral epithelium, fungal pathogens
With the advent of the AIDS epidemic, with modern medical interventions such as cancer therapy and allogenic transplantation, we have witnessed a rapid rise in the prevalence of fungal infections. This is the combined result of reducing the CD4+ lymphocyte population of the cell-mediated immune system and using immunosuppressive therapies (i.e., steroids). The predisposition of certain patient groups to a multitude of opportunistic fungal infections led to a notable increase in basic research into pathogenic fungi, predominantly on Candida sp., Cryptococcus neoformans, and Aspergillus fumigatus (Odds, 2000). The outcome of this research has led to the unraveling of many fundamental biological processes that take place during host-fungal interactions, particularly with regard to Candida albicans.
Epithelial Innate Immunity and C. albicans
Innate immunity plays the vital role of gatekeeper of the mucosal epithelia. It is required to maintain homeostatic function and coordinate immunologic reactions against commensal and pathogenic microbes. The specialized interaction among resident microbes, epithelial cells, and local immune cells results in either a degree of mutualism (commensalism) or a breach of the mucosal barrier and subsequent cell injury (pathogenicity). Of particular interest are “opportunistic” microbes such as C. albicans, the most common fungal pathogen of humans. Candida spp. reside as commensal organisms in the normal oral, vaginal, and gut flora in approximately half the population. However, if the balance of the normal flora is disrupted or the immune defenses are compromised, Candida spp. often become pathogenic, causing mucosal disease in a significant proportion of immunosuppressed individuals and women of fertile age (Sobel, 1992). The majority of patients experience only superficial forms of mucosal candidiasis—most commonly, thrush. Determining exactly how this transformation from “commensal to pathogen” takes place and how it can be prevented is a continuing challenge. Although many virulence attributes have been suggested to contribute to Candida infections (Naglik et al., 2003), hyphal formation and subsequent invasion have been the most widely studied and most compelling (Calderone and Fonzi, 2001; Zakikhany et al., 2007). Currently, one of the most fundamental immunologic issues is to determine how the host distinguishes between commensal colonization and virulent infection and between the yeast and hyphal form of this fungus. Here, we examine the mechanisms by which the major constituent of any mucosal surface, the epithelial cell, responds to the human fungal pathogen C. albicans.
Pattern Recognition of C. albicans
Detection of pathogens at mucosal surfaces is primarily an innate immune-mediated event, using a multitude of detection systems. Detection almost invariably involves recognition of pathogen-associated molecular patterns—either secreted by or present on the surface of microbes—by a large group of receptors termed pattern recognition receptors (PRRs) (Table). These PRRs include the well-known Toll-like receptors (TLRs), the C-type lectin receptors, and the Nod-like receptors (Kumagai et al., 2008; Hollmig et al., 2009) and have varying cellular locations. Although these 3 families constitute the majority of the currently known PRRs, others exist and more are being discovered each year. Thus, the PRRs form a large arsenal of receptors, all detecting different aspects of a microbial organism.
Table.
