

Abstract
Bone is a highly vascularized tissue, but the function of angiogenesis in bone modeling and remodeling is still poorly defined, and the molecular mechanisms that regulate angiogenesis in bone are only partially elucidated. Genetic manipulations in mice have recently highlighted the critical role of the hypoxia-inducible-factor/vascular endothelial growth factor pathway in coupling angiogenesis and osteogenesis. In this brief perspective, we review the current understanding of the mechanisms responsible for this coupling. Elucidation of such mechanisms will expand our knowledge of bone development and homeostasis, and it may aid in the design of new therapies for accelerating bone regeneration and repair.
Keywords: hypoxia inducible factor, vascular endothelial growth factor, osteoblast, angiogenesis
INTRODUCTION
Bone is a highly vascularized and heterogeneous tissue that forms through at least two independent mechanisms: intramembranous and endochondral ossification.(1) The first, in which mesenchymal cells develop directly into osteoblasts, is involved in the formation of the flat bones of the skull. The second, accounting for the development of most other bones, involves a two-stage mechanism, whereby chondrocytes form a matrix template, the growth plate, which is replaced by bone. During endochondral bone development, growth plate chondrocytes undergo well-ordered and controlled phases of cell proliferation, maturation, and death. This unique differentiation process is followed by blood vessel invasion and replacement of the cartilaginous matrix with bone.(2–5)
Osteoblasts or bone-forming cells are thought to originate from undifferentiated mesenchymal cells whose commitment to osteoblasts is regulated by at least two transcription factors: Runx2 and Osterix.(6,7) According to the current model, committed osteoprogenitors proliferate, differentiate into postmitotic osteoblasts that synthesize and mineralize bone matrix, and finally become either terminally differentiated osteocytes encased into the bony matrix or bone-lining cells. The identification of cells that are osteoprogenitors has been difficult, but their presence in the bone marrow stroma can be confirmed by their functional capacity to divide and differentiate in vitro into bone nodule–forming osteoblasts.(8)
Blood vessel invasion is a critical event in the replacement of cartilage by bone and in the formation of the bone marrow cavity. Vascular endothelial growth factor (VEGF)-A is one of the critical mediators of blood vessel invasion of the cartilaginous mold. Five VEGF-A isoforms have been identified in humans, whereas there are three major isoforms in the mouse (VEGF120, VEGF164, and VEGF188). VEGF-A binds to and activates two tyrosine kinase receptors, VEGFR1 (Flt-1) and VEGFR2 (KDR/Flk-1), which regulate both physiological and pathological angiogenesis.(9) In the embryo, VEGF signaling is essential for angiogenesis, because the deletion of even a single copy of the VEGF-A gene results in embryonic lethality because of defective vascular development.(10,11) During endochondral bone development, VEGF-A is produced by both chondrocytes, particularly in their later stages of terminal differentiation, and by osteoblasts.(12–14) Altering the expression or the levels of VEGF has a profound impact on vascular invasion of the cartilaginous mold. Mice expressing only the soluble form of VEGF, VEGF120, but lacking VEGF188 and VEGF164 exhibit delayed blood vessel invasion during endochondral bone development.(15,16) Similarly, administration of the VEGF inhibitor mFlt(1–3)–IgG completely blocked neoangiogenesis in the growth plates of 24-day-old mice.(17)
Whereas cartilage is an avascular and hypoxic mesenchymal tissue,(18–22) bone is highly vascularized, although the bone marrow is relatively hypoxic compared with other adult organs (see below).(23) It is obvious to assume that blood vessels are critical in the biology of bone as providers of nutrients. However, it is also becoming progressively evident that the biological role of blood vessels in bone goes beyond being a mere source of nutrients. For example, progenitors of osteoblasts have been reported to be present in the wall of human bone marrow blood vessels.(24) All in all, the function of angiogenesis in bone modeling and remodeling is still poorly defined, and the molecular mechanisms that regulate angiogenesis in bone are only partially elucidated.
In recent years, it has been shown that hypoxia is a major driving force for angiogenesis and VEGF-A expression by stabilizing the hypoxia inducible factors (HIFs) protein.(25) Hypoxia is not an absolute concept, but it is rather a relative decrease of O2 availability. The definition of “physiologically” normoxic conditions for either embryonic or adult cells varies significantly. Before the circulatory system is established, mammalian development proceeds in a relatively low O2 environment of ∼3%.(26,27) Moreover, studies that have used small-molecule hypoxia markers have shown the existence of specific regions of moderate to severe hypoxia in the developing embryos.(28,29) In the majority of normal adult tissues, oxygen (O2) levels vary between 2% and 9% (compared with ambient air that contains 21% O2).(23) In contrast, O2 concentrations in regions of the bone marrow, cartilage, kidney medulla, and thymus are <1% O2.(23) Hypoxia is not only a critical factor in fetal development and differentiation but is also a pathophysiological component of many human disorders, including cancer and ischemic diseases.(20,28–30)
HIF-1, a ubiquitously expressed transcription factor, is a major regulator of cellular adaptation to hypoxia.(31–35) It is a heterodimeric DNA-binding complex that consists of two basic helix-loop-helix (bHLH) proteins of the PER/ARNT/SIM (PAS) subfamily: HIF-1α and HiF-1β.(36) HIF-1α and HIF-1 β mRNAs are ubiquitously expressed.(37) In general, α-class members of the PAS subfamily respond to environmental signals, whereas β-class molecules aid in _targeting the heterodimer to their nuclear _targets.(38) In the HIF-1 system, HIF-1α levels increase exponentially as O2 levels drop below 5%.(39–44) On the other hand, HIF-1β (also known as aryl hydrocarbon nuclear translocator or ARNT) is non–oxygen responsive. On heterodimerization with HIF-1α, the HIF-1α:HIF-1β complex binds to a specific sequence 5′-RCGTG-3′ (where R denotes a purine residue) termed hypoxia response elements (HREs) and transactivates _target genes containing HREs.(45) HIF-1α does not directly sense variations of O2 tension(46); a class of 2-oxoglutarate–dependent and Fe2+-dependent dioxygenases are the O2 sensors.(39) Two types of O2 sensors are involved in HIF-1α action: prolyl-hydroxylase domain proteins (PHDs) and an asparaginyl hydroxylase, respectively. PHDs hydroxylate two prolyl residues (P402 and P564) in the HIF-1α region referred to as the O2-dependent degradation domain (ODDD).(47) This modification occurs in normoxic conditions and mediates the binding of the von Hippel-Lindau tumor suppressor protein (pVHL), which is an E3 ubiquitin ligase, to HIF-1α. HIF-1α is marked with polyubiquitin chains and _targeted for degradation by the proteasome. In well-oxygenated tissues, where O2 tension is >5%, HIF-1α displays one of the shortest half-lives (<5 min) among cellular proteins. Conversely, under hypoxic conditions, the activity of the PHDs is largely impaired, and proline hydroxylation cannot occur. As a result, HIF-1α protein accumulates, and this initiates a multistep pathway that includes nuclear translocation of HIF-1α, dimerization with its partner HIF-1β, recruitment of transcriptional co-activators, and binding to HREs within the promoters of hypoxia-responsive genes.(48) The second type of O2 sensor is an asparaginyl hydroxylase called factor inhibiting HIF-1 (FIH-1).(49,50) This enzyme hydroxylates an asparagine residue (N803) in the carboxy-terminal transcriptional activation domain (C-TAD) of HIF-1α. This covalent modification blocks C-TAD interaction with transcriptional co-activators, such as p300 and CBP. Thus, the two O2 sensors, PHD and FIH, by regulating the destruction and activity of HIF-1α, respectively, ensure the repression of the HIF-1 pathway in well-oxygenated cells.
To date, >100 putative HIF-1 _target genes have been identified.(51–54) They are involved in a wide variety of biological processes including energy metabolism, angiogenesis, erythropoiesis, cell survival, apoptosis, redox, and pH regulation.(53,55) Mouse embryos lacking HIF-1α exhibit multiple morphological defects as early as embryonic day E8.5 and die in utero by E10.5.(56–58) Many malignant cancers contain regions of severe hypoxia, resulting in high levels of HIF-1α that drive tumor progression,(32,35) and inhibition of HIF-1α has been proposed as a potentially powerful approach.(59)
pVHL is expressed in most tissues and cells.(60) Heterozygous germline missense mutations of the VHL gene are the cause of von Hippel Lindau syndrome,(61,62) a disease characterized by a dominant predisposition to develop pheochromocytomas and highly vascular tumors of the kidney, central nervous system, and retina.(61,62) Tumorigenesis results from the loss or inactivation of the wildtype allele.(61,62) The importance of pVHL for proteolysis of HIF-1α is underscored by the finding that cells lacking functional pVHL have dramatically reduced ability to degrade this transcription factor, resulting in accumulation of high levels of HIF-1α under normoxic conditions.(61,62)
Stimuli other than hypoxia also cause HIF-1α to accumulate in normoxic cells. For example, growth factors such as IGF-1 can induce HIF-1α synthesis through activation of the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signal transduction pathway.(63–66)
Besides HIF-1α, two proteins with sequence similarity to HIF-1α have been characterized: HIF-2α and HIF-3α.(67) HIF-1α and HIF-2α have a similar protein structure and undergo the same oxygen-dependent proteolysis. This may indicate that they are functionally redundant, at least in some settings.(68) However, the pattern of expression of HIF-2α is largely restricted to blood vessels, neural crest, and distinct cell populations in the brain, heart, lung, kidney, liver, pancreas, and intestine,(69) whereas HIF-1α is expressed in all cells. Moreover, mice that are null for HIF-1α die at early stages of embryonic development, but mice deficient in HIF-2α survive until mid-to-late gestation or, depending on the strain, until birth.(56–58,70–74) The two isoforms therefore seem to have distinct developmental functions. Last, some genes are activated by either HIF-1α or HIF-2α, whereas others are only activated by one or the other.(23,75–77) HIF-3α is not closely related to HIF-1α and HIF-2α.(78) Alternative splicing of the HIF-3α primary RNA transcript produces mRNAs that encode at least six different protein isoforms,(79) one of which is an inhibitory protein that contains the N-terminal bHLH and PAS domains but lacks the C-TAD.(80) This protein acts as a negative regulator of HIF-mediated gene expression.
The next two sections of this brief perspective will summarize our current knowledge about the role of the transcription factor HIF-1α as an essential modulator of osteoblast-angiogenic coupling, particularly in the trabecular compartment of the long bones.
HIFS AND ANGIOGENESIS/OSTEOGENESIS COUPLING IN BONE DEVELOPMENT
Hypoxia is likely one of the major drivers of the tight coupling between angiogenesis and bone formation. Osteoblasts, like all other nucleated metazoan cells, express components of the HIF-1 pathway. Studies in the late 1990s have shown that hypoxia is a potent stimulator of VEGF-A mRNA expression in osteoblastic cells.(81) More recently, manipulation of the HIF-1α pathway in osteoblasts has led to altered VEGF-A levels and dramatic changes in bone mass.(82) Indeed, mutant mice that lack VHL in fully differentiated osteoblasts (ΔVHL) and thus overexpress HIFs have a strikingly increased bone volume, which was secondary, at least at early stages, to an increase of osteoblast number and of bone formation rate in absence of detectable changes in osteoclast number and/or activity. Conversely, lack of HIF-1α in osteoblasts (ΔHIF-1α) negatively impacts bone volume. The amount of bone in both ΔVHL and ΔHIF-1α mice is directly proportional to the degree of skeletal vascularization. This suggests that the regulation of bone mass in these mutants may be secondary to changes in VEGF-A levels and angiogenesis. Consistent with this idea, VEGF-A mRNA expression is upregulated in trabecular bone of ΔVHL mice. In addition, in an ex vivo assay, ΔVHL metatarsals exhibit a dramatic increase in endothelial sprouting, which is entirely reversed by preincubation with an anti-VEGF neutralizing antibody. However, the putative mechanisms responsible for coupling angiogenesis to osteogenesis physiologically, as well as in both ΔVHL and ΔHIF-1α mice, remain to be determined. It has been proposed that the bone marrow vascular setting provides a true niche for pericytic mesenchymal stem cell (MSC)-like cells and could be a source of osteoprogenitors or of MSCs with osteogenic potential.(24,83) Thus, the VEGF-dependent increase in angiogenesis observed in ΔVHL mice may lead to more bone volume by providing a larger pool of MSCs. Not mutually exclusive, HIF stabilization or inactivation may also affect osteoblasts directly and independently of angiogenesis. Although cell autonomous effects were not detected by in vitro assays of proliferation, differentiation, and apoptosis, prolonged alterations in HIF activity in vivo may modulate cellular metabolism, matrix formation, or autophagy as proposed for other cell types.(20) Moreover, VEGF-A itself has also been reported to have a direct action on osteoblast differentiation. In particular, mice that express only the VEGF120 isoform exhibit both delayed invasion of vessels into the primary ossification center and altered osteoblastic differentiation in vitro.(16) Interestingly, however, transient hypoxia has been shown to be an inhibitor of osteoblast differentiation in vitro,(84) which further suggests that the dramatic increase in bone volume in mice lacking pVHL in osteoblasts is not a cell autonomous effect but rather results from the increase in blood vessels mediated by the increased VEGF levels.
Numerous factors other than hypoxia increase HIF protein levels in osteoblasts, which consequently leads to increased VEGF-A expression; an example is IGF-1. In human osteoblast-like cells, IGF-1 induces a rapid, 3-fold increase in VEGF-A mRNA.(85) This is accompanied by an increase in HIF-2α protein without a corresponding change in HIF-2α mRNA expression.(85) IGF-I also stimulates the phosphorylation of Akt, an effect that is abolished by pretreating the cells with the phosphatidylinositol-3 kinase (PI3K) inhibitor LY294002. Treatment with this inhibitor also significantly reduced HIF-2 α accumulation and the induction of VEGF mRNA expression by IGF-1. Thus, IGF-1 seems to induce VEGF-A expression in osteoblasts by increasing accumulation of HIF-2α protein levels in a PI3K-dependent fashion.(85) These findings highlight a potential role for HIF-2α in osteoblasts, a finding that needs to be verified in vivo.
Interestingly, manipulation of HIF levels in mature osteoblasts does not noticeably influence the formation of the flat bones of the skull.(82) The calvarial bones are formed through an intramembranous process in which mesenchymal cells differentiate directly into osteoblasts without an intermediate avascular cartilaginous template. It is possible that signals from cranial sutures and/or from the dura induce the angiogenic response necessary for intramembranous ossification or that VEGF is regulated by other factors than HIF in calvarial osteoblasts. This would explain the lack of both blood vessel and bone phenotypes in the skull of ΔVHL and ΔHIF-1α mutant mice.
HIFS AND ANGIOGENESIS/OSTEOGENESIS COUPLING IN REGENERATION AND REPAIR
Angiogenesis is essential for bone repair. It has been proposed that, at fracture sites, mechanical stimuli and inflammatory signals, along with hypoxia, which results when the vascular and nutrient supply is interrupted, initiate the events that lead to bone repair.(86) When angiogenesis is delayed, chondrocytic cells rather than osteoblasts make up the healing tissue. This suggests that Hifs play a role in allocating mesenchymal lineage during repair.(87)
Distraction osteogenesis (DO) is a valuable model for examining the cellular mechanisms that couple angiogenesis and bone formation during repair and regeneration. In DO, intramembranous bone formation is induced by the application of an external fixation device that applies gradual mechanical distraction across an osteotomy.(88) This procedure leads to a close temporal and spatial relationship between bone formation and angiogenesis.(86) DO has also been used to investigate the role of HIF-1α in bone healing. In ΔVHL mice, DO is characterized by increases in HIF-1α protein, in VEGF-A mRNA and protein, and in a number of endothelial cells, leading to more blood vessels and more dense woven bone.(89) At DO sites in ΔHIF-1α mice, the opposite takes place, namely deficient angiogenesis and delayed bone consolidation.(89) Additionally, the mRNA and protein expressions of VEGF-A and of osteoblast markers (Runx2, alkaline phosphatase, and osteocalcin) are decreased in this animal model, and, conversely, increased in ΔVHL mice.(90) Perhaps not surprisingly, desferrioxamine, a small molecule that when administered directly into the distraction gap blocks PHD activity and thus elevates HIF-1α can improve healing in a manner virtually identical with that seen when HIF-1α is activated.(89) These studies provide proof of principle that a therapeutical approach that modulates the HIF pathway may speed bone healing.
Numerous studies have highlighted the role of VEGF-A receptor signaling in bone repair and regeneration. Both receptors, which have different affinities for the VEGF-A ligands as well as different downstream effects,(91) are expressed by osteoblasts.(92,93) During normal DO, both VEGFR1 and VEGFR2 and all three VEGF-A isoform mRNAs are induced. Moreover, inhibition of VEGF-A activity in the distraction gap by antibody blockade of VEGFR1 and VEGFR2 leads to a dramatic decrease of bone formation and a smaller number of blood vessels.(94) Of note, the VEGF-A homolog placental growth factor (PlGF), which binds VEGFR1 as well, probably contributes, because fracture healing is impaired in mice lacking PlGF.(95)
CONCLUSION
A growing body of evidence shows that angiogenesis plays a critical role in skeletal development and repair. It has been suggested that increasing numbers of blood vessels introduce more osteoblast progenitors that mature and increase bone formation (Fig. 1). It is also possible that signals emanating from vascular cells hasten osteogenesis (Fig. 1). Further elucidation of the mechanisms that are responsible for the osteoblast-angiogenesis coupling will deepen our understanding of bone development and homeostasis, and it may also aid in the design of new therapies for accelerating bone regeneration and repair.
FIG. 1.
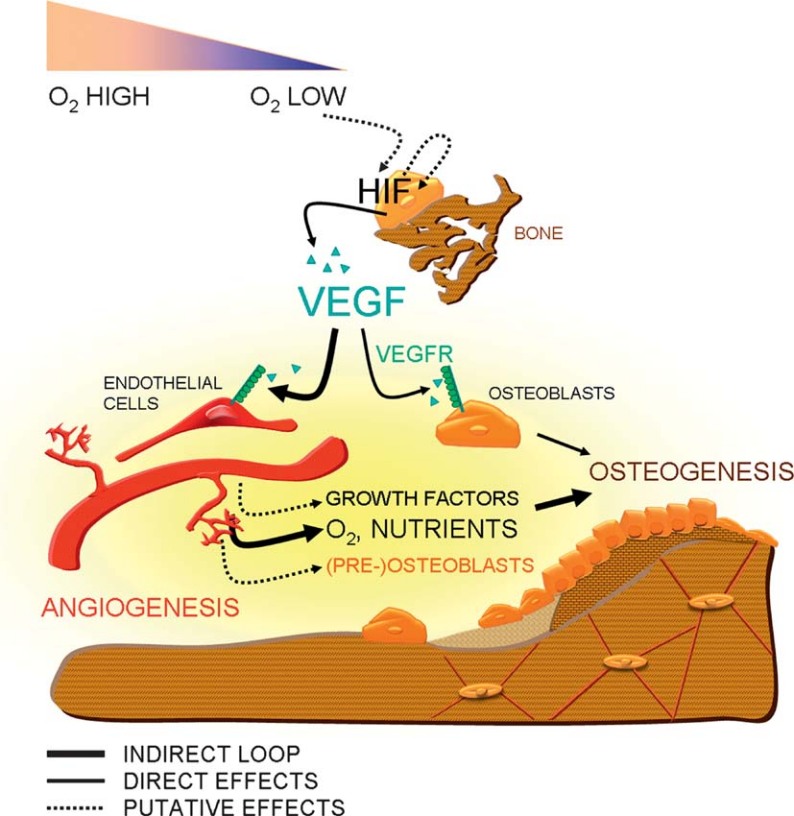
Regulation of osteogenesis-angiogenesis coupling by HIF and VEGF. Mature osteoblasts located on the bone surface express HIFs and respond to HIF activation, which may be induced in response to low oxygen tensions in the bone and marrow environment. Cell-autonomous effects of HIF may affect bone formation (osteogenesis), but a critical effect of HIF stabilization in mature osteoblasts is the increased accumulation of VEGF. VEGF can act through its receptors (VEGFR) on endothelial cells to induce angiogenesis and thereby indirectly increase the supply of oxygen and nutrients required for osteogenesis. Increased vascularization may also lead to a higher input of putative skeletal stem cells and/or (pre)osteoblasts and to elevated levels of endothelium-derived osteogenic growth factors or anabolic signals. In addition, VEGF can affect osteogenesis through direct interactions with osteoblasts that also express VEGF receptors. Altogether, the HIF–VEGF pathway is likely to be critically important in coupling the processes of angiogenesis and osteogenesis.
REFERENCES
- 1.Karsenty The complexities of skeletal biology. Nature. 2003;423:316–318. doi: 10.1038/nature01654. [DOI] [PubMed] [Google Scholar]
- 2.Kronenberg H. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 3.Zelzer E, Olsen B. The genetic basis for skeletal diseases. Nature. 2003;423:343–348. doi: 10.1038/nature01659. [DOI] [PubMed] [Google Scholar]
- 4.Provot S, Schipani E. Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun. 2005;328:658–665. doi: 10.1016/j.bbrc.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 5.Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75:200–212. doi: 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- 6.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 7.Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13:1025–1036. doi: 10.1101/gad.13.8.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bianco P, Robey P, Simmons P. Mesenchymal stem cells: Revisiting history, concepts and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–478. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 11.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by _targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 12.Pfander D, Kobayashi T, Knight M, Zelzer E, Chan D, Olsen B, Giaccia A, Johnson R, Haase V, Schipani E. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development. 2004;131:2497–2508. doi: 10.1242/dev.01138. [DOI] [PubMed] [Google Scholar]
- 13.Zelzer E, Mamluk R, Ferrara N, Johnson R, Schipani E, Olsen B. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171. doi: 10.1242/dev.01053. [DOI] [PubMed] [Google Scholar]
- 14.Zelzer E, Olsen B. Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth and repair. Curr Top Dev Biol. 2005;65:169–187. doi: 10.1016/S0070-2153(04)65006-X. [DOI] [PubMed] [Google Scholar]
- 15.Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, Bouillon R, Carmeliet G. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111:61–73. doi: 10.1016/s0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 16.Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D'Amore PA, Olsen BR. Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- 17.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 18.Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Provot S, Zinyk D, Gunes Y, Khatri R, Le Q, Kronenberg H, Johnson R, Longaker M, Giaccia A, Schipani E. HIF-1alpha regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol. 2007;177:451–464. doi: 10.1083/jcb.200612023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Provot S, Schipani E. Fetal growth plate: A developmental model of cellular adaptation to hypoxia. Ann N Y Acad Sci. 2007;1117:26–39. doi: 10.1196/annals.1402.076. [DOI] [PubMed] [Google Scholar]
- 21.Maes C, Stockmans I, Moermans K, Looveren RV, Smets N, Carmeliet P, Bouillon R, Carmeliet G. Soluble VEGF isoforms are essential for establishing epiphyseal vascularization and regulating chondrocyte development and survival. J Clin Invest. 2004;113:188–199. doi: 10.1172/JCI19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schipani E. Hypoxia and HIF-1 alpha in chondrogenesis. Semin Cell Dev Biol. 2005;16:539–546. doi: 10.1016/j.semcdb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacchetti B, Funari A, Michienzi S, DiCesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey P, Riminucci M, Bianco P. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–326. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 25.Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19:12–16. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell JA, Yochim JM. Intrauterine oxygen tension during the estrous cycle in the rat: Its relation to uterine respiration and vascular activity. Endocrinology. 1968;83:701–705. doi: 10.1210/endo-83-4-701. [DOI] [PubMed] [Google Scholar]
- 27.Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80:283–285. [PubMed] [Google Scholar]
- 28.Chen EY, Fujinaga M, Giaccia AJ. Hypoxic microenvironment within an embryo induces apoptosis and is essential for proper morphological development. Teratology. 1999;60:215–225. doi: 10.1002/(SICI)1096-9926(199910)60:4<215::AID-TERA6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp Mol Med. 2004;36:1–12. doi: 10.1038/emm.2004.1. [DOI] [PubMed] [Google Scholar]
- 30.Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: The role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004;18:2183–2194. doi: 10.1101/gad.1243304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- 32.Giaccia A, Siim B, Johnson R. HIF-1 as a _target for drug development. Nat Rev Drug Discov. 2003;2:803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- 33.Kaelin W. How oxygen makes its presence felt. Genes Dev. 2002;16:1441–1445. doi: 10.1101/gad.1003602. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–497. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 35.Semenza GL. _targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 36.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenger RH, Rolfs A, Kvietikova I, Spielmann P, Zimmermann DR, Gassmann M. The mouse gene for hypoxia-inducible factor-1alpha–genomic organization, expression and characterization of an alternative first exon and 5′ flanking sequence. Eur J Biochem. 1997;246:155–165. doi: 10.1111/j.1432-1033.1997.t01-1-00155.x. [DOI] [PubMed] [Google Scholar]
- 38.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: Sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 39.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 40.Chan D, Suthphin P, Denko N, Giaccia A. Role of prolyl hydroxylation in oncogenically stabilized hyoxia-inducible factor-1alpha. J Biol Chem. 2002;277:40112–40117. doi: 10.1074/jbc.M206922200. [DOI] [PubMed] [Google Scholar]
- 41.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara J, Lane W, Kaelin W. HIFalpha _targeted for VHL-mediated destruction by proline hydroxylation: Imlications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 42.Jaakkola P, Mole D, Tian Y, Wilson M, Gielbert J, Gakell S, Kriegsheim A, Heberstreit H, Mukherji M, Schofield C, Maxwell P, Ratcliffe P. _targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 43.Min J, Yang H, Ivan M, Gertler F, Jr, WK, Pavletich N. Structure of an HIF-1alpha-pVHL complex: Hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 44.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci USA. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan D, Sutphin P, Yen S, Giaccia A. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2005;25:6415–6426. doi: 10.1128/MCB.25.15.6415-6426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kallio PJ, Wilson WJ, O'Brien S, Makino Y, Poellinger L. Regulation of the Hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- 49.Mahon PC, Hirota K, Semenza GL. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leo C, Giaccia A, Denko N. The hypoxic tumor microenvironment and gene expression. Semin Radiat Oncol. 2004;14:207–214. doi: 10.1016/j.semradonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Wykoff C, Pugh C, Maxwell P, Harris A, Ratcliffe P. Identification of novel hypoxia dependent and independent _target genes of the von Hippel Lindau (VHL) tumor suppressor by mRNA differential expression profiling. Oncogene. 2000;19:6297–6305. doi: 10.1038/sj.onc.1204012. [DOI] [PubMed] [Google Scholar]
- 53.Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, Meijer GA, van de Wiel MA, Belien JA, van Diest PJ, van der Wall E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1) J Pathol. 2005;206:291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 54.Bishop T, Lau K, Epstein A, Kim S, Jiang M, O'Rourke D, Pugh C, Gleadle J, Taylor M, Hodgkin J, Ratcliffe P. Genetic analysis of pathways regulated by the von hippel-lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004;2:e289. doi: 10.1371/journal.pbio.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maxwell P, Wiesener M, Chang G, Clifford S, Vaux E, Cockman M, Wykoff C, Pugh C, Maher E, Ratcliffe P. The tumour suppressor protein VHL _targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 56.Ryan HE, Lo J, Johnson RS. HIF 1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iyer N, Kotch L, Agani F, Leung S, Laughner E, Wenger R, Gassmann M, Gearhart J, Lawler A, Yu A, Semenza G. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, Carmeliet P. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor −1alpha. Cardiovasc Res. 2003;60:569–579. doi: 10.1016/j.cardiores.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 59.Chan DA, Krieg AJ, Turcotte S, Giaccia AJ. HIF gene expression in cancer therapy. Methods Enzymol. 2007;435:323–345. doi: 10.1016/S0076-6879(07)35016-7. [DOI] [PubMed] [Google Scholar]
- 60.Haase VH. The VHL tumor suppressor in development and disease: Functional studies in mice by conditional gene _targeting. Semin Cell Dev Biol. 2005;16:564–574. doi: 10.1016/j.semcdb.2005.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaelin W, Maher E. The VHL tumor-suppressor gene paradigm. Trends Genet. 1998;14:423–426. doi: 10.1016/s0168-9525(98)01558-3. [DOI] [PubMed] [Google Scholar]
- 62.Ratcliffe P, Pugh C, Maxwell P. _targeting tumors through the HIF system. Nat Med. 2000;12:1315–1316. doi: 10.1038/82113. [DOI] [PubMed] [Google Scholar]
- 63.Zelzer E, Levy Y, Kahana C, Shilo B, Rubinstein M, Cohen B. Insulin induces transcription of _target genes through the hypoxia-inducible factor 1 alpha. EMBO J. 1998;17:5085–5094. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 65.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 67.Gordon J, Simon M. Hypoxia-inducible factors: Central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–77. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park SK, Dadak AM, Haase VH, Fontana L, Giaccia AJ, Johnson RS. Hypoxia-induced gene expression occurs solely through the action of hypoxia-inducible factor 1alpha (HIF-1alpha): Role of cytoplasmic trapping of HIF-2alpha. Mol Cell Biol. 2003;23:4959–4971. doi: 10.1128/MCB.23.14.4959-4971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17:271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- 70.Carmeliet P, Dor Y, Herbert J, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshet E. Role of HIF-1alpha in hypoxia mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 71.Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 73.Peng J, Zhang L, Drysdale L, Fong GH. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci USA. 2000;97:8386–8391. doi: 10.1073/pnas.140087397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 75.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005;65:3299–3306. doi: 10.1158/0008-5472.CAN-04-4130. [DOI] [PubMed] [Google Scholar]
- 78.Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621–627. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 79.Maynard MA, Qi H, Chung J, Lee EH, Kondo Y, Hara S, Conaway RC, Conaway JW, Ohh M. Multiple splice variants of the human HIF-3 alpha locus are _targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem. 2003;278:11032–11040. doi: 10.1074/jbc.M208681200. [DOI] [PubMed] [Google Scholar]
- 80.Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–554. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 81.Steinbrech DS, Mehrara BJ, Saadeh PB, Chin G, Dudziak ME, Gerrets RP, Gittes GK, Longaker MT. Hypoxia regulates VEGF expression and cellular proliferation by osteoblasts in vitro. Plast Reconstr Surg. 1999;104:738–747. doi: 10.1097/00006534-199909030-00019. [DOI] [PubMed] [Google Scholar]
- 82.Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert S, Bouxsein M, Faugere M, Guldberg R, Gerstenfeld L, Haase V, Johnson R, Schipani E, Clemens T. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117:1616–1626. doi: 10.1172/JCI31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maes C, Kobayashi T, Kronenberg HM. A novel transgenic mouse model to study the osteoblast lineage in vivo. Ann NY Acad Sci. 2007;1116:149–164. doi: 10.1196/annals.1402.060. [DOI] [PubMed] [Google Scholar]
- 84.Salim A, Nacamuli R, Morgan E, Giaccia A, Longaker M. Transient changes in oxygen tension inhibit osteogenic differentiation and Runx2 expression in osteoblasts. J Biol Chem. 2004;279:40007–40016. doi: 10.1074/jbc.M403715200. [DOI] [PubMed] [Google Scholar]
- 85.Akeno N, Robins J, Zhang M, Czyzyk-Krzeska MF, Clemens TL. Induction of vascular endothelial growth factor by IGF-I in osteoblast-like cells is mediated by the PI3K signaling pathway through the hypoxia-inducible factor-2alpha. Endocrinology. 2002;143:420–425. doi: 10.1210/endo.143.2.8639. [DOI] [PubMed] [Google Scholar]
- 86.Danis A. [Mechanism of bone lengthening by the Ilizarov technique] Bull Mem Acad R Med Belg. 2001;156:107–112. [PubMed] [Google Scholar]
- 87.Choi I, Ahn J, Chung C, Cho T. Vascular proliferation and blood supply during distraction osteogenesis: A scanning electron microscopic observation. J Orthop Res. 2000;18:698–705. doi: 10.1002/jor.1100180504. [DOI] [PubMed] [Google Scholar]
- 88.Ilizarov GA. Clinical application of the tension-stress effect for limb lengthening. Clin Orthop Relat Res. 1990:8–26. [PubMed] [Google Scholar]
- 89.Wan C, Gilbert SR, Wang Y, Cao X, Shen X, Ramaswamy G, Jacobsen KA, Alaql ZS, Eberhardt AW, Gerstenfeld LC, Einhorn TA, Deng L, Clemens TL. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc Natl Acad Sci USA. 2008;105:686–691. doi: 10.1073/pnas.0708474105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu XD, Deng LF, Wang J, Qi J, Zhou Q, Wang JS, Wei L, Zhu YP. [Regulation of hypoxia inducible factor-1alpha on osteoblast function in osteogenesis] Zhonghua Yi Xue Za Zhi. 2007;87:3357–3361. [PubMed] [Google Scholar]
- 91.Carano RA, Filvaroff EH. Angiogenesis and bone repair. Drug Discov Today. 2003;8:980–989. doi: 10.1016/s1359-6446(03)02866-6. [DOI] [PubMed] [Google Scholar]
- 92.Street J, Bao M, deGuzman L, Bunting S, Peale FV, Jr, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, van Bruggen N, Redmond HP, Carano RA, Filvaroff EH. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci USA. 2002;99:9656–9661. doi: 10.1073/pnas.152324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harper J, Gerstenfeld LC, Klagsbrun M. Neuropilin-1 expression in osteogenic cells: Down-regulation during differentiation of osteoblasts into osteocytes. J Cell Biochem. 2001;81:82–92. doi: 10.1002/1097-4644(20010401)81:1<82::aid-jcb1025>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 94.Jacobsen KA, Al-Aql ZS, Wan C, Fitch JL, Stapleton SN, Mason ZD, Cole RM, Gilbert SR, Clemens TL, Morgan EF, Einhorn TA, Gerstenfeld LC. Bone formation during distraction osteogenesis is dependent on both VEGFR1 and VEGFR2 signaling. J Bone Miner Res. 2008;23:596–609. doi: 10.1359/JBMR.080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maes C, Coenegrachts L, Stockmans I, Daci E, Luttun A, Petryk A, Gopalakrishnan R, Moermans K, Smets N, Verfaillie CM, Carmeliet P, Bouillon R, Carmeliet G. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest. 2006;116:1230–1242. doi: 10.1172/JCI26772. [DOI] [PMC free article] [PubMed] [Google Scholar]