

Abstract
The first mammalian cell lineage commitment is the formation of the trophectoderm (TE) and the inner cell mass (ICM) lineages during preimplantation development. Proper development of the TE and ICM lineages is dependent upon establishment of specific transcriptional programs. However, the epigenetic mechanisms that functionally contribute to establish TE- and ICM-specific transcriptional programs are poorly understood. Here, we show that proper development of the TE and ICM lineages is coordinated via combinatorial regulation of embryonic ectoderm development (EED) and lysine-specific demethylase 6B (KDM6B). During blastocyst formation, the relative levels of EED and KDM6B expression determine altered polycomb repressor 2 (PRC2) complex recruitment and incorporation of the repressive histone H3 lysine 27 trimethylation (H3K27Me3) mark at the chromatin domains of TE-specific master regulators CDX2 and GATA3, leading to their activation in the TE lineage and repression in the ICM lineage. Furthermore, ectopic gain of EED along with depletion of KDM6B in preimplantation mouse embryos abrogates CDX2 and GATA3 expression in the nascent TE lineage. The loss of CDX2 and GATA3 in the nascent TE lineage results in improper TE development, leading to failure in embryo implantation to the uterus. Our study delineates a novel epigenetic mechanism that orchestrates proper development of the first mammalian cell lineages.
INTRODUCTION
Successful reproduction in placental mammals requires proper specification of the trophectoderm (TE) and the inner cell mass (ICM) lineages in preimplantation embryos. The ICM develops to the embryo proper, whereas the TE is essential for embryo implantation to the uterus and the origin of trophoblast cells of the placenta. During preimplantation mouse development, beginning at the 8-cell stage, blastomeres are polarized and allocated to inside and outside positions. During subsequent development, distinct transcriptional programs ensure the commitment of outside cells to the TE lineage and the inside cells to the ICM lineage (1–4). The TE is crucial for embryo implantation and develops into parts of the placenta. Improper TE specification results in either impaired preimplantation development or defective embryo implantation (5–8), which are the leading causes of infertility and early pregnancy failure.
Several key transcription factors, like CDX2 and GATA3, have been implicated in proper development of the TE lineage (9–11). Gene-knockout studies in mice showed that another transcription factor, TEAD4, is essential for the establishment of the TE-specific transcriptional program and maturation of embryos to the blastocyst stage (12, 13). Interestingly, these key TE regulators have dynamic expression patterns during the course of preimplantation development. For example, transcription factors CDX2 and GATA3 are ubiquitously expressed in the blastomeres of an 8-cell mouse embryo. However, upon cell polarization, they are predominantly expressed in the outside cells (9, 14). This pattern is maintained at the blastocyst stage, and these factors are expressed only in the outer TE lineage. Similarly, we recently showed (1) that even though TEAD4 is ubiquitously expressed in all blastomeres of 4- to 8-cell mouse embryos, at the blastocyst stage, TEAD4 mRNA expression is higher in the TE lineage than the ICM lineage.
Differential expression of a key TE regulator during preimplantation development indicates an altered nucleoprotein structure at its chromatin domain, a region of chromatin with boundary elements and with distinct sets of epigenetic modifications. The transcriptionally permissive epigenetic modifications allow expression of TE regulators in outer/TE cells, whereas a repressive chromatin prevents their expression in the inner/ICM cells. Epigenetic regulation at a chromatin domain could be mediated by alteration in DNA methylation and covalent histone modifications (15). However, recent studies indicated that initial segregation of TE and ICM lineages is not dependent upon altered DNA methylation (16). Thus, alteration of histone modifications could be the major epigenetic mechanism that contributes to cell-type-specific transcription during TE versus ICM lineage specification. In recent years, a number of studies have been performed to characterize the histone modification patterns, especially H3 lysine 27 trimethylation (H3K27Me3), in the TE and ICM lineages as well as in TE-derived trophoblast stem cells (TSCs) and ICM-derived embryonic stem cells (ESCs) (17–19). These studies revealed a quantitative difference in the global H3K27Me3 level in the TE versus ICM of a mouse blastocyst and indicated that, in general, the promoter regions of several TE-specific genes are enriched with the repressive H3K27Me3 modification in the ICM lineage or in ICM-derived ESCs. Thus, it has been predicted that the altered levels of the repressive H3K27Me3 modification contribute to the TE-specific expression of these key regulators. However, the functional importance of the altered H3K27Me3 levels and the mechanisms underlying the differential H3K27Me3 levels at the chromatin domains of TE regulators have not been studied in detail.
In order to understand the transcriptional mechanisms that regulate expression of key TE-regulators, we selected Cdx2, Gata3, and Tead4 as model genes and asked whether an alteration in the H3K27Me3 modification is functionally important for regulating their expression during preimplantation development. We found that deposition of the H3K27Me3 mark at the Cdx2, Gata3, and Tead4 chromatin domains is regulated in a spatiotemporal manner and that a chromatin domain-specific mechanism rather than global levels of the H3K27Me3 mark is associated with this dynamic deposition. Our mechanistic analyses in mouse TSCs (mTSCs), mouse ESCs (mESCs), and preimplantation embryos revealed that differential H3K27Me3 deposition at the chromatin domains of TE regulators is coordinately regulated by the altered function of embryonic ectoderm development (EED) and lysine-specific demethylase 6B (KDM6B). We show that reduced expression of EED leads to impaired recruitment of polycomb repressor complex 2 (PRC2) at the chromatin domains of TE regulators in mTSCs, whereas KDM6B reduces global H3K27Me3 levels and also functions at the chromatin domains of TE regulators to activate their transcription. Interestingly, even though the H3K27Me3 mark is differentially