Classes of Pattern Recognition Receptors
| PRR | Location | Recognition Molecules | Role in Fungal Responses |
|---|---|---|---|
| TLR | |||
| 1/2 | Surface | Lipoproteins; triacylated lipopeptides; zymosan? | Possibly |
| 2 | Surface | Peptidoglycan; lipoproteins; glycolipids; lipoaribomannan; zymosan | Yes |
| 3 | Endosomal | dsRNA; poly (I:C) | Possibly |
| 4 | Surface/cytoplasmic | Lipopolysacharide; lipid A; fungal O-mannan | Yes |
| 5 | Surface | Flagellin | No |
| 6/2 | Surface | Diacylated lipopeptides; zymosan? | Possibly |
| 7 | Endosomal | ssRNA; imidazoquinolines | No |
| 8 | Endosomal | ssRNA; imidazoquinolines | No |
| 9 | Endosomal | Unmethylated CpG DNA, fungal DNA? | Yes? |
| 10 | Surface? | Unknown | No |
| Lectins | |||
| C-typea | Surface/extracellular | Variable; fungal β-glucans and mannan | Yes |
| Galectins | Cytoplasm/nucleus; extracellular | β-Galactosides (variety of complex N-glycans) | Yes |
| Siglecs | Surface | Variable; sialic acids | No |
| Pentraxinsb | Body fluids | Variable; bind and activate the complement system | Probably |
| LBP | Extracellular | Lipopolysacharide | Possibly |
| NLR | |||
| NOD1/2 | Cytoplasmic | Muramyl dipeptide | Possibly |
| NALP1 | Cytoplasmic | Variable | Probably |
| NALP3 | Cytoplasmic | Variable | Probably |
| RLH | |||
| RIG-I | Cytoplasmic | 5′-triphosphate ssRNA | No |
| MDA-5 | Cytoplasmic | ssRNA | No |
Poly (I:C), polyriboinosinic:polyribocytidylic acid; LBP, lipopolysacharide binding protein; NOD1/2, nucleotide-binding oligomerization domain containing 1 or 2; NALP1/3, NACHT, LRR, and pyrin domain containing protein 1 or 3; RLH, retinoic acid–inducible gene I–like helicases; RIG-I, retinoic acid–inducible gene I; MDA-5, melanoma differentiation-associated gene 5.
Dectin-1/2, MR, DC-SIGN.
PTX3 (Pentraxin 3), CRP (C-reactive protein), SAP (serum amyloid protein), MBL (mannose binding lectin), C1q and C3 (complement components).
The known PRRs utilized by myeloid cells to detect C. albicans pathogen-associated molecular patterns are reasonably well defined—they include TLR2 (phospholipomannan), TLR4 (O-bound mannans), mannose receptor (N-bound mannans), dectin-1 (β-glucans), dectin-2 (hypha-specific high-mannose containing structures), DC-SIGN (dendritic cell–specific intercellular adhesion molecule 3–grabbing nonintegrin: α-linked mannans), galectin-3 (β-1,2 mannosides), and potentially, TLR9 (fungal DNA) (Netea et al., 2008; Roeder et al., 2004a). However, recognition is complex, and although each receptor can function individually, synergy exists between these receptors. In particular, TLR2 and dectin-1 work in conjunction to regulate cytokine responses and phagocytosis (Gantner et al., 2003). However, the PRR-mediated recognition systems that initiate innate responses in epithelial cells are still largely unknown or undefined.
Prr Epithelial Responses to C. albicans
We and others have shown that epithelial cells express a range of PRRs, including TLR 1-6 and 8-10, dectin-1 and galectins, as well as adaptors and coreceptors, such as CD14 and MyD88 (Weindl et al., 2007; Backhed and Hornef, 2003; Hornef and Bogdan, 2005). In further work, we compared TLR expression profiles in oral (TR146) and vaginal (A431) epithelial cells and discovered that the range of TLR expression was similar in both types (unpublished data). TLR 2 and 5 were expressed at high levels, which is notable because stimulation of these TLRs has been associated with epithelial repair, growth, and survival (Shaykhiev et al., 2008; Rhee et al., 2005). In comparison with vaginal epithelial cells (A431 epidermal lineage), oral epithelial cells (TR146 buccal lineage) generally expressed higher levels of most TLRs—particularly, TLR 1, 3, and 6. Interestingly, TLR4 was undetectable or expressed at extremely low levels in both epithelial cells, confirming published reports (Backhed and Hornef, 2003) and indicating that mucosal tissues might be unresponsive to TLR4 stimulation. Indeed, we have demonstrated that oral and vaginal epithelial cells are unresponsive to Escherichia coli lipopolysaccharide (unpublished data). Infection of epithelial cell monolayers and reconstituted human epithelium with C. albicans resulted in strong downregulation of most TLRs, specifically in oral epithelial cells. TLR2 was the only upregulated TLR but only in oral epithelial cells and not vaginal cells. Because the TLRs are generally downregulated and TLR2 has been shown to exhibit a negative or suppressive role (Netea et al., 2004), the data suggest that C. albicans may negatively regulate TLR-mediated sensing in oral epithelial cells. This induction of negative regulation by C. albicans does not occur in myeloid cells, which suggests a specific response in mucosal tissues.
Although epithelial TLR profiles may be modified by C. albicans, the PRRs that epithelial cells utilize to initially recognize C. albicans are currently undefined, although several have been suggested. We have demonstrated that initial activation of epithelial cells is unlikely to be through TLR2, TLR4, or dectin-1, because blocking of these receptors with antibodies does not appear to alter the epithelial cytokine profile (Weindl et al., 2007). Given that mannans and β-glucans are the classic fungal moieties recognized by TLR2, TLR4, or dectin-1 in myeloid cells, these data further confirm that epithelial cell PRR responsiveness is different from that of myeloid cells.
Prr-Induced Epithelial Signalling
PRR ligation of Candida pathogen-associated molecular patterns results in the activation of intracellular signaling pathways, leading to transcriptional activation of genes in the nucleus and, ultimately, the production of novel proteins (Fig. 1). Initiation of signaling involves several potential mechanisms, depending on the PRR (Ivashkiv, 2008; Kenny and O’Neill, 2008; O’Neill, 2008; Tsoni and Brown, 2008). TLRs, for example, contain a Toll/IL-1 receptor domain in their cytoplasmic region that interacts with adaptor proteins—most commonly, MyD88 (myeloid differentiation 88) but also TRIF (Toll/IL-1 receptor domain–containing adapter-inducing interferon β), TRAM (TRIF-related adaptor molecule), and MAL (MyD88 adapter-like protein). This interaction leads to the activation of the IRAK (IL-1 receptor-associated kinase) proteins and TRAF6 (TNF receptor-associated factor 6). In turn, this leads to activation of the NF-κB and MAPK pathways (respectively, nuclear factor kappa light chain enhancer of activated B cells and mitogen-activated protein kinase), as well as IRF (interferon regulatory factor) signaling via IRF 3, 5, and 7.
Fig. 1.
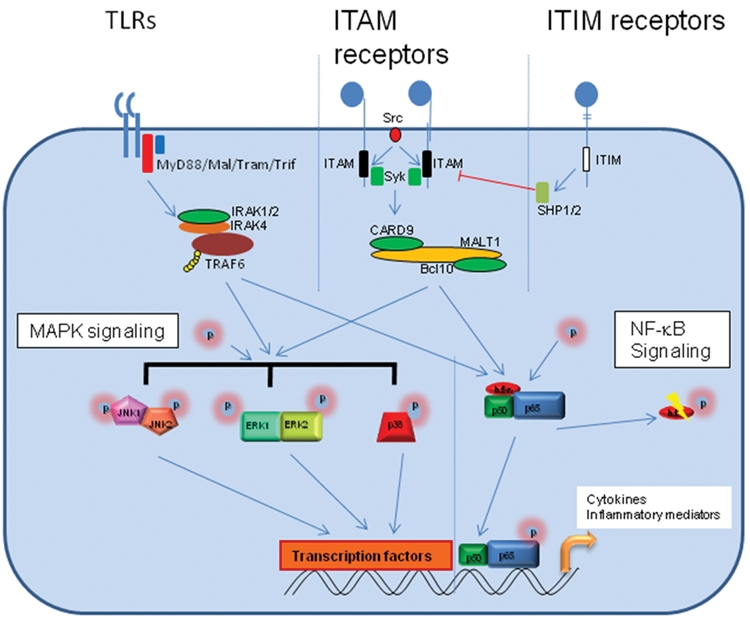
Signal pathway activation by different surface PRRs. All known PRRs activate MAPK and NF-κB signal pathways but may differ in the extent to which they activate these pathways or which combination of these pathways are activated, thus allowing differences in the responses generated. Here we show the different upstream mechanisms by which different families of PRRs activate these pathways. TLRs are the best-described family and utilize Toll/IL-1 receptor domain-containing adapter proteins such as MyD88, MAL, TRAM, and TRIF. The other surface PRRs usually signal using ITAM and ITIM domains. Examples of ITAM domain-containing PRRs include the C-type lectin dectin-1. These proteins either contain an ITAM domain within their cytoplasmic region (as with dectin-1) or associate with an ITAM-containing transducing protein (e.g., dectin-2 with FcRγ). Dectin-1 utilizes Src kinases and Syk kinase to activate a complex containing CARD9, MALT1, and Bcl10 to activate the downstream signal pathways. ITIM domain-containing PRRs are generally considered to be inhibitory, activating the inhibitory phosphatases SHP1 and SHP2, although there is some evidence to suggest that they may also play a role in activation. Examples of these proteins include the S-type lectins or Siglecs.
The MAPK pathways constitute 3 arms: p38, JNK (c-Jun N-terminal kinase), and ERK1/2 (extracellular signal-regulated kinase 1/2). However, C-type lectin receptors are thought to signal through activation of ITAM/ITIM cytoplasmic domains (immunoreceptor tyrosine-based activation/inhibition motif)—through their own (e.g., dectin-1) or through a coreceptor, such as DAP12 (DNAX activation protein of 12 kDa) and FcRγ (Fc gamma chain receptor; e.g., dectin-2). Ligation of these receptors leads to activation of a different set of early adaptors— predominantly, Src family kinases such as Src, Lyn, and Fyn. In the case of dectin-1, this leads to activation of Syk kinase and the downstream activation of the CARD9/Bcl10/MALT1 signaling complex (caspase recruitment domain family/B-cell CLL-lymphoma 10/mucosa-associated lymphoid tissue lymphoma translocation gene 1). Irrespective of the C-type lectin receptor pathways and adapters used, the result is the activation of the NF-κB and MAPK pathways.
Further regulation of these pathways is achieved by activation of other signal-associated proteins. For example, specific regulation of MAPK responses occurs via the action of dual specificity phosphatases, also known as MKPs (MAPK phosphatases; Liu et al., 2007). In the case of ITIM activation, the SHP protein tyrosine phosphatases are activated, which generally suppresses signaling. Which of these pathways and proteins are induced depends on the PRR activated and the heterogeneity of responses generated by the different PRRs will manifest through differences in the activation of alternative elements of these pathways. For example, one pathway may lead to the activation of p38 and JNK but not ERK1/2 MAPK signaling, whereas another may lead to JNK and ERK1/2 but not p38 signaling, with similar differences in the NF-κB elements activated. However, irrespective of the signal pathways activated, there may be cell-specific differences resulting from different expression levels of the various proteins. The majority of studies defining the PRR-mediated pathways were performed using myeloid or lymphoid cells, but detailed analysis of PRR-mediated pathways in other cell types—specifically, epithelial cells—may yet identify novel and unusual mechanisms of pathogen recognition and control at mucosal surfaces.
To this end, the majority of epithelial cell work has been bacterial based and has identified NF-κB and MAPK as the main pathways activated (Kinane et al., 2008; Handfield et al., 2008). NF-κB is also thought to be a likely signaling candidate in the activation of epithelial cells to Candida (Pivarcsi et al., 2003; Roeder et al., 2004b). However, in a recent study using oral epithelial cells we demonstrated that the innate response to C. albicans is via both NF-κB and MAPK pathways (Moyes et al., 2010). We found that NF-κB activation appears to be independent of morphology and results from generic recognition of fungal moieties present on all Candida spp. and, probably, fungi in general. The MAPK network appears to elicit the “danger response” and is dependent on hyphal recognition and fungal dose. Further detailed characterization of these signaling pathways in epithelial cell types is required, which will advance our understanding of how human mucosal surfaces recognize, respond to, and control these important fungal pathogens.
C. albicans–Induced Effector Responses
The terminal stages of PRR ligation is the activation of an “effector response” profile in the _target cell, which depends on the cell type. In the case of myeloid cells, it usually results in the activation of immune-specific responses, including production of either proinflammatory or anti-inflammatory cytokines. For example, activation of TLR4 on macrophages by lipopolysaccharide leads to the release of IL-1α, TNFα, and IFNγ (Iwasaki and Medzhitov, 2004). Recognition of Candida is similar in this respect, with IL-12, TNFα, and IL-1 (as well as other commonly induced cytokines) being released after stimulation of immune cells (Netea et al., 2008). Also, activation via TLR2 (potentially in conjunction with dectin-1) on macrophages leads to the secretion of IL-12 and TNFα.
With respect to epithelial cells, we and others have shown the secretion of a general profile of proinflammatory cytokines and chemokines after stimulation with C. albicans, which includes IL-1α/β, IL-6, G-CSF, GM-CSF, and TNFα, as well as the chemokines IL-8 and RANTES (Weindl et al., 2007; Schaller et al., 2004; Villar et al., 2005; Dongari-Bagtzoglou and Fidel, 2005). Unlike myeloid and lymphoid cells, epithelial cells do not produce IL-12, IFNγ, IL-4, or IL-13. The effect that these cytokines have on epithelial function and protective responses against Candida is currently unclear. However, it is unwise to assume that epithelial cells work independently from other cells. The mucosal surface, though predominantly made up of epithelial cells, includes a variety of other immune cells, such as dendritic cells and neutrophils. Indeed, the naturally high levels of IL-8 released by epithelial cells will actively recruit neutrophils into mucosal tissues, which may constitute an immune surveillance mechanism. The importance of this becomes apparent in the light of recent work that we have described with our collaborators, demonstrating a protective interaction between these 2 cell types against oral candidiasis (Weindl et al., 2007).
Using 3-dimensional organotypic oral (TR146) models, when applied alone, C. albicans induces a proinflammatory cytokine response but fails to signal through or significantly modulate TLR expression. However, addition of polymorphonuclear (PMN) cells to the C. albicans–infected model strongly upregulates TLR4 (~100-fold) and protects the epithelium against fungal invasion and cell damage. No alteration in TLR4 expression or protection was observed when PMN cells were added in the absence of C. albicans. Of note, C. albicans–induced epithelial cell damage was abolished in the presence of PMN cells irrespective of whether the PMN cells were applied apically or basally. This demonstrated that (1) PMN-dependent protection against C. albicans infection was independent of PMN migration or direct cell-cell contact with oral epithelial cells and (2) 3-way communication among all 3 cell types (pathogen, epithelial cell, and immune cell) was essential for TLR4 upregulation and subsequent protection. Furthermore, Candida invasion and cell injury could be restored by TLR4 blockade (antibody) or “knockdown” of TLR4 using siRNA (short interfering RNA), even in the presence of PMN cells, demonstrating a direct role of epithelial TLR4 in antifungal protective responses. Our data indicate that although epithelial TLR4 does not seem to be utilized in the initiation of responses to C. albicans, it is nevertheless involved in mediating a secondary protective effect against C. albicans infection in the presence of PMN cells. This is the first description of such a PMN-dependent TLR4-mediated protective mechanism at epithelial surfaces (Fig. 2.). It also indicates that, far from being passive cells, epithelial cells can play a major part in host defense against invading pathogens and may provide significant insights into how fungal infections are managed and controlled in the oral mucosa.
Fig. 2.
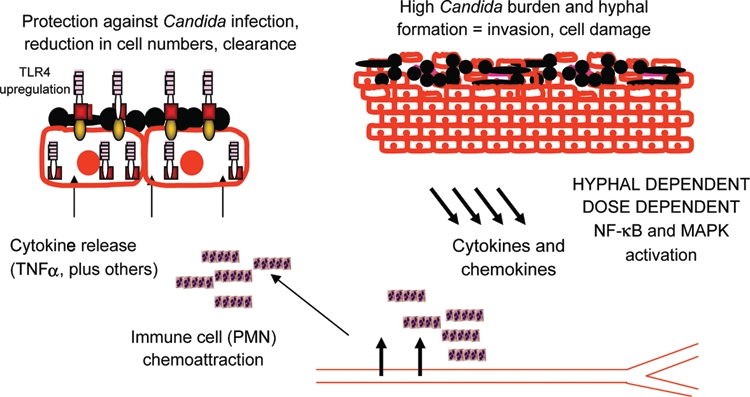
Model for protective innate immune responses against C albicans at oral epithelial surfaces in vivo. C albicans induces cytokine and chemokine production from the epithelium, predominantly through activation of NF-κB and MAPK signaling pathways. This is dose dependent and relies on hypha formation and probably invasion. PMN cells are recruited from blood vessels and secrete TNFα (and other cytokines), which leads to the upregulation of epithelial TLR4 and, subsequently, the protection against C albicans tissue damage and invasion through an as-yet-unknown mechanism.
Conclusions
The host epithelial mechanisms enabling discrimination between the commensal and pathogenic states of this fungus are not fully understood. Different Candida spp. and different morphologic forms of C. albicans appear to induce a variety of signaling pathways and effector profiles in epithelial cells. We believe that hypha formation and cell invasion are key determinants in activating the MAPK-based danger response pathways, whereas NF-κB activation constitutes generic recognition of fungal moieties present on all Candida spp. and is independent of hypha formation. Further work into how the yeast and hyphal phases of C. albicans induce MAPK-based responses will be crucial to determine whether activation of these signaling networks results in a protective host phenotype in vivo. Likewise, it will be vital to identify the yeast- and hypha-specific moieties that subvert or activate protective host mechanisms against C. albicans. This will significantly advance our understanding of how epithelial cells respond to this and, potentially, other fungal mucosal pathogens. Ultimately, the successful _targeting of specific epithelial signaling networks or novel fungal processes/proteins resulting in the prevention or control of infection could provide a basis for novel therapeutic strategies against a multitude of mucosal pathogens.
Footnotes
This work is supported by the National Institute of Dental and Craniofacial Research (DE017514). We also acknowledge financial support from the Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London.
References
- Backhed F, Hornef M. (2003). Toll-like receptor 4-mediated signaling by epithelial surfaces: necessity or threat? Microbes Infect 5:951-959 [DOI] [PubMed] [Google Scholar]
- Calderone RA, Fonzi WA. (2001). Virulence factors of Candida albicans. Trends Microbiol 9:327-335 [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Fidel PL., Jr (2005). The host cytokine responses and protective immunity in oropharyngeal candidiasis. J Dent Res 84:966-977 [DOI] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. (2003). Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 197:1107-1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handfield M, Baker HV, Lamont RJ. (2008). Beyond good and evil in the oral cavity: insights into host-microbe relationships derived from transcriptional profiling of gingival cells. J Dent Res 87:203-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmig ST, Ariizumi K, Cruz PD., Jr (2009). Recognition of non-self- polysaccharides by C-type lectin receptors dectin-1 and dectin-2. Glycobiology 19:568-575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornef MW, Bogdan C. (2005). The role of epithelial Toll-like receptor expression in host defense and microbial tolerance. J Endotoxin Res 11:124-128 [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB. (2008). A signal-switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. Nat Rev Immunol 8:816-822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. (2004). Toll-like receptor control of the adaptive immune responses. Nat Immunol 5:987-995 [DOI] [PubMed] [Google Scholar]
- Kenny EF, O’Neill LA. (2008). Signalling adaptors used by Toll-like receptors: an update. Cytokine 43:342-349 [DOI] [PubMed] [Google Scholar]
- Kinane DF, Galicia JC, Gorr SU, Stathopoulou PG, Benakanakere M. (2008). P gingivalis interactions with epithelial cells. Front Biosci 13:966-984 [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Takeuchi O, Akira S. (2008). Pathogen recognition by innate receptors. J Infect Chemother 14:86-92 [DOI] [PubMed] [Google Scholar]
- Liu Y, Shepherd EG, Nelin LD. (2007). MAPK phosphatases regulating the immune response. Nat Rev Immunol 7:202-212 [DOI] [PubMed] [Google Scholar]
- Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj S, et al. (2010). A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in Epithelial Cells. Cell Host and Microbe 8:225-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naglik JR, Challacombe SJ, Hube B. (2003). Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 67:400-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Brown GD, Kullberg BJ, Gow NA. (2008). An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 6:67-78 [DOI] [PubMed] [Google Scholar]
- Netea MG, Sutmuller R, Hermann C, Van Der Graaf CA, Van Der Meer JW, van Krieken JH, et al. (2004). Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol 172:3712-3718 [DOI] [PubMed] [Google Scholar]
- Odds FC. (2000). Pathogenic fungi in the 21st century. Trends Microbiol 8:200-201 [DOI] [PubMed] [Google Scholar]
- O’Neill LA. (2008). “Fine tuning” TLR signaling. Nat Immunol 9:459-461 [DOI] [PubMed] [Google Scholar]
- Pivarcsi A, Bodai L, Rethi B, Kenderessy-Szabo A, Koreck A, Szell M, et al. (2003). Expression and function of Toll-like receptors 2 and 4 in human keratinocytes. Int Immunol 15:721-730 [DOI] [PubMed] [Google Scholar]
- Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. (2005). Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci U S A 102:13610-13615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder A, Kirschning CJ, Rupec RA, Schaller M, Weindl G, Korting HC. (2004a). Toll-like receptors as key mediators in innate antifungal immunity. Med Mycol 42:485-498 [DOI] [PubMed] [Google Scholar]
- Roeder A, Kirschning CJ, Schaller M, Weindl G, Wagner H, Korting HC, et al. (2004b). Induction of nuclear factor- kappa B and c-Jun/activator protein-1 via toll-like receptor 2 in macrophages by antimycotic-treated Candida albicans. J Infect Dis 190:1318-1326 [DOI] [PubMed] [Google Scholar]
- Schaller M, Boeld U, Oberbauer S, Hamm G, Hube B, Korting HC. (2004). Polymorphonuclear leukocytes (PMNs) induce protective Th1-type cytokine epithelial responses in an in vitro model of oral candidosis. Microbiology 150(pt 9):2807-2813 [DOI] [PubMed] [Google Scholar]
- Shaykhiev R, Behr J, Bals R. (2008). Microbial patterns signaling via Toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS ONE 3:e1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel JD. (1992). Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clin Infect Dis 14(suppl 1):148-153 [DOI] [PubMed] [Google Scholar]
- Tsoni SV, Brown GD. (2008). Beta-glucans and dectin-1. Ann N Y Acad Sci 1143:45-60 [DOI] [PubMed] [Google Scholar]
- Villar CC, Kashleva H, Mitchell AP, Dongari-Bagtzoglou A. (2005). Invasive phenotype of Candida albicans affects the host proinflammatory response to infection. Infect Immun 73:4588-4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, et al. (2007). Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J Clin Invest 117:3664-3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakikhany K, Naglik JR, Schmidt-Westhausen A, Holland G, Schaller M, Hube B. (2007). In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell Microbiol 9:2938-2954 [DOI] [PubMed] [Google Scholar]