

There is now strong evidence that mutation not only abrogates p53 tumor-suppressive functions, but in some instances can also endow mutant proteins with novel activities. Such neomorphic p53 proteins are capable of dramatically altering tumor cell behavior, primarily through their interactions with other cellular proteins and regulation of cancer cell transcriptional programs. Different missense mutations in p53 may confer unique activities and thereby offer insight into the mutagenic events that drive tumor progression. In this review by Prives and colleague, the mechanisms by which mutant p53 exerts its cellular effects—with a particular focus on the burgeoning mutant p53 transcriptome—and the biological and clinical consequences of mutant p53 gain-of-function activities are discussed.
Keywords: gain of function, mutant p53, oncogenic, p53, transcriptome, tumor suppression
Abstract
There is now strong evidence that mutation not only abrogates p53 tumor-suppressive functions, but in some instances can also endow mutant proteins with novel activities. Such neomorphic p53 proteins are capable of dramatically altering tumor cell behavior, primarily through their interactions with other cellular proteins and regulation of cancer cell transcriptional programs. Different missense mutations in p53 may confer unique activities and thereby offer insight into the mutagenic events that drive tumor progression. Here we review mechanisms by which mutant p53 exerts its cellular effects, with a particular focus on the burgeoning mutant p53 transcriptome, and discuss the biological and clinical consequences of mutant p53 gain of function.
The TP53 gene, which resides on chromosome 17p13.1 and encodes the p53 protein, is the most frequent _target for mutation in human cancer, with greater than half of all tumors exhibiting mutation at this locus (Vogelstein et al. 2000; Petitjean et al. 2007b). In this review, we present an overview of the current understanding of the significance of p53 mutations in cancer. At the outset, we would like to point out that there are several excellent reviews on this subject (Brosh and Rotter 2009; Kim et al. 2009; Oren and Rotter 2010; Goh et al. 2011; Goldstein et al. 2011). Our goal is to provide an update on these reviews, focusing in particular on the means by which mutant p53 proteins regulate the gene expression patterns of the tumor cells that they inhabit.
The discovery of the p53 ‘proto-oncogene'
The p53 protein was first identified in a complex with the simian virus 40 (SV40) large T-antigen (Lane and Crawford 1979; Linzer and Levine 1979). It was subsequently demonstrated that many tumors produce abundant levels of this protein, a phenomenon that was not observed in normal tissue, suggesting that p53 might act as a cellular oncogene (DeLeo et al. 1979; Rotter 1983). This notion was reinforced when ectopic expression of newly cloned p53 cDNAs was shown to cooperate with oncogenic Ras to transform primary cells in culture (Eliyahu et al. 1984; Parada et al. 1984). Furthermore, overexpression of p53 was demonstrated to increase tumorigenicity in otherwise p53-null cells (Wolf et al. 1984). Thus, throughout the first decade after its discovery, p53 was generally acknowledged as a proto-oncogene (Levine and Oren 2009).
However, multiple early findings called into question the role of p53 as a pro-oncogenic factor. For example, the murine Trp53 gene was shown to be inactivated by retroviral insertions in several tumor models (Wolf and Rotter 1984; Ben David et al. 1988). Discrepant findings were reported regarding the ability of p53 to transform primary cells, and when several groups compared the sequences of their cloned p53 cDNAs, the striking result was that each clone differed in sequence from the others (Levine and Oren 2009). It was soon recognized that these early experiments demonstrating that p53 overexpression could transform cells and promote in vivo tumor growth were actually performed with mutated versions of p53 that had been isolated from tumor cells (Hinds et al. 1989, 1990; Levine and Oren 2009). Thus, instead of describing the function of wild-type p53, they were in fact detailing the role of mutant p53 in tumor biology (the importance of which becomes apparent below).
In 1989, seminal findings overturned the widely accepted notion that p53 acts as a proto-oncogene. Vogelstein and colleagues (Baker et al. 1989) investigated genetic alterations in colorectal carcinomas and demonstrated that >50% of these tumors exhibit loss of heterozygosity (LOH), a hallmark of tumor suppressor genes, at the TP53 locus (most commonly, mutation of one TP53 allele and deletion of the corresponding TP53 allele). Work from Levine's and Oren's groups (Eliyahu et al. 1989; Finlay et al. 1989) demonstrated that overexpression of wild-type p53 was actually sufficient to suppress oncogenic transformation. These and subsequent findings described below firmly established p53 as a tumor suppressor gene. Looking back, however, one should never discount seemingly contradictory results. It turns out that we can still gain deep insight into the role of p53 in tumor biology from these early “artifacts” studying mutant p53.
p53 and tumor suppression
Soon after this paradigm shift took place in the p53 field, several studies served to confirm the role of wild-type p53 as a tumor suppressor and establish this protein as one of the most important players in cancer biology. In the late 1960s, a number of extremely cancer-prone families were identified in the United States and Europe (Li and Fraumeni 1969a,b). This familial cancer syndrome came to be known as Li-Fraumeni syndrome (LFS), a rare autosomal-dominant disorder that predisposes individuals to breast cancer, sarcomas, and other neoplasms. LFS was later shown to be caused by germline mutations in TP53 (Malkin et al. 1990; Srivastava et al. 1990). A mouse model in which p53 was disrupted by homologous recombination revealed that although p53−/− mice were developmentally normal (for the most part), they were extremely cancer-prone (Donehower et al. 1992; Attardi and Jacks 1999). Mice devoid of p53 exhibited an extreme susceptibility to developing tumors, primarily lymphomas and sarcomas, with three out of every four p53−/− mice having developed at least one obvious neoplasm by 6 mo of age. In contrast, wild-type littermates failed to develop any tumors by 9 mo of age. In addition to the early findings by the Vogelstein group (Vogelstein et al. 2000; Petitjean et al. 2007b), literally thousands of studies have now confirmed that TP53 mutations are not restricted to colorectal cancer, but are present in >50% of all human tumors, although the extent of p53 mutation varies with the tumor type. It is now widely acknowledged that p53 mutations are the most common genetic event in human cancer (Levine and Oren 2009).
Indeed, it has been hypothesized that p53 function is compromised in most human tumors (Polager and Ginsberg 2009). While at least half of all tumors exhibit mutation of p53, in those that retain wild-type p53, its activity can be attenuated by several other mechanisms. For example, many DNA tumor viruses encode proteins that can inactivate p53; SV40 large T-antigen, adenovirus E1B-55-kDa protein, and the E6 oncoprotein of human papilloma virus (HPV) types 16 and 18 all bind to p53 and inactivate its function (Levine 2009). The biological relevance of these interactions is highlighted by the fact that HPV types 16 and 18 have been implicated in cervical carcinogenesis (Ferenczy and Franco 2002). Another mechanism by which tumors inactivate p53 is through the up-regulation or activation of negative regulators of p53. Mdm2, an E3 ubiquitin ligase, is the major negative regulator of p53 and serves to keep p53 levels in check under unstressed conditions (Poyurovsky and Prives 2006; Manfredi 2010; Marine and Lozano 2010). Additionally, a homolog of Mdm2, MdmX (also known as Mdm4), also serves as a negative regulator of p53 (Marine et al. 2007). Not surprisingly, both Mdm2 and MdmX are overexpressed in a variety of neoplasms (Marine et al. 2006).
Wild-type p53 has now secured its place as a critical player in cancer biology, but before we move on to the role of mutant p53 in tumors, it is important to briefly review the current view of how p53 acts as a tumor suppressor.
Function of wild-type p53
Wild-type p53 can be activated by a number of cellular stressors, including DNA damage, hypoxia, and oncogene activation (Vousden and Lu 2002). Following activation, wild-type p53 normally functions as a sequence-specific transcription factor to inhibit cell cycle progression, promote senescence, or induce apoptotic cell death (Prives and Hall 1999; Vousden and Lu 2002; Vousden and Prives 2009).
The p53 protein possesses an acidic N-terminal transactivation domain (now recognized to be two distinct transactivation subdomains), a proline-rich domain, and a centrally located sequence-specific DNA-binding domain, followed by an oligomerization domain and a basic C-terminal regulatory domain (Fig. 1A; Laptenko and Prives 2006). Wild-type p53 functions as a homotetramer in cells, binding to p53 response elements composed of two decamers separated by a spacer of 0–14 nucleotides (5′-RRRCWWGYYYn0–14RRRCWWGYYY-3′) [(A) adenine; (T) thymine; (C) cytosine; (G) guanine; (R) purine; (Y) pyrimidine; (W) A/T; (n) any nucleotide] (el-Deiry et al. 1992; Funk et al. 1992; Riley et al. 2008). A myriad of genes have been shown to be transcriptional _targets of wild-type human p53. The products of p53 _target genes mediate the downstream cellular outcomes of p53 activation such as cell cycle arrest (CDKN1A, MIR34A, etc.), senescence (CDKN1A, PAI1, etc.), apoptosis (PUMA, BAX, etc.), and metabolic processes (TIGAR, SCO2, GLS2, etc.) (Prives and Hall 1999; Vousden and Lu 2002; Riley et al. 2008; Vousden and Prives 2009; Vousden and Ryan 2009).
Figure 1.

TP53 mutational spectrum in human cancers. (A) TP53 missense mutation data for human cancer patients (N = 19,262) were obtained from the p53 International Agency for Research on Cancer (IARC) database and plotted as a function of amino acid position. Schematic of the p53 protein with domain structures illustrated. (TAD) Transactivation domain (1–42); (PRD) proline-rich domain (40–92), which also contains a second transactivation domain; (DBD) DNA-binding domain (101–306); (OD) oligomerization domain (307–355), also contains a nuclear export signal; (CTD) C-terminal regulatory domain (356–393), also contains three nuclear localization signals (data adapted from http://p53.free.fr). (B) Table for the six “hot spot” residues in p53 with corresponding frequency of any mutation at a given residue. TP53 mutation data for human cancer patients (N = 25,902) were obtained from the p53 database (http://p53.free.fr). (C) Table for the most common missense mutations at “hot spot” residues in p53 with corresponding frequency of particular mutation. TP53 mutation data for human cancer patients (N = 25,902) were obtained from the p53 database (http://p53.free.fr).
Each of these cellular outcomes has been shown to be important for the tumor-suppressive ability of wild-type p53. There is substantial evidence to support a role for apoptosis in the tumor-suppressive function of p53. For example, mice in which the proline-rich domain of p53 had been deleted (this mutant protein lacks the ability to induce cell cycle arrest but retains the ability to induce programmed cell death) are still efficiently protected from spontaneous tumor development, suggesting that the ability to induce apoptosis is critical to the tumor-suppressive ability of p53 (Toledo et al. 2006). However, a second mouse model argues that cell cycle arrest plays at least a partial role in tumor-suppressive function: A rare tumor-derived mutant of p53 (p53-R175P) was identified that can induce cell cycle arrest, but not apoptosis (Rowan et al. 1996). (This nomenclature will be used throughout this review. The first letter represents the amino acid present in the wild-type protein, the number represents the amino acid position number counting from the N terminus, and the last letter represents the amino acid present in the mutated protein. In this case, R175P designates an arginine mutated to a proline at position 175 in the p53 protein.) Mice expressing the murine equivalent to this p53 mutant (p53-R172P) demonstrate a delay in spontaneous tumor formation, suggesting that cell cycle arrest and chromosome stability also protect against tumor development (Liu et al. 2004). Possibly all p53 activities contribute to its ability to suppress tumorigenesis in different contexts. These and many other data support the notion that wild-type p53 is unequivocally a tumor suppressor. However, biology, it seems, is never satisfied with a simple answer.
Gain-of-function hypothesis
Findings within the p53 field have emerged to help explain many of the seemingly contradictory results from the first decade of p53 research, when p53 was widely considered to be an oncogene. Unlike most tumor suppressor genes, which are predominantly inactivated as a result of deletion or truncation (Weinberg 1991), the vast majority of cancer-associated mutations in TP53 are missense mutations, single base-pair substitutions that result in the translation of a different amino acid in that position in the context of the full-length protein. The great majority of these missense mutations are clustered within the central most conserved region of p53 that spans the DNA-binding domain, and among these are a small number (approximately six) of “hot spot” residues that occur with unusually high frequency (Fig. 1A,B; Harris and Hollstein 1993; Cho et al. 1994; Petitjean et al. 2007a). This is in striking contrast to the majority of tumor suppressors (examples include RB1, APC, NF1, NF2, and VHL), the primary mutations in which are deletion or nonsense, leading to little or no expression of the respective proteins (Levine et al. 1995). While wild-type p53 under unstressed conditions is a very short-lived protein, these missense mutations lead to the production of full-length altered p53 protein with a prolonged half-life (Strano et al. 2007). Mutant p53 protein stability is discussed at greater length later in this review. Many of these stable mutant forms of p53 can exert a dominant-negative effect on the remaining wild-type allele, serving to abrogate the ability of wild-type p53 to inhibit cellular transformation, particularly when the mutant protein is expressed in excess of its wild-type counterpart (Brosh and Rotter 2009; Oren and Rotter 2010). Such dominant-negative activity may be effected by either formation of mutant/wild-type p53 cotetramers (Chan et al. 2004) or the incorporation of wild-type p53 into mutant p53 supratetrameric aggregates (Xu et al. 2011). Importantly, missense mutations in p53 in human tumors are usually followed by LOH at the corresponding locus, suggesting that there is a selective advantage conferred by losing the remaining wild-type p53, even after one allele has been mutated (Baker et al. 1990; Brosh and Rotter 2009). These observations, among others, have led to the “gain-of-function” hypothesis, which states that mutation of TP53 is not equivalent to simply losing wild-type p53 function; rather, the strong selection for maintained expression of a select group of mutant p53 proteins suggests a positive role for certain p53 mutants in tumorigenesis.
While inactivating missense mutations in p53 may be selected for during tumor progression due to their ability to act as dominant-negative inhibitors of wild-type p53, there is also clear evidence that mutant p53 can exert oncogenic or gain-of-function activity independent of its effects on wild-type p53 (Sigal and Rotter 2000). For example, when tumor-derived mutants of p53 are expressed in nontransformed, otherwise p53-null cells, this greatly increases their ability to form tumors in nude mice when compared with the parental cells (Wolf et al. 1984; Dittmer et al. 1993). As was the case in confirming the tumor-suppressive role of wild-type p53, mouse models were essential in demonstrating that mutation of p53 is not equivalent to simply losing wild-type p53 function. Almost a decade after the original p53 knockout mice were generated, two “knock-in” mouse models of LFS were generated (Lang et al. 2004; Olive et al. 2004). These groups used mice expressing tumor-derived mutants of p53, in which the murine equivalents of two of the most frequent “hot spot” mutations in p53 were inserted into the endogenous Trp53 locus using homologous recombination, in an attempt to better recapitulate the human disease. Remarkably, both knock-in mutations produce an altered tumor spectrum in addition to more metastatic tumors (Liu et al. 2000; Lang et al. 2004; Olive et al. 2004). Similarly, in an analysis of LFS patients, germline missense mutations in TP53 have been shown to be associated with an earlier age of onset (∼9 years) when compared with germline deletions in TP53, suggesting a gain-of-function effect of missense p53 mutants in human tumors (Bougeard et al. 2008). In addition, multiple studies have also demonstrated that the mutational status of p53 is a strong predictor for poor outcomes in many types of human tumors, particularly breast cancer (Elledge et al. 1993; Olivier et al. 2006; Langerod et al. 2007; Petitjean et al. 2007a).
Not only data from mice and human epidemiology but also cell-based assays have implicated mutant p53 in diverse aspects of tumorigenesis. These include DNA synthesis and proliferation (Bossi et al. 2006; Di Agostino et al. 2006), survival (Stambolsky et al. 2010), chemoresistance (Blandino et al. 1999; Bristow et al. 2003; Irwin et al. 2003; Scian et al. 2005), abnormal centrosomes and spindle checkpoints (Gualberto et al. 1998; Wang et al. 1998), gene amplification (El-Hizawi et al. 2002), somatic cell reprogramming and stem cell characteristics (Mizuno et al. 2010; Sarig et al. 2010), disruption of tissue architecture (Freed-Pastor et al. 2012), and angiogenesis (Fontemaggi et al. 2009), as well as migration, invasion, and metastasis (Hsiao et al. 1994; Adorno et al. 2009; Muller et al. 2009; Noll et al. 2011).
While the concept of mutant p53 gain of function is now well established, the exact criteria for what constitutes “gain of function” can still be quite confusing (Oren and Rotter 2010). Our current understanding of mutant p53 gain of function comes largely from experiments in which researchers have overexpressed mutant versions of p53 in cell culture or in mouse tumor models. As discussed above, many cancer-associated p53 mutants can exert dominant-negative effects on wild-type p53, so any “gain-of-function” experiments performed in the presence of wild-type p53 must be interpreted with extreme caution. To thoroughly demonstrate gain of function, two main approaches are now used: (1) expression of mutant forms of p53 in an otherwise p53-null background, or (2) RNAi to deplete the levels of an endogenous mutant p53 from cancer cells that have lost the wild-type p53 allele. This is an important point to make, as any experiments performed in cells that harbor wild-type p53 do not necessarily prove a gain of function, but may simply reflect a dominant-negative effect.
Pro-oncogenic roles of mutant p53
Recent years have seen an explosion of proposed roles for p53 mutations in tumorigenesis and diverse mechanisms to explain these roles for mutant p53 proteins. It is important to keep in mind that while the term “gain of function” has grown in popularity to explain observed effects of mutant p53 proteins, this may not necessarily be the best terminology, as seen below. There are three primary categories of proposed mechanisms to explain the pro-oncogenic effects of mutant p53 in tumor biology, which are not mutually exclusive (Brosh and Rotter 2009). First, tumors may select for mutation of p53 solely for loss of wild-type p53 tumor-suppressive activity. Although missense mutations comprise ∼75% of all mutant forms of p53 in humans, p53-null mice are extremely tumor-prone, and a subset (∼15%) of mutant p53-bearing human tumors sustains either frameshift or nonsense mutations (see the International Agency for Research on Cancer [IARC] database, http://www-p53.iarc.fr). So, loss of wild-type p53 activity can be a significant component of tumor predisposition. Second, mutants of p53 may lose certain tumor-suppressive functions of wild-type p53, while retaining and/or exaggerating other aspects of normal wild-type p53 function. Indeed, yeast and mammalian cell-based assays have revealed a remarkable complexity in the ability of tumor-derived mutant forms of p53 to activate transcription from known wild-type p53 response elements (Di Como and Prives 1998; Kato et al. 2003; Resnick and Inga 2003; Jordan et al. 2008). Finally, these mutant p53 proteins may acquire truly neomorphic or gain-of-function activities that actively promote tumor growth.
Such activities of mutant p53 are now well-established and are commonly attributed to one of two primary mechanisms: (1) an interaction between mutant p53 and cellular proteins, or (2) mutant p53-mediated regulation of novel _target genes (Fig. 2). Of course, these two options are not mutually exclusive; for example, tumor-derived mutants of p53 have been shown to interact with several cellular partners, including cellular transcription factors (such as NF-Y, Sp1, VDR [vitamin D receptor], SREBP, and Ets-1), and regulate their transcriptional activity. This is discussed in greater detail in a later section.
Figure 2.

Mechanisms of mutant p53 gain of function. There are multiple proposed mechanisms that account for different mutant p53 gain-of-function activities. These include both transcriptional and nontranscriptional mechanisms: (A) Physical interaction with p53 family members p63 and p73 to inhibit transactivation of their respective _target genes. (B) Interaction with and recruitment by cellular transcription factors to their cognate binding sites, leading to more robust transactivation of their respective _target genes. Examples of this mechanism include NF-Y, SREBP-2, Sp1, Ets-1, and VDR. (C) Structure-specific DNA binding by mutant p53, resulting in transcriptional regulation of the relevant promoter. The most well-described example is the interaction of mutant p53 and matrix attachment regions (MARs). (D) “Direct” recruitment of mutant p53 to unique sequence-specific DNA elements and/or unique chromatin landscapes. The dashed gray arrow indicates that there is currently little evidence to support this proposed mechanism. (E) Physical interaction with other cellular proteins that are not transcription factors, such as TopB1, Pin1, MRE11, PML, and others.
Mutant p53-interacting partners
While wild-type p53 acts predominantly as a transcription factor, a number of the described gain-of-function activities of mutant p53 are mediated through nontranscriptional processes (Brosh and Rotter 2009). Mutant p53 can form aberrant protein complexes with several interacting partners, perturbing their activity (Table 1). A few of the more prominent examples are reviewed below.
Table 1.
Select list of mutant p53-interacting partners
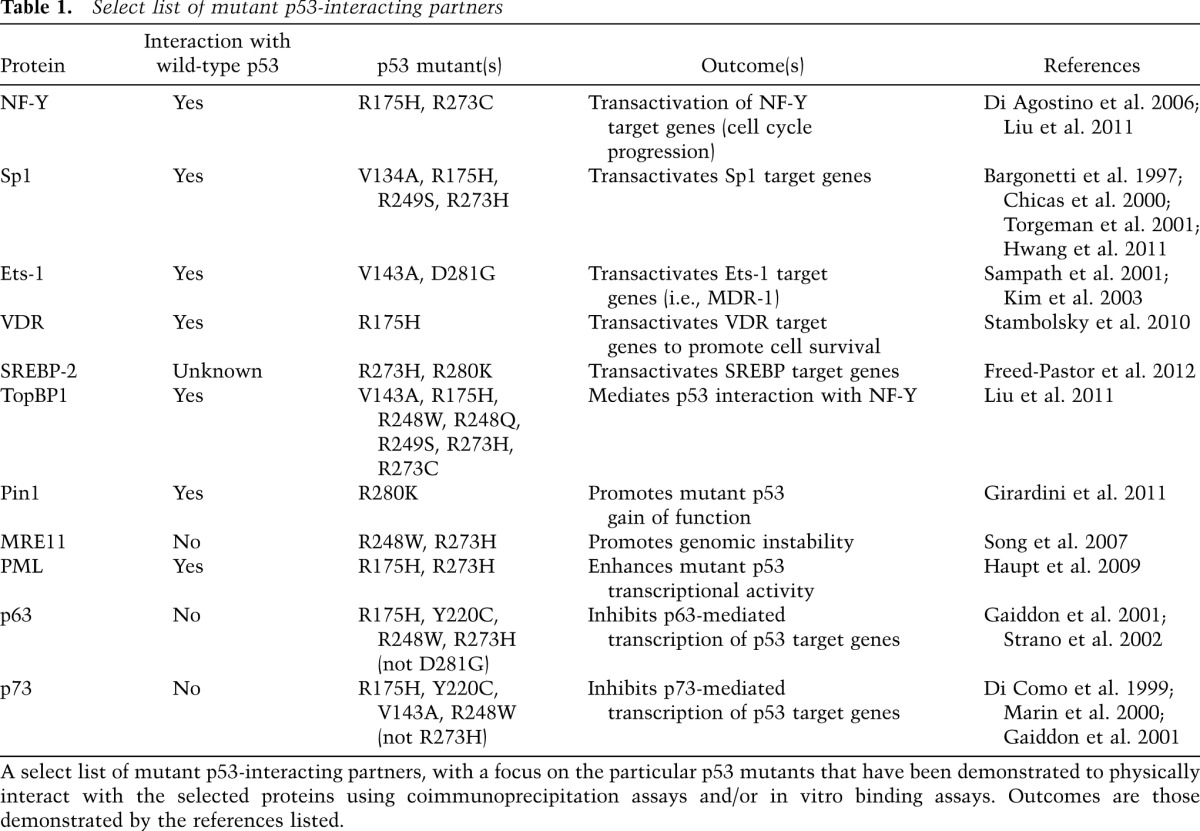
MRE11 was recently identified as a novel interacting partner with two hot spot p53 mutants (p53-R248W and p53-R273H) that had been “knocked in” to a humanized p53 allele in mice (p53hki) (Song et al. 2007). The humanized (“Hupki”) p53 mouse model encodes a human/mouse chimeric p53 protein—amino acids 33–332 are human, while the N and C termini are composed of murine p53 (Luo et al. 2001). The interaction between the nuclease MRE11 and mutant p53 was demonstrated to confer a gain of function by promoting genetic instability in tumors and pretumorigenic lesions in the Hupki knock-in mice. This interaction impaired the recruitment of the Mre11–Rad50–NBS1 (MRN) complex to double-strand breaks and diminished the activation of ataxia telangiectasia-mutated (ATM), enabling the persistence of unrepaired DNA breaks (Song et al. 2007).
Two recently described mutant p53-interacting partners enhance the oncogenic activity of tumor-derived p53 mutants. The promyelocytic leukemia (PML) protein is the major component of nuclear PML bodies, which have been implicated in proliferation and programmed cell death. Mutant p53 physically associates with PML, and this interaction facilitates the transcriptional activity of mutant p53 (Haupt et al. 2009). In addition, mutant p53 interacts with the prolyl isomerase Pin1, which binds to phosphorylated serine/threonine residues next to a proline and isomerizes the intervening peptide bond, resulting in conformational changes that affect protein stability and activity. Pin1 was demonstrated to cooperate with mutant p53 to promote a “proaggressiveness” transcriptional program that increased the migration and invasion of breast cancer cells (Girardini et al. 2011). Furthermore, this interaction was shown to be required for certain mutant p53 gain-of-function activities, since Pin1 was necessary for oncogenic transformation of primary mouse fibroblasts by mutant p53 in cooperation with oncogenic H-RasG12V, and deletion of Pin1 attenuated this mutant p53 gain-of-function phenotype in mice (Girardini et al. 2011).
Mutant p53 regulates p63 and p73
While the mutant p53 interactome is rapidly expanding, the most widely studied mutant p53-interacting partners remain the p53 family members p63 and p73. The p53 family consists of three proteins—p53, p63, and p73—that are homologous at the amino acid level in the three primary domains of p53: transactivation domain, DNA-binding domain, and C-terminal oligomerization domain (Li and Prives 2007). The highest degree of homology exists in the DNA-binding domains; p63 and p73 exhibit 60% and 63% homology with the DNA-binding domain of p53, respectively (Irwin and Kaelin 2001). The N-terminal transactivation domain is only 30% identical between p53 and p73 and 22% identical between p53 and p63, while the oligomerization domains of both p63 and p73 exhibit 38% identity with that of p53 (Irwin and Kaelin 2001). Both p63 and p73 are expressed as multiple isoforms, through either alternate promoter usage (N-terminal TA or ΔN forms for both p63 and p73) or through alternative splicing of the C termini (p63-α, p63-β, and p63-γ, and p73-α, p73-β, p73-γ, p73-δ, p73-ɛ, and p73-ζ) (Pietsch et al. 2008). To complicate matters further, it is now recognized that p53 itself is also expressed as multiple isoforms (Machado-Silva et al. 2010), and it will be interesting in future studies to discern whether and how this is relevant to activities of full-length mutant p53 proteins.
While p63 and p73 were shown to form homotetramers and heterotetramers with each other, neither p63 nor p73 forms heterotetramers with wild-type p53 (Davison et al. 1999). It was therefore somewhat surprising when several tumor-derived mutants of p53 were demonstrated to interact with both p63 and p73 (Di Como et al. 1999; Marin et al. 2000; Gaiddon et al. 2001; Strano et al. 2001, 2002; Irwin et al. 2003). This interaction has been invoked to explain many of the gain-of-function effects of mutant p53. For example, binding to p63 and/or p73 has been linked to the ability of certain tumor-derived mutants of p53 to promote chemoresistance, migration, invasion, and metastasis (Sampath et al. 2001; Adorno et al. 2009; Muller et al. 2009). As mentioned above, the altered tumorigenic phenotypes observed in mutant p53 knock-in mice (p53R172H/− and p53R270H/−, compared with p53−/−) provide some of the most compelling evidence for a mutant p53 gain of function in tumorigenesis (Olive et al. 2004). Interestingly, both p53+/−/p63+/− and p53+/−/p73+/− double-mutant mice display an altered tumor spectrum and a more metastatic phenotype, highly reminiscent of the phenotype observed in the mutant p53 knock-in models (Flores et al. 2005). This is in line with the idea that gain-of-function p53 mutants act at least in part by inhibiting the function of p63/p73.
Consistent with findings that the p53 tetramerization domains cannot oligomerize with the corresponding regions from p63 or p73, early reports demonstrated that the mutant p53 interaction with the other p53 family members was mediated through their DNA-binding domains and that the core domain of mutant p53 is sufficient to interact with p63 or p73 in coimmunoprecipitation assays (Gaiddon et al. 2001; Strano et al. 2001, 2002). These observations provide a potential explanation for the specificity of the interaction between p63/p73 and tumor-derived mutants of p53 and not wild-type p53, given that the tumor-derived missense mutations in p53 occur within the core DNA-binding domain and many of the hot spot mutants of p53 have been shown to produce either local or global conformational changes (Wong et al. 1999). However, it should be noted that p63/p73 also interact with “DNA contact” mutants of p53 (albeit less effectively than with “conformational” mutants), which are reported not to dramatically alter the structure of the mutant p53 protein (Li and Prives 2007). There are multiple lines of evidence that support the central core domain as the site of interaction, although the debate is not yet settled. First, there is a strong correlation between the observed ability of a tumor-derived mutant to bind to p63/p73 and the ability of that p53 mutant to be recognized by PAb240, which recognizes a cryptic epitope within the core domain of p53 (Li and Prives 2007). Second, wild-type p53, when denatured, was demonstrated to acquire the ability to interact with p73 (Bensaad et al. 2003). Finally, two peptides derived from the p73 DNA-binding domain were demonstrated to disrupt the interaction between p73 and tumor-derived mutants of p53 (Di Agostino et al. 2008).
Despite the evidence that it is the central region of mutant p53 that is involved in its ability to regulate p63 and p73, the region of mutant p53 required for its interactions with p53 siblings is still not fully clarified. For one thing, it was reported that a well-studied single-nucleotide polymorphism (SNP) at codon 72 within the N terminus of p53 (this residue can be either arginine or proline) alters the ability of mutant p53 to interact with p73, as mutant p53 with an arginine at this residue has a much higher affinity for p73 than a mutant p53 with a proline (Marin et al. 2000). Moreover, the C terminus has also been shown to be a determinant of the interaction between mutant p53 and p63/p73, at least in the case of certain tumor-derived mutants (Muller et al. 2009; Xu et al. 2011).
Since p63 and p73 are both sequence-specific transcription factors, mutant p53 binding may alter the activity of these family members at their _target gene promoters. Mutant p53 has traditionally been assumed to play an inhibitory role toward its family members, particularly when assayed using reporter constructs with a consensus p53 response element (Gaiddon et al. 2001). However, as more and more p63 and/or p73 _target genes are uncovered, many of which are independent of wild-type p53, it remains to be seen whether the relationship between mutant p53 and p63/p73 is always antagonistic.
Mutant p53 and transcription
Wild-type p53 is a potent transcriptional activator, and the transactivation domain (TAD) of p53 can be subdivided into two subdomains: TAD1 contained within residues 1–40, and TAD2 contained within residues 41–61 (Chang et al. 1995; Walker and Levine 1996). In fact, it has been shown that wild-type p53 is dependent on four critical hydrophobic amino acids in its N terminus for transactivation activity: Leu 22, Trp 23, Trp 53, and Phe 54 (Lin et al. 1994; Candau et al. 1997; Zhu et al. 1998; Venot et al. 1999). When these four residues are mutated to polar amino acids (L22Q/W23S/W53Q/F54S), the transactivation capability of wild-type p53 is completely abolished. The transactivation subdomains have also been shown to be essential for binding to coactivators of p53, such as p300 (Teufel et al. 2007) and TATA-binding protein (Chang et al. 1995), in addition to mediating other regulatory interactions, such as with Mdm2 (Kussie et al. 1996; Lin et al. 1994) and Mdm4 (Shvarts et al. 1996).
Just as wild-type p53 primarily functions as a transcription factor, mutant p53 proteins have been shown to transactivate myriad genes involved in many different aspects of tumorigenesis (Strano et al. 2007; Weisz et al. 2007b; Brosh and Rotter 2009). In addition, several studies have implicated the transactivation domains of p53 as critical for pro-oncogenic functions of p53 mutants (Lin et al. 1995; Frazier et al. 1998; Matas et al. 2001; Yan and Chen 2010; Freed-Pastor et al. 2012). While the transactivation _targets of wild-type p53 tend to promote either cell cycle arrest/senescence or programmed cell death, the _targets of tumor-derived p53 mutants are quite varied in terms of their biological effects. We list here a number of _target genes that serve to increase proliferation, inhibit apoptosis, promote chemoresistance, and impinge on key metabolic or cell–cell/cell–ECM signaling pathways (Table 2).
Table 2.
Select list of mutant p53 transcriptionally activated genes
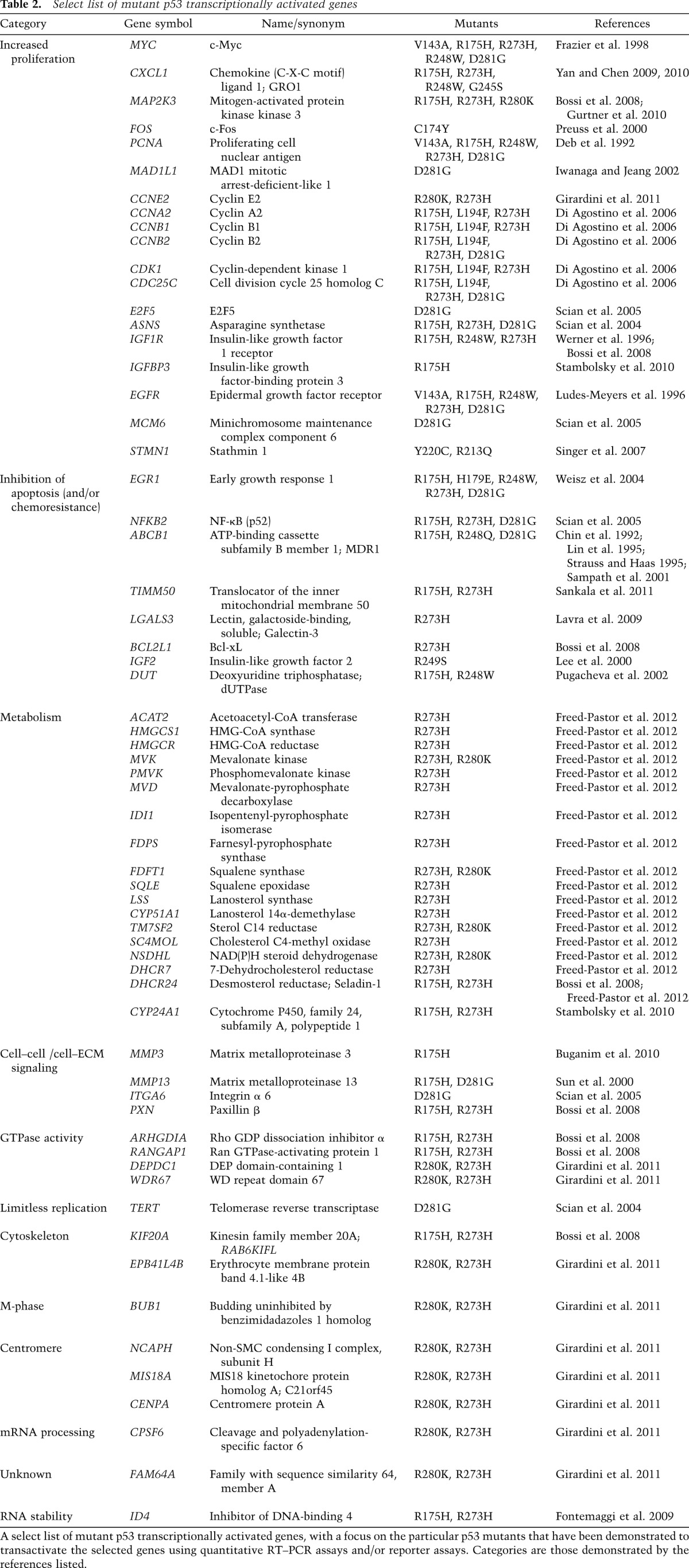
Tumor-derived p53 mutants have been demonstrated to transactivate MYC (Frazier et al. 1998), CXCL1 (Yan and Chen 2009), PCNA (Deb et al. 1992), MAP2K3 (Bossi et al. 2008), CCNA, CCNB, CDK1, CDC25C (Di Agostino et al. 2008), ASNS (Scian et al. 2004), E2F5, MCM6 (Scian et al. 2005), IGF1R (Werner et al. 1996), STMN1 (Singer et al. 2007), and EGFR (Ludes-Meyers et al. 1996), all of which can promote proliferation of cancer cells. Additionally, mutant p53 proteins can up-regulate genes, the protein products of which serve to inhibit apoptosis or promote chemoresistance. For example, p53 mutants can transactivate EGR1 (Weisz et al. 2004), ABCB1 (also known as MDR1 or P-glycoprotein) (Chin et al. 1992; Lin et al. 1995; Strauss and Haas 1995; Sampath et al. 2001), IGF2 (Lee et al. 2000), DUT (Pugacheva et al. 2002), BCL2L1 (also known as Bcl-xL) (Bossi et al. 2008), TIMM50 (Sankala et al. 2011), LGALS3 (Lavra et al. 2009), and NFKB2 (Scian et al. 2005; Vaughan et al. 2012), all of which can inhibit cell death.
Finally, p53 mutants can up-regulate genes with a variety of other cellular effects, such as limitless replication (TERT) (Scian et al. 2004), matrix degradation (MMP3 and MMP13) (Sun et al. 2000; Buganim et al. 2010), sterol biosynthesis (17 genes, including HMGCR; HMG-CoA reductase) (Freed-Pastor et al. 2012), and Rho GTPase signaling (ARHGDIA) (Bossi et al. 2008).
To date, there is no unifying hypothesis to explain the ability of mutant p53 proteins to regulate such a wide variety of _target genes, and a defined mutant p53 response element has yet to be identified. Nevertheless, there have been several proposed mechanisms that account for individual genes or classes of genes regulated by p53 mutants. One of the more commonly proposed mechanisms posits that mutant forms of p53 can interact with other sequence-specific transcription factors and thereby be recruited to their respective cognate binding sites and either strengthen or dampen the _target response. To this end, at least three tumor-derived p53 variants have been demonstrated to interact with the CCAAT-binding factor NF-Y, and this complex serves to up-regulate NF-Y _target genes such as CCNA, CCNB, CDK1, and CDC25C (encoding cyclins and cyclin-dependent kinases that serve to promote cell cycle progression) following adriamycin (also known as doxorubicin) treatment (Di Agostino et al. 2006). Furthermore, the transcriptional activity of mutant p53 toward NF-Y _target genes is enhanced by p53 interacting with PML (Haupt et al. 2009). Recently, it was also shown that the interaction between mutant p53 and NF-Y is mediated by TopBP1 (Liu et al. 2011). The ability of mutant p53 to up-regulate ABCB1 (also known as MDR1), a transmembrane protein characterized by an ATP-binding cassette that can efflux a variety of structurally and functionally distinct substrates and thus promote tumor chemoresistance (Bush and Li 2002), has been shown to be dependent on a functional interaction with Ets-1 (Sampath et al. 2001). Mutant p53 can also interact with Sp1 when bound to the consensus Sp1 response elements in the HIV-LTR and augment its activity (Chicas et al. 2000). Additionally, NF-κB _target genes have been found to be significantly overrepresented in a chromatin immunoprecipitation (ChIP)-on-chip analysis of potential mutant p53-binding sites, and a tumor-derived p53 mutant can enhance the transcriptional activity of NF-κB in response to TNF-α (Weisz et al. 2007a; Dell'Orso et al. 2011). Mutant p53 has also been shown to bind to VDR and serve to synergistically up-regulate vitamin D-responsive genes such as IGFBP3 and CYP24A1 (Stambolsky et al. 2010). Finally, our recent work has shown that mutant p53 can transcriptionally regulate the expression of mevalonate pathway genes (whose products allow cells to make de novo cholesterol and other key metabolites) through an interaction with the SREBP family of transcription factors (Freed-Pastor et al. 2012).
Interestingly, wild-type p53 has been shown to interact with many of the same transcription factors: NF-Y, Sp1, and VDR (Bargonetti et al. 1997; Imbriano et al. 2005; Stambolsky et al. 2010). Likewise, wild-type p53 regulates many of the same _target genes as described for mutant p53. However, the outcome of wild-type p53 binding is often exactly reciprocal to that mediated by mutant p53. Whereas mutant p53 augments the expression of NF-Y, Sp1, NF-κB, Ets-1, and VDR _target genes, wild-type p53 most commonly represses these same _target genes (Werner et al. 1996; Sun et al. 2000; Sampath et al. 2001; Di Agostino et al. 2006; Peart and Prives 2006; Lane and Levine 2010; Stambolsky et al. 2010). In partial explanation of this, Di Agostino et al. (2006) have shown that in response to adriamycin, wild-type p53 and mutant p53 recruit different cofactors: the histone deacetylase HDAC1 in the case of wild-type p53, and the histone acetyltransferase p300 in the case of mutant p53. While the mechanism for the recruitment of opposing histone-modifying enzymes is as yet unclear, in the presence of mutant p53 proteins, this switch in epigenetic modifiers serves to increase histone acetylation and thus results in increased transcription. It will be interesting to examine whether the dichotomy between wild-type and mutant p53 is a universal theme in relation to newly uncovered p53-interacting partners.
While coopting cellular transcription factors to promote the transactivation of their _target genes provides some explanation for the activity of mutant p53, a critical question still remains: In cells in which mutant p53 can be shown to interact with a particular transcription factor (e.g., NF-Y, Sp1, Ets-1, etc.), why is only a subset of their _target genes affected by mutant p53? One can speculate that perhaps this is due to a necessary cofactor (in addition to mutant p53 and its partner transcription factor) and that the relevant complex is only present on certain promoters at any given time. Alternatively, perhaps the chromatin landscapes of certain promoters are particularly amenable to mutant p53 binding.
While a subset of mutant p53 _target gene regulation can be explained by association with sequence-specific transcription factors, the different promoters activated by p53 mutants share little to no sequence homology. This has led to the hypothesis that mutant p53 recruitment may be determined by DNA structural motifs as opposed to recognition of a sequence-specific mutant p53 response element (Kim and Deppert 2004, 2007). It has also been shown that several tumor-derived mutants of p53 have a high affinity for nuclear matrix attachment regions (MARs), DNA sequences that bind with high affinity to the nuclear matrix, a salt and detergent-insoluble proteinacious structure (Muller et al. 1996; Will et al. 1998; Koga and Deppert 2000). MARs are highly AT-rich regions that promote structural alterations within chromatin and often adopt non-B DNA conformations (Kim and Deppert 2004). In support of this hypothesis, mutant p53 proteins have been shown to bind to non-B DNA in vitro and in vivo (Gohler et al. 2005; Brazdova et al. 2009). Mutant p53 binding to DNA structure-specific motifs is presumably a remnant of a wild-type p53 function, as it has now been shown that, in addition to sequence specificity, wild-type p53 binds to DNA via multiple sequence-nonspecific interactions (Kim and Deppert 2003). For example, wild-type p53 exhibits high-affinity binding to dsDNA, ssDNA, secondary DNA structures, and mismatched bases and DNA bulges (Ahn and Prives 2001). In line with this, the C terminus of p53 can recognize DNA in a non-sequence-specific manner, and the structure selectivity of both wild-type and mutant p53 requires its C terminus, which is rarely mutated in human tumors (Petitjean et al. 2007a,b). Finally, it is intriguing that numerous pediatric adrenal cortical carcinoma patients in southern Brazil harbor a specific germline mutation in p53 (R337H) that lies within its tetramerization domain (Ribeiro et al. 2001). It would be interesting to determine whether this inherited mutation could influence p53 interactions with structural motifs in DNA.
The findings that mutant p53 proteins can interact with _target gene promoters through other transcription factors and in a DNA structure-specific manner have been used to justify the absence of a well-described consensus mutant p53 sequence-specific response element. However, these attributes may in fact mask the existence of a mutant p53 response element, if one exists. In other words, using genome-wide analyses to identify mutant p53-binding sequences, one could not distinguish between sequences that are bound through other transcription factors, through DNA structural motifs, or directly through a (hypothetical) mutant p53 response element.
What can we learn from the mutational spectra of TP53?
Mutagenic events in DNA are generally accepted to arise from either external events (environmental mutagens) or internal events (resulting from replication errors, depurination, errors in repair, etc.). The nature of mutations arising from external (exogenous) events is determined primarily by the mutagenic agent. For example, ultraviolet (UV) light commonly induces pyrimidine dimers in adjacent nucleotides, a common mutational event in skin carcinogenesis (Basset-Seguin et al. 1994). On the other hand, internal (endogenous) mutations arise from spontaneous events and preferentially occur at CpG dinucleotides. This is likely due to the fact that the cytosine in this dinucleotide is frequently methylated; spontaneous deamination can occur, changing the cytosine to uracil, and the resulting U:G mispair can give rise to a C:G-to-T:A transition (Barnes and Lindahl 2004).
As mentioned above, there are certain “hot spot” missense mutations in the TP53 gene, residues that are mutated at a much higher frequency than expected by chance. Together, these account for nearly one-third of all p53 mutations. This fact is often cited to support a gain-of-function hypothesis, as it is suggestive of a selective advantage of having certain mutations. The amino acids generally regarded as hot spots (R175, G245, R248, R249, R273, and R282) are found mutated in many malignancies. Endogenous mutations commonly arise from spontaneous deamination of methylated CpG dinucleotides, and interestingly, the codons at four of these hot spot residues contain CpG dinucleotides. A transition (C to T or G to A) can occur following deamination of the 5-methylcytosine, and not surprisingly, this is exactly the type of nucleotide change observed at these hot spots in p53 (Caron de Fromentel and Soussi 1992). However, the p53 gene contains many other CpG dinucleotides that are rarely found mutated in cancer, supporting the notion that either these hot spots are functionally important to inactivate wild-type p53 or these substitutions offer distinct activities to the neomorphic protein.
Complicating this further is the fact that p53 mutations vary in the frequency with which they occur in specific tumor types, suggesting that environmental mutagens leave their mark on p53 in a tumor- and tissue-selective manner. Lung, liver, and skin cancer all bear unique mutation distributions in TP53, which may be a result of carcinogens. Perhaps the most dramatic example of this is the finding that hepatocellular carcinomas in certain developing nations have a much higher frequency of p53-R249S mutations than other tumor types or even liver cancers in developed nations (Staib et al. 2003). In addition to the fact that this mutation is deleterious to p53 function, it has a strong association with exposure to Aflatoxin B1, a carcinogenic agent present in a fungal species (Aspergillus flavus) and a common contaminant of food supplies in developing areas of sub-Saharan Africa and Asia. In support of this association playing a causal role in hepatocellular carcinogenesis, Aflatoxin B1 has been shown to induce the same R249S mutation in TP53 when cultured with hepatocytes in vitro (Staib et al. 2003). Similarly, a strong association has been demonstrated between benzo(a)pyrene exposure (found in cigarette smoke) and G:C-to-T:A transversions. These often occur in lung cancer at amino acid positions in p53 that are not otherwise frequently mutated (i.e., 157 and 158) (Toyooka et al. 2003). In fact, it has been proposed that the mutational spectra for different tumor types might represent a “fingerprint” for the mutational agent responsible for tumorigenesis (Lasky and Silbergeld 1996).
The many faces of mutant p53
While many groups choose to use the generic term “mutant p53” to designate any tumor-derived p53 mutant, it is important to recognize that not all p53 mutants are equal. Some mutants of p53 will exert gain-of-function effects, but many other p53 mutants will be selected for during tumorigenesis simply because they abolish wild-type p53 activity. p53 mutations can generally be classified as either “conformational” or “DNA contact” mutants (Fig. 1C; Cho et al. 1994). DNA contact (or class I) mutants, exemplified by p53-R273H, are missense mutations in the amino acid residues that normally make direct contact with _target DNA sequences. Conformational (or class II) mutants, typified by p53-R175H, are those missense mutations that disrupt the structure of the p53 protein on either a local or global level (Sigal and Rotter 2000; Soussi and Lozano 2005).
This distinction is potentially important, particularly when one attempts to make generalizations about mutant p53 proteins. Many mutant p53-binding partners bind with a higher affinity to a subset of tumor-derived p53 mutants (Table 1). Additionally, some _target genes have been identified in the context of particular missense mutations in p53 (Table 2), even though they are now referred to as mutant p53 _target genes. In fact, even different amino acid substitutions at the same position in the p53 protein can have dramatically different phenotypic effects. For example, p53-R248Q, but not p53-R248W (both of which are common tumor-derived mutants), is able to confer invasive ability when overexpressed in p53-null cells (Yoshikawa et al. 2010). Additionally, as already mentioned, p53-R175H and p53-R175P have dramatically different properties, which is highlighted by the different phenotypes they produce when “knocked in” to the endogenous locus in mice (Donehower and Lozano 2009). Furthermore, a retrospective analysis of human breast cancer patients that examined the prognostic impact of TP53 mutations in breast cancer found that specific missense mutations (R248W and mutations at codon 179) actually correlated with a significantly poorer prognosis compared with other missense mutations (Olivier et al. 2006). Therefore, as TP53 mutational status moves closer to clinical practice, it will be important to keep in mind that not all p53 mutations are equivalent.
This is particularly true in the case of attempting to identify a “mutant p53 response element” (if one can be identified), as it is likely that each contact mutant of p53 may have slightly different sequence specificity, since these mutations affect the amino acid residues that directly contact the DNA backbone (Brosh and Rotter 2009).
Stabilization of mutant p53 in tumors
Mutant p53 proteins are often found at extremely high levels in tumors. In fact, positive immunohistochemical staining of p53 in tissue sections is commonly used as a surrogate for detecting a missense mutation in TP53 (although this is an imperfect marker) (Bartek et al. 1990; Alsner et al. 2008). Wild-type p53 is maintained at very low levels in most cells due primarily to tight regulation by the Mdm2 E3 ubiquitin ligase, itself a p53 _target gene, thus creating a negative feedback loop (Poyurovsky and Prives 2006). Mutant forms of p53, on the other hand, are quite stable and often accumulate in tumor cells. Until recently, the prevailing hypothesis to explain the observation of high levels of mutant p53 in tumors was that p53 mutation broke this feedback loop by rendering the p53 mutant protein incapable of transactivating MDM2 (Oren and Rotter 2010). Arguing against this hypothesis is the fact that in the mutant p53 knock-in mouse models, although all tissues contained the mutant allele, these mice did not accumulate mutant p53 in most normal tissues (Lang et al. 2004; Olive et al. 2004). On the other hand, mutant p53 was observed at high levels in a subset of the tumors in these same mouse models (Terzian et al. 2008). Interestingly, when the mutant p53 knock-in mice were crossed into an Mdm2-null background, the mutant p53 protein was stabilized in some normal tissues, albeit not all (Terzian et al. 2008). Together, these findings and others (Suh et al. 2011) strongly suggest that levels of mutant p53 can be regulated by mechanisms similar to that of the wild-type p53 protein, but additional events occur during tumorigenesis that are necessary to abrogate this regulation and lead to accumulation of mutant p53.
In line with this idea, at least one report has demonstrated an enhanced mutant p53 gain-of-function phenotype following common chemotherapeutic agents (Di Agostino et al. 2006). Similarly, it was recently reported that in a large randomized clinical trial to investigate the effects of cisplatin-based adjuvant chemotherapy in non-small-cell lung carcinoma (NSCLC) patients following tumor resection, TP53 mutational status had a significant predictive impact for response to therapy. While TP53 status had no prognostic significance in the absence of adjuvant chemotherapy, patients bearing a mutant p53 had a significantly reduced disease-free interval and overall survival only after undergoing treatment with cisplatin (Goldstein et al. 2011). These findings may have important clinical repercussions, not just in the setting of traditional chemotherapeutics, but particularly as Mdm2 antagonists are under investigation as therapeutic agents (Prives and White 2008).
As high levels of mutant p53 have been shown to be important for many aspects of tumorigenesis, one potential therapeutic approach is to _target cellular proteins responsible for mutant p53 stabilization. One recent development has done just that in _targeting HSP90. It has long been observed that mutant forms of p53 interact with heat-shock proteins in tumors (Sepehrnia et al. 1996) and that this interaction affects Mdm2-mediated turnover of mutant p53 (Peng et al. 2001). Recently, RNAi-mediated depletion or pharmacological inhibition of HSP90 has been shown to destabilize mutant p53, counteracting many of its gain-of-function effects (Li et al. 2011a,b).
p53 and therapeutic approaches
Importantly, not only has p53 been demonstrated to be inactivated in most tumors, but several in vivo studies have demonstrated that reactivation of wild-type p53 in p53-null or p53 mutant tumors is sufficient to lead to tumor stasis/regression (Christophorou et al. 2005, 2006; Xue et al. 2007; Kenzelmann Broz and Attardi 2010; Wang et al. 2011). This observation has paved the way for multiple approaches to activate wild-type p53 in tumor cells.
Among such strategies that bolster or release wild-type p53 activity in tumors are small molecules that inhibit the interaction between p53 and its negative regulators (Mdm2 and/or MdmX), gene therapy to deliver wild-type p53 to tumors, and small molecules that block the activity of cellular factors that inhibit wild-type functionality, such as members of the sirtuin family. Since the above approaches are not pertinent to the topic of this review, we do not delve further into this area, but excellent reviews on this topic are available (Chen et al. 2010; Brown et al. 2011; Di et al. 2011; Vu and Vassilev 2011). More relevantly, significant effort has been mounted to identify means to counteract mutant forms of p53 in tumors. These include small molecules that either reactivate mutant p53 to a wild-type conformation or destabilize mutant p53, as well as therapeutic approaches that _target downstream mediators of mutant p53 gain of function. Several other strategies are currently in development that seek to take advantage of p53 mutation in a large proportion of tumors. These are discussed below.
For some time now, an actively sought goal has been to somehow convert existing mutant p53 proteins in tumors into forms that exhibit wild-type p53 functions, thereby allowing p53 to carry out its tumor-suppressive functions in cancer cells (Brown et al. 2011). The hope was that since mutant proteins are often present at very high levels in tumors, it might be possible to recover enough wild-type activity to induce arrest or, optimally, death in such cells. One of the first approaches involved the use of short peptides corresponding to the C terminus that were considered capable of activating the DNA-binding properties of some mutant forms of p53 (Selivanova et al. 1997). Another potentially promising avenue using small peptides has involved a short (nine-amino-acid) peptide that interacts with the p53 core domain and functions as a chaperone to stabilize the wild-type conformation of mutant proteins (Friedler et al. 2002). Since small peptides themselves have limitations as drugs, small molecules have been actively pursued that might similarly directly interact with and alter the activity of mutant p53 proteins. The first such reported molecule, CP31398, allows mutant p53-expressing cells to induce the expression of canonical p53 _target genes or drive expression from a p53 reporter construct and even impair tumor growth (Foster et al. 1999). Despite this promising start, it was later discovered that instead of binding directly to mutant p53, CP31398 intercalates into DNA (Rippin et al. 2002). However, this setback has not dismayed the many groups looking to identify compounds that restore p53 activity in mutant p53 tumors. A derivative of a naturally occurring compound, 9-hydroxy-ellipticine, can produce wild-type p53 activities (arrest and death) in cells containing but not lacking mutant p53 proteins, although the mechanism by which it does so is apparently not yet understood (Sugikawa et al. 1999). Of the small molecules that _target mutant p53, the most well-advanced so far has been PRIMA-1 (also known as APR-246), which has been shown to reactivate missense mutants of p53 to regain at least some functions of wild-type p53 and thus halt tumor growth and is currently in a phase I clinical trial (Bykov et al. 2002; Wiman 2010). However, PRIMA-1 has also been shown to exert p53-independent activities, potentially complicating its use in the clinic (Brown et al. 2011).
As the interaction between mutant p53 and p63/p73 has been incriminated in many pro-oncogenic effects of mutant p53, one of the more intriguing therapeutic approaches may be to disrupt this interaction, thus allowing p63 and/or p73 to function. RETRA (reactivation of transcriptional reporter activity) is a small molecule reported to do just that. In a screen hoping to identify small molecules that could activate transcription from a wild-type p53 reporter construct, the top compound, RETRA, was actually found to disrupt the mutant p53:p73 complex, allowing p73 to activate transcription from this reporter construct. This was elegantly demonstrated, as RETRA-mediated activity was completely impaired when the cells bearing mutant p53 were depleted of p73 using RNAi and then treated with RETRA. This compound could also activate transcription of p53 _target genes in mutant p53-bearing tumors in vivo and prevent xenografted tumor cell growth (Kravchenko et al. 2008). While this compound may not “reactivate” wild-type p53, it is tempting to speculate that perhaps inhibiting some of the gain-of-function effects of mutant p53 might be sufficient as a therapeutic approach for mutant p53 tumors.
Likewise, another approach involves inhibiting factors that function to stabilize mutant p53 in tumors. As mentioned above, recent work has further implicated the HDAC6–Hsp90 axis in stabilizing p53 mutants in tumors. In fact, deletion of HSF1, a master regulator of the heat-shock response, dramatically impacts tumor formation and overall survival in the mutant p53 knock-in mouse model (Dai et al. 2007), and inhibition of either Hsp90 or HDAC6 is sufficient to destabilize mutant p53 in cancer cell lines and thereby decrease tumorigenicity (Li et al. 2011a,b). Excitingly, mutant p53 destabilization could be achieved using an FDA-approved HDAC inhibitor (SAHA) (Li et al. 2011a), suggesting that this approach may be quickly amenable to translation into the clinic.
While the above-mentioned compounds _target multiple p53 mutants, one of the more impressive approaches from recent years has been to develop a structure-based “mutant-specific” drug. PhiKan083 is a small molecule that binds to a small cleft in the C terminus of p53-Y220C, stabilizing the conformation of the core domain and restoring transactivation of p53 _target genes (Boeckler et al. 2008). While it will take time to determine the utility of PhiKan083 as a clinical agent, it is a beautiful proof of principle of rational drug discovery based on the crystal structure of a mutant p53. Although it must be noted that this cleft is probably unique to Y220C, a related approach to design drugs specific to hot spot mutants of p53 may be a promising avenue in the future. Somewhat relevant to the concept of rational drug discovery, second-site suppressor mutations have been identified that restore wild-type DNA binding and transactivation to tumor-derived mutant forms of p53 (Baroni et al. 2004; Suad et al. 2009).
The final therapeutic approach seeks to take advantage of the large proportion of p53 mutations in human cancer by identifying common mechanisms by which tumor-derived mutants of p53 promote tumor growth and use small molecules to _target these downstream pathways. One recent advance in this area came about with the identification that certain mutants of p53 promote up-regulation of many genes in the mevalonate pathway, by which cells make de novo cholesterol (Freed-Pastor et al. 2012). HMG-CoA reductase, the rate-limiting enzyme within the mevalonate pathway, is the famous _target of the statin class of drugs (Wong et al. 2002). As HMG-CoA reductase was one of the genes up-regulated by mutant p53, we and our colleagues (Freed-Pastor et al. 2012) treated breast cancer cell lines bearing mutant p53 with statins and showed that inhibition of the mevalonate pathway had dramatic effects on the invasion and survival of mutant p53-expressing breast cancer cells using a three-dimensional culture model, and statins served to inhibit tumor growth in vivo. It is too early to say whether statins will be a useful adjunctive therapy in mutant p53-expressing tumors, but the idea of _targeting downstream mediators of mutant p53 gain of function is another approach that may yield clinical benefits.
Conclusion
As our understanding of the biology surrounding the p53 tumor suppressor continues to grow, we are only beginning to grasp its complexity. Here we reviewed the importance of the p53 tumor suppressor gene in human cancer and highlighted growing evidence that missense mutations in p53 not only abrogate tumor suppressor function, but can actually endow p53 with oncogenic properties. While the gain-of-function hypothesis has existed almost as long as the p53 field, recent years have seen a renewed interest in pro-oncogenic properties of mutant p53 following the seminal finding that mutant p53 knock-in mice exhibit an altered tumor spectrum compared with p53 knockout mice and that mutant p53 confers a metastatic phenotype not observed in p53-null mice. Over the last 5 years alone, p53 mutants have been found to actively contribute to tumor proliferation, survival, limitless replication, somatic cell reprogramming, genomic instability, inflammation, disruption of tissue architecture, migration, invasion, angiogenesis, and metastasis. Amazingly, this places mutant p53 in a central role in tumorigenesis, impacting nearly all of the 10 (updated) “hallmarks of cancer” proposed by Hanahan and Weinberg (2011).
The recognition that not all p53 mutants are equivalent is important not merely as a conceptual distinction, but may also have practical implications. As cancer therapy inches closer and closer to personalized medicine, it will be important to not only differentiate between wild-type and mutant p53 tumors, but it may also prove beneficial to delineate the particular mutations that a patient's tumor bears. This may provide prognostic information (Olivier et al. 2006), and as more groups begin to look for therapeutics _targeting individual mutant p53 proteins, specific p53 mutations may even guide therapeutic decisions.
Throughout this review, we posed questions that are relevant to what is already known about mutant p53. But there are clearly more avenues to pursue, and more important questions still abound. How do mutant forms of p53 regulate the chromatin landscape of their _target genes? What keeps mutant p53 proteins so high in tumors, and what prevents Mdm2 from destabilizing them? Does the fact that, like p63 and p73, p53 is also expressed as different isoforms impact mutant p53 function in tumors? Are there sequences within the genome to which mutant proteins bind directly? Do protein modifications affect mutant p53 functions in cells? Can the immune system be harnessed to destroy mutant p53-expressing tumor cells? Thus, the findings in mutant p53 biology that we reviewed herein are likely only the beginning of the journey that will hopefully end with compounds or strategies that can be used to treat the large numbers of cancer patients whose tumors harbor mutant p53.
Acknowledgments
We are grateful to members of the Prives laboratory for helpful discussions and comments. This work was supported by grants CA87497 and CA77742 from the National Cancer Institute.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.190678.112.
References
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. 2009. A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137: 87–98 [DOI] [PubMed] [Google Scholar]
- Ahn J, Prives C 2001. The C-terminus of p53: The more you learn the less you know. Nat Struct Biol 8: 730–732 [DOI] [PubMed] [Google Scholar]
- Alsner J, Jensen V, Kyndi M, Offersen BV, Vu P, Borresen-Dale AL, Overgaard J 2008. A comparison between p53 accumulation determined by immunohistochemistry and TP53 mutations as prognostic variables in tumours from breast cancer patients. Acta Oncol 47: 600–607 [DOI] [PubMed] [Google Scholar]
- Attardi LD, Jacks T 1999. The role of p53 in tumour suppression: Lessons from mouse models. Cell Mol Life Sci 55: 48–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, et al. 1989. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244: 217–221 [DOI] [PubMed] [Google Scholar]
- Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B 1990. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res 50: 7717–7722 [PubMed] [Google Scholar]
- Bargonetti J, Chicas A, White D, Prives C 1997. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell Mol Biol (Noisy-le-grand) 43: 935–949 [PubMed] [Google Scholar]
- Barnes DE, Lindahl T 2004. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38: 445–476 [DOI] [PubMed] [Google Scholar]
- Baroni TE, Wang T, Qian H, Dearth LR, Truong LN, Zeng J, Denes AE, Chen SW, Brachmann RK 2004. A global suppressor motif for p53 cancer mutants. Proc Natl Acad Sci 101: 4930–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Iggo R, Gannon J, Lane DP 1990. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene 5: 893–899 [PubMed] [Google Scholar]
- Basset-Seguin N, Moles JP, Mils V, Dereure O, Guilhou JJ 1994. TP53 tumor suppressor gene and skin carcinogenesis. J Invest Dermatol 103: 102S–106S [DOI] [PubMed] [Google Scholar]
- Ben David Y, Prideaux VR, Chow V, Benchimol S, Bernstein A 1988. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene 3: 179–185 [PubMed] [Google Scholar]
- Bensaad K, Le Bras M, Unsal K, Strano S, Blandino G, Tominaga O, Rouillard D, Soussi T 2003. Change of conformation of the DNA-binding domain of p53 is the only key element for binding of and interference with p73. J Biol Chem 278: 10546–10555 [DOI] [PubMed] [Google Scholar]
- Blandino G, Levine AJ, Oren M 1999. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 18: 477–485 [DOI] [PubMed] [Google Scholar]
- Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR 2008. _targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci 105: 10360–10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A 2006. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 25: 304–309 [DOI] [PubMed] [Google Scholar]
- Bossi G, Marampon F, Maor-Aloni R, Zani B, Rotter V, Oren M, Strano S, Blandino G, Sacchi A 2008. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle 7: 1870–1879 [DOI] [PubMed] [Google Scholar]
- Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, Brugieres L, Chompret A, de Paillerets BB, Stoppa-Lyonnet D, et al. 2008. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J Med Genet 45: 535–538 [DOI] [PubMed] [Google Scholar]
- Brazdova M, Quante T, Togel L, Walter K, Loscher C, Tichy V, Cincarova L, Deppert W, Tolstonog GV 2009. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res 37: 1486–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow RG, Peacock J, Jang A, Kim J, Hill RP, Benchimol S 2003. Resistance to DNA-damaging agents is discordant from experimental metastatic capacity in MEF ras-transformants-expressing gain of function MTp53. Oncogene 22: 2960–2966 [DOI] [PubMed] [Google Scholar]
- Brosh R, Rotter V 2009. When mutants gain new powers: News from the mutant p53 field. Nat Rev Cancer 9: 701–713 [DOI] [PubMed] [Google Scholar]
- Brown CJ, Cheok CF, Verma CS, Lane DP 2011. Reactivation of p53: From peptides to small molecules. Trends Pharmacol Sci 32: 53–62 [DOI] [PubMed] [Google Scholar]
- Buganim Y, Solomon H, Rais Y, Kistner D, Nachmany I, Brait M, Madar S, Goldstein I, Kalo E, Adam N, et al. 2010. p53 Regulates the Ras circuit to inhibit the expression of a cancer-related gene signature by various molecular pathways. Cancer Res 70: 2274–2284 [DOI] [PubMed] [Google Scholar]
- Bush JA, Li G 2002. Cancer chemoresistance: The relationship between p53 and multidrug transporters. Int J Cancer 98: 323–330 [DOI] [PubMed] [Google Scholar]
- Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G 2002. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 8: 282–288 [DOI] [PubMed] [Google Scholar]
- Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis TD, Berger SL 1997. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 15: 807–816 [DOI] [PubMed] [Google Scholar]
- Caron de Fromentel C, Soussi T 1992. TP53 tumor suppressor gene: A model for investigating human mutagenesis. Genes Chromosomes Cancer 4: 1–15 [DOI] [PubMed] [Google Scholar]
- Chan WM, Siu WY, Lau A, Poon RY 2004. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol 24: 3536–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Kim DH, Lee SW, Choi KY, Sung YC 1995. Transactivation ability of p53 transcriptional activation domain is directly related to the binding affinity to TATA-binding protein. J Biol Chem 270: 25014–25019 [DOI] [PubMed] [Google Scholar]
- Chen F, Wang W, El-Deiry WS 2010. Current strategies to _target p53 in cancer. Biochem Pharmacol 80: 724–730 [DOI] [PubMed] [Google Scholar]
- Chicas A, Molina P, Bargonetti J 2000. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem Biophys Res Commun 279: 383–390 [DOI] [PubMed] [Google Scholar]
- Chin KV, Ueda K, Pastan I, Gottesman MM 1992. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 255: 459–462 [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP 1994. Crystal structure of a p53 tumor suppressor–DNA complex: Understanding tumorigenic mutations. Science 265: 346–355 [DOI] [PubMed] [Google Scholar]
- Christophorou MA, Martin-Zanca D, Soucek L, Lawlor ER, Brown-Swigart L, Verschuren EW, Evan GI 2005. Temporal dissection of p53 function in vitro and in vivo. Nat Genet 37: 718–726 [DOI] [PubMed] [Google Scholar]
- Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI 2006. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443: 214–217 [DOI] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S 2007. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130: 1005–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH 1999. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem 274: 18709–18714 [DOI] [PubMed] [Google Scholar]
- Deb S, Jackson CT, Subler MA, Martin DW 1992. Modulation of cellular and viral promoters by mutant human p53 proteins found in tumor cells. J Virol 66: 6164–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ 1979. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci 76: 2420–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Orso S, Fontemaggi G, Stambolsky P, Goeman F, Voellenkle C, Levrero M, Strano S, Rotter V, Oren M, Blandino G 2011. ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. OMICS 15: 305–312 [DOI] [PubMed] [Google Scholar]
- Di J, Zhang Y, Zheng J 2011. Reactivation of p53 by inhibiting Mdm2 E3 ligase: A novel antitumor approach. Curr Cancer Drug _targets 11: 987–994 [DOI] [PubMed] [Google Scholar]
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G 2006. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10: 191–202 [DOI] [PubMed] [Google Scholar]
- Di Agostino S, Cortese G, Monti O, Dell'Orso S, Sacchi A, Eisenstein M, Citro G, Strano S, Blandino G 2008. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 7: 3440–3447 [DOI] [PubMed] [Google Scholar]
- Di Como CJ, Prives C 1998. Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene 16: 2527–2539 [DOI] [PubMed] [Google Scholar]
- Di Como CJ, Gaiddon C, Prives C 1999. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol 19: 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ 1993. Gain of function mutations in p53. Nat Genet 4: 42–46 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Lozano G 2009. 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer 9: 831–841 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B 1992. Definition of a consensus binding site for p53. Nat Genet 1: 45–49 [DOI] [PubMed] [Google Scholar]
- El-Hizawi S, Lagowski JP, Kulesz-Martin M, Albor A 2002. Induction of gene amplification as a gain-of-function phenotype of mutant p53 proteins. Cancer Res 62: 3264–3270 [PubMed] [Google Scholar]
- Eliyahu D, Raz A, Gruss P, Givol D, Oren M 1984. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 312: 646–649 [DOI] [PubMed] [Google Scholar]
- Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M 1989. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci 86: 8763–8767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge RM, Fuqua SA, Clark GM, Pujol P, Allred DC, McGuire WL 1993. Prognostic significance of p53 gene alterations in node-negative breast cancer. Breast Cancer Res Treat 26: 225–235 [DOI] [PubMed] [Google Scholar]
- Ferenczy A, Franco E 2002. Persistent human papillomavirus infection and cervical neoplasia. Lancet Oncol 3: 11–16 [DOI] [PubMed] [Google Scholar]
- Finlay CA, Hinds PW, Levine AJ 1989. The p53 proto-oncogene can act as a suppressor of transformation. Cell 57: 1083–1093 [DOI] [PubMed] [Google Scholar]
- Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F, Jacks T 2005. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7: 363–373 [DOI] [PubMed] [Google Scholar]
- Fontemaggi G, Dell'Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, Terrenato I, Mottolese M, Muti P, Domany E, et al. 2009. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol 16: 1086–1093 [DOI] [PubMed] [Google Scholar]
- Foster BA, Coffey HA, Morin MJ, Rastinejad F 1999. Pharmacological rescue of mutant p53 conformation and function. Science 286: 2507–2510 [DOI] [PubMed] [Google Scholar]
- Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP 1998. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol 18: 3735–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. 2012. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148: 244–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, Proctor MR, Rudiger S, Fersht AR 2002. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci 99: 937–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk WD, Pak DT, Karas RH, Wright WE, Shay JW 1992. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol 12: 2866–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C 2001. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21: 1874–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. 2011. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20: 79–91 [DOI] [PubMed] [Google Scholar]
- Goh AM, Coffill CR, Lane DP 2011. The role of mutant p53 in human cancer. J Pathol 223: 116–126 [DOI] [PubMed] [Google Scholar]
- Gohler T, Jager S, Warnecke G, Yasuda H, Kim E, Deppert W 2005. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res 33: 1087–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P 2011. Understanding wild-type and mutant p53 activities in human cancer: New landmarks on the way to _targeted therapies. Cancer Gene Ther 18: 2–11 [DOI] [PubMed] [Google Scholar]
- Gualberto A, Aldape K, Kozakiewicz K, Tlsty TD 1998. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc Natl Acad Sci 95: 5166–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtner A, Starace G, Norelli G, Piaggio G, Sacchi A, Bossi G 2010. Mutant p53-induced up-regulation of mitogen-activated protein kinase kinase 3 contributes to gain of function. J Biol Chem 285: 14160–14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Harris CC, Hollstein M 1993. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med 329: 1318–1327 [DOI] [PubMed] [Google Scholar]
- Haupt S, di Agostino S, Mizrahi I, Alsheich-Bartok O, Voorhoeve M, Damalas A, Blandino G, Haupt Y 2009. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res 69: 4818–4826 [DOI] [PubMed] [Google Scholar]
- Hinds P, Finlay C, Levine AJ 1989. Mutation is required to activate the p53 gene for cooperation with the ras oncogene and transformation. J Virol 63: 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds PW, Finlay CA, Quartin RS, Baker SJ, Fearon ER, Vogelstein B, Levine AJ 1990. Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: A comparison of the ‘hot spot' mutant phenotypes. Cell Growth Differ 1: 571–580 [PubMed] [Google Scholar]
- Hsiao M, Low J, Dorn E, Ku D, Pattengale P, Yeargin J, Haas M 1994. Gain-of-function mutations of the p53 gene induce lymphohematopoietic metastatic potential and tissue invasiveness. Am J Pathol 145: 702–714 [PMC free article] [PubMed] [Google Scholar]
- Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, Korner S, Wang W, Boccaccio C, Thorgeirsson SS, Comoglio PM, Hermeking H, et al. 2011. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci 108: 14240–14245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M, Dobbelstein M, Del Sal G, Piaggio G, Mantovani R 2005. Direct p53 transcriptional repression: In vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol 25: 3737–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MS, Kaelin WG Jr 2001. Role of the newer p53 family proteins in malignancy. Apoptosis 6: 17–29 [DOI] [PubMed] [Google Scholar]
- Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr 2003. Chemosensitivity linked to p73 function. Cancer Cell 3: 403–410 [DOI] [PubMed] [Google Scholar]
- Iwanaga Y, Jeang KT 2002. Expression of mitotic spindle checkpoint protein hsMAD1 correlates with cellular proliferation and is activated by a gain-of-function p53 mutant. Cancer Res 62: 2618–2624 [PubMed] [Google Scholar]
- Jordan JJ, Menendez D, Inga A, Noureddine M, Bell DA, Resnick MA 2008. Noncanonical DNA motifs as transactivation _targets by wild type and mutant p53. PLoS Genet 4: e1000104 doi: 10.1371/journal.pgen.1000104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C 2003. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci 100: 8424–8429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Attardi LD 2010. In vivo analysis of p53 tumor suppressor function using genetically engineered mouse models. Carcinogenesis 31: 1311–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Deppert W 2003. The complex interactions of p53 with _target DNA: We learn as we go. Biochem Cell Biol 81: 141–150 [DOI] [PubMed] [Google Scholar]
- Kim E, Deppert W 2004. Transcriptional activities of mutant p53: When mutations are more than a loss. J Cell Biochem 93: 878–886 [DOI] [PubMed] [Google Scholar]
- Kim E, Deppert W 2007. Interactions of mutant p53 with DNA: Guilt by association. Oncogene 26: 2185–2190 [DOI] [PubMed] [Google Scholar]
- Kim E, Gunther W, Yoshizato K, Meissner H, Zapf S, Nusing RM, Yamamoto H, Van Meir EG, Deppert W, Giese A 2003. Tumor suppressor p53 inhibits transcriptional activation of invasion gene thromboxane synthase mediated by the proto-oncogenic factor ets-1. Oncogene 22: 7716–7727 [DOI] [PubMed] [Google Scholar]
- Kim E, Giese A, Deppert W 2009. Wild-type p53 in cancer cells: When a guardian turns into a blackguard. Biochem Pharmacol 77: 11–20 [DOI] [PubMed] [Google Scholar]
- Koga H, Deppert W 2000. Identification of genomic DNA sequences bound by mutant p53 protein (Gly245 → Ser) in vivo. Oncogene 19: 4178–4183 [DOI] [PubMed] [Google Scholar]
- Kravchenko JE, Ilyinskaya GV, Komarov PG, Agapova LS, Kochetkov DV, Strom E, Frolova EI, Kovriga I, Gudkov AV, Feinstein E, et al. 2008. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci 105: 6302–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP 1996. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274: 948–953 [DOI] [PubMed] [Google Scholar]
- Lane DP, Crawford LV 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278: 261–263 [DOI] [PubMed] [Google Scholar]
- Lane D, Levine A 2010. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2: a000893 doi: 10.1101/cshperspect.a000893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119: 861–872 [DOI] [PubMed] [Google Scholar]
- Langerod A, Zhao H, Borgan O, Nesland JM, Bukholm IR, Ikdahl T, Karesen R, Borresen-Dale AL, Jeffrey SS 2007. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res 9: R30 doi: 10.1186/bcr1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Prives C 2006. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ 13: 951–961 [DOI] [PubMed] [Google Scholar]
- Lasky T, Silbergeld E 1996. P53 mutations associated with breast, colorectal, liver, lung, and ovarian cancers. Environ Health Perspect 104: 1324–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavra L, Ulivieri A, Rinaldo C, Dominici R, Volante M, Luciani E, Bartolazzi A, Frasca F, Soddu S, Sciacchitano S 2009. Gal-3 is stimulated by gain-of-function p53 mutations and modulates chemoresistance in anaplastic thyroid carcinomas. J Pathol 218: 66–75 [DOI] [PubMed] [Google Scholar]
- Lee YI, Lee S, Das GC, Park US, Park SM 2000. Activation of the insulin-like growth factor II transcription by aflatoxin B1 induced p53 mutant 249 is caused by activation of transcription complexes; implications for a gain-of-function during the formation of hepatocellular carcinoma. Oncogene 19: 3717–3726 [DOI] [PubMed] [Google Scholar]
- Levine AJ 2009. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology 384: 285–293 [DOI] [PubMed] [Google Scholar]
- Levine AJ, Oren M 2009. The first 30 years of p53: Growing ever more complex. Nat Rev Cancer 9: 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Wu MC, Chang A, Silver A, Attiyeh EF, Lin J, Epstein CB 1995. The spectrum of mutations at the p53 locus. Evidence for tissue-specific mutagenesis, selection of mutant alleles, and a ‘gain of function' phenotype. Ann N Y Acad Sci 768: 111–128 [DOI] [PubMed] [Google Scholar]
- Li FP, Fraumeni JF Jr 1969a. Rhabdomyosarcoma in children: Epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 43: 1365–1373 [PubMed] [Google Scholar]
- Li FP, Fraumeni JF Jr 1969b. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 71: 747–752 [DOI] [PubMed] [Google Scholar]
- Li Y, Prives C 2007. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 26: 2220–2225 [DOI] [PubMed] [Google Scholar]
- Li D, Marchenko ND, Moll UM 2011a. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6–Hsp90 chaperone axis. Cell Death Differ 18: 1904–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, Moll UM 2011b. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res 9: 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Chen J, Elenbaas B, Levine AJ 1994. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev 8: 1235–1246 [DOI] [PubMed] [Google Scholar]
- Lin J, Teresky AK, Levine AJ 1995. Two critical hydrophobic amino acids in the N-terminal domain of the p53 protein are required for the gain of function phenotypes of human p53 mutants. Oncogene 10: 2387–2390 [PubMed] [Google Scholar]
- Linzer DI, Levine AJ 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17: 43–52 [DOI] [PubMed] [Google Scholar]
- Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M, Mims B, El-Naggar AK, Lozano G 2000. High metastatic potential in mice inheriting a _targeted p53 missense mutation. Proc Natl Acad Sci 97: 4174–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G 2004. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36: 63–68 [DOI] [PubMed] [Google Scholar]
- Liu K, Ling S, Lin WC 2011. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol Cell Biol 31: 4464–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludes-Meyers JH, Subler MA, Shivakumar CV, Munoz RM, Jiang P, Bigger JE, Brown DR, Deb SP, Deb S 1996. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol Cell Biol 16: 6009–6019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M 2001. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: A new biomedical research tool. Oncogene 20: 320–328 [DOI] [PubMed] [Google Scholar]
- Machado-Silva A, Perrier S, Bourdon JC 2010. p53 family members in cancer diagnosis and treatment. Semin Cancer Biol 20: 57–62 [DOI] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. 1990. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233–1238 [DOI] [PubMed] [Google Scholar]
- Manfredi JJ 2010. The Mdm2–p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 24: 1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin MC, Jost CA, Brooks LA, Irwin MS, O'Nions J, Tidy JA, James N, McGregor JM, Harwood CA, Yulug IG, et al. 2000. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet 25: 47–54 [DOI] [PubMed] [Google Scholar]
- Marine JC, Lozano G 2010. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ 17: 93–102 [DOI] [PubMed] [Google Scholar]
- Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G 2006. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 13: 927–934 [DOI] [PubMed] [Google Scholar]
- Marine JC, Dyer MA, Jochemsen AG 2007. MDMX: From bench to bedside. J Cell Sci 120: 371–378 [DOI] [PubMed] [Google Scholar]
- Matas D, Sigal A, Stambolsky P, Milyavsky M, Weisz L, Schwartz D, Goldfinger N, Rotter V 2001. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J 20: 4163–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Spike BT, Wahl GM, Levine AJ 2010. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci 107: 22745–22750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller BF, Paulsen D, Deppert W 1996. Specific binding of MAR/SAR DNA-elements by mutant p53. Oncogene 12: 1941–1952 [PubMed] [Google Scholar]
- Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. 2009. Mutant p53 drives invasion by promoting integrin recycling. Cell 139: 1327–1341 [DOI] [PubMed] [Google Scholar]
- Noll JE, Jeffery J, Al-Ejeh F, Kumar R, Khanna KK, Callen DF, Neilsen PM 2011. Mutant p53 drives multinucleation and invasion through a process that is suppressed by ANKRD11. Oncogene doi: 10.1038/onc.2011.456 [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119: 847–860 [DOI] [PubMed] [Google Scholar]
- Olivier M, Langerod A, Carrieri P, Bergh J, Klaar S, Eyfjord J, Theillet C, Rodriguez C, Lidereau R, Bieche I, et al. 2006. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res 12: 1157–1167 [DOI] [PubMed] [Google Scholar]
- Oren M, Rotter V 2010. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2: a001107 doi: 10.1101/cshperspect.a001107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada LF, Land H, Weinberg RA, Wolf D, Rotter V 1984. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312: 649–651 [DOI] [PubMed] [Google Scholar]
- Peart MJ, Prives C 2006. Mutant p53 gain of function: The NF-Y connection. Cancer Cell 10: 173–174 [DOI] [PubMed] [Google Scholar]
- Peng Y, Chen L, Li C, Lu W, Chen J 2001. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 276: 40583–40590 [DOI] [PubMed] [Google Scholar]
- Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M 2007a. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 26: 2157–2165 [DOI] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M 2007b. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629 [DOI] [PubMed] [Google Scholar]
- Pietsch EC, Sykes SM, McMahon SB, Murphy ME 2008. The p53 family and programmed cell death. Oncogene 27: 6507–6521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D 2009. p53 and E2f: Partners in life and death. Nat Rev Cancer 9: 738–748 [DOI] [PubMed] [Google Scholar]
- Poyurovsky MV, Prives C 2006. Unleashing the power of p53: Lessons from mice and men. Genes Dev 20: 125–131 [DOI] [PubMed] [Google Scholar]
- Preuss U, Kreutzfeld R, Scheidtmann KH 2000. Tumor-derived p53 mutant C174Y is a gain-of-function mutant which activates the fos promoter and enhances colony formation. Int J Cancer 88: 162–171 [DOI] [PubMed] [Google Scholar]
- Prives C, Hall PA 1999. The p53 pathway. J Pathol 187: 112–126 [DOI] [PubMed] [Google Scholar]
- Prives C, White E 2008. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev 22: 1259–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva EN, Ivanov AV, Kravchenko JE, Kopnin BP, Levine AJ, Chumakov PM 2002. Novel gain of function activity of p53 mutants: Activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene 21: 4595–4600 [DOI] [PubMed] [Google Scholar]
- Resnick MA, Inga A 2003. Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc Natl Acad Sci 100: 9934–9939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, DeLacerda L, Rabin M, Cadwell C, Sampaio G, et al. 2001. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci 98: 9330–9335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A 2008. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9: 402–412 [DOI] [PubMed] [Google Scholar]
- Rippin TM, Bykov VJ, Freund SM, Selivanova G, Wiman KG, Fersht AR 2002. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21: 2119–2129 [DOI] [PubMed] [Google Scholar]
- Rotter V 1983. p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc Natl Acad Sci 80: 2613–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, Oren M, Vousden KH 1996. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J 15: 827–838 [PMC free article] [PubMed] [Google Scholar]
- Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD 2001. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 276: 39359–39367 [DOI] [PubMed] [Google Scholar]
- Sankala H, Vaughan C, Wang J, Deb S, Graves PR 2011. Upregulation of the mitochondrial transport protein, Tim50, by mutant p53 contributes to cell growth and chemoresistance. Arch Biochem Biophys 512: 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarig R, Rivlin N, Brosh R, Bornstein C, Kamer I, Ezra O, Molchadsky A, Goldfinger N, Brenner O, Rotter V 2010. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med 207: 2127–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scian MJ, Stagliano KE, Deb D, Ellis MA, Carchman EH, Das A, Valerie K, Deb SP, Deb S 2004. Tumor-derived p53 mutants induce oncogenesis by transactivating growth-promoting genes. Oncogene 23: 4430–4443 [DOI] [PubMed] [Google Scholar]
- Scian MJ, Stagliano KE, Anderson MA, Hassan S, Bowman M, Miles MF, Deb SP, Deb S 2005. Tumor-derived p53 mutants induce NF-κB2 gene expression. Mol Cell Biol 25: 10097–10110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanova G, Iotsova V, Okan I, Fritsche M, Strom M, Groner B, Grafstrom RC, Wiman KG 1997. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med 3: 632–638 [DOI] [PubMed] [Google Scholar]
- Sepehrnia B, Paz IB, Dasgupta G, Momand J 1996. Heat shock protein 84 forms a complex with mutant p53 protein predominantly within a cytoplasmic compartment of the cell. J Biol Chem 271: 15084–15090 [DOI] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, et al. 1996. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J 15: 5349–5357 [PMC free article] [PubMed] [Google Scholar]
- Sigal A, Rotter V 2000. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res 60: 6788–6793 [PubMed] [Google Scholar]
- Singer S, Ehemann V, Brauckhoff A, Keith M, Vreden S, Schirmacher P, Breuhahn K 2007. Protumorigenic overexpression of stathmin/Op18 by gain-of-function mutation in p53 in human hepatocarcinogenesis. Hepatology 46: 759–768 [DOI] [PubMed] [Google Scholar]
- Song H, Hollstein M, Xu Y 2007. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 9: 573–580 [DOI] [PubMed] [Google Scholar]
- Soussi T, Lozano G 2005. p53 mutation heterogeneity in cancer. Biochem Biophys Res Commun 331: 834–842 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH 1990. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348: 747–749 [DOI] [PubMed] [Google Scholar]
- Staib F, Hussain SP, Hofseth LJ, Wang XW, Harris CC 2003. TP53 and liver carcinogenesis. Hum Mutat 21: 201–216 [DOI] [PubMed] [Google Scholar]
- Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, Shiff I, Kogan I, Shay M, Kalo E, et al. 2010. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 17: 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G 2001. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem 276: 15164–15173 [DOI] [PubMed] [Google Scholar]
- Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M, et al. 2002. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem 277: 18817–18826 [DOI] [PubMed] [Google Scholar]
- Strano S, Dell'Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blandino G 2007. Mutant p53: An oncogenic transcription factor. Oncogene 26: 2212–2219 [DOI] [PubMed] [Google Scholar]
- Strauss BE, Haas M 1995. The region 3′ to the major transcriptional start site of the MDR1 downstream promoter mediates activation by a subset of mutant P53 proteins. Biochem Biophys Res Commun 217: 333–340 [DOI] [PubMed] [Google Scholar]
- Suad O, Rozenberg H, Brosh R, Diskin-Posner Y, Kessler N, Shimon LJ, Frolow F, Liran A, Rotter V, Shakked Z 2009. Structural basis of restoring sequence-specific DNA binding and transactivation to mutant p53 by suppressor mutations. J Mol Biol 385: 249–265 [DOI] [PubMed] [Google Scholar]
- Sugikawa E, Hosoi T, Yazaki N, Gamanuma M, Nakanishi N, Ohashi M 1999. Mutant p53 mediated induction of cell cycle arrest and apoptosis at G1 phase by 9-hydroxyellipticine. Anticancer Res 19: 3099–3108 [PubMed] [Google Scholar]
- Suh YA, Post SM, Elizondo-Fraire AC, Maccio DR, Jackson JG, El-Naggar AK, Van Pelt C, Terzian T, Lozano G 2011. Multiple stress signals activate mutant p53 in vivo. Cancer Res 71: 7168–7175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Cheung JM, Martel-Pelletier J, Pelletier JP, Wenger L, Altman RD, Howell DS, Cheung HS 2000. Wild type and mutant p53 differentially regulate the gene expression of human collagenase-3 (hMMP-13). J Biol Chem 275: 11327–11332 [DOI] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G 2008. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 22: 1337–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel DP, Freund SM, Bycroft M, Fersht AR 2007. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc Natl Acad Sci 104: 7009–7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Krummel KA, Lee CJ, Liu CW, Rodewald LW, Tang M, Wahl GM 2006. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2–Mdm4–p53 regulatory network. Cancer Cell 9: 273–285 [DOI] [PubMed] [Google Scholar]
- Torgeman A, Mor-Vaknin N, Zelin E, Ben-Aroya Z, Lochelt M, Flugel RM, Aboud M 2001. Sp1–p53 heterocomplex mediates activation of HTLV-I long terminal repeat by 12-O-tetradecanoylphorbol-13-acetate that is antagonized by protein kinase C. Virology 281: 10–20 [DOI] [PubMed] [Google Scholar]
- Toyooka S, Tsuda T, Gazdar AF 2003. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat 21: 229–239 [DOI] [PubMed] [Google Scholar]
- Vaughan CA, Singh S, Windle B, Sankala HM, Graves PR, Andrew Yeudall W, Deb SP, Deb S 2012. p53 mutants induce transcription of NF-κB2 in H1299 cells through CBP and STAT binding on the NF-κB2 promoter and gain of function activity. Arch Biochem Biophys 518: 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venot C, Maratrat M, Sierra V, Conseiller E, Debussche L 1999. Definition of a p53 transactivation function-deficient mutant and characterization of two independent p53 transactivation subdomains. Oncogene 18: 2405–2410 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ 2000. Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X 2002. Live or let die: The cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C 2009. Blinded by the light: The growing complexity of p53. Cell 137: 413–431 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM 2009. p53 and metabolism. Nat Rev Cancer 9: 691–700 [DOI] [PubMed] [Google Scholar]
- Vu BT, Vassilev L 2011. Small-molecule inhibitors of the p53–MDM2 interaction. Curr Top Microbiol Immunol 348: 151–172 [DOI] [PubMed] [Google Scholar]
- Walker KK, Levine AJ 1996. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci 93: 15335–15340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Greenhalgh DA, Jiang A, He D, Zhong L, Medina D, Brinkley BR, Roop DR 1998. Expression of a p53 mutant in the epidermis of transgenic mice accelerates chemical carcinogenesis. Oncogene 17: 35–45 [DOI] [PubMed] [Google Scholar]
- Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, Quintas-Cardama A, Bankson JA, El-Naggar AK, Lozano G 2011. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest 121: 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA 1991. Tumor suppressor genes. Science 254: 1138–1146 [DOI] [PubMed] [Google Scholar]
- Weisz L, Zalcenstein A, Stambolsky P, Cohen Y, Goldfinger N, Oren M, Rotter V 2004. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res 64: 8318–8327 [DOI] [PubMed] [Google Scholar]
- Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, Kalis M, Levrero M, Strano S, Gorgoulis VG, et al. 2007a. Mutant p53 enhances nuclear factor κB activation by tumor necrosis factor α in cancer cells. Cancer Res 67: 2396–2401 [DOI] [PubMed] [Google Scholar]
- Weisz L, Oren M, Rotter V 2007b. Transcription regulation by mutant p53. Oncogene 26: 2202–2211 [DOI] [PubMed] [Google Scholar]
- Werner H, Karnieli E, Rauscher FJ, LeRoith D 1996. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc Natl Acad Sci 93: 8318–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will K, Warnecke G, Wiesmuller L, Deppert W 1998. Specific interaction of mutant p53 with regions of matrix attachment region DNA elements (MARs) with a high potential for base-unpairing. Proc Natl Acad Sci 95: 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiman KG 2010. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene 29: 4245–4252 [DOI] [PubMed] [Google Scholar]
- Wolf D, Rotter V 1984. Inactivation of p53 gene expression by an insertion of Moloney murine leukemia virus-like DNA sequences. Mol Cell Biol 4: 1402–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Harris N, Rotter V 1984. Reconstitution of p53 expression in a nonproducer Ab-MuLV-transformed cell line by transfection of a functional p53 gene. Cell 38: 119–126 [DOI] [PubMed] [Google Scholar]
- Wong KB, DeDecker BS, Freund SM, Proctor MR, Bycroft M, Fersht AR 1999. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc Natl Acad Sci 96: 8438–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WW, Dimitroulakos J, Minden MD, Penn LZ 2002. HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 16: 508–519 [DOI] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. 2011. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 7: 285–295 [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Chen X 2009. Identification of GRO1 as a critical determinant for mutant p53 gain of function. J Biol Chem 284: 12178–12187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Chen X 2010. Characterization of functional domains necessary for mutant p53 gain of function. J Biol Chem 285: 14229–14238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Hamada J, Tada M, Kameyama T, Nakagawa K, Suzuki Y, Ikawa M, Hassan NM, Kitagawa Y, Moriuchi T 2010. Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomed Res 31: 401–411 [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhou W, Jiang J, Chen X 1998. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J Biol Chem 273: 13030–13036 [DOI] [PubMed] [Google Scholar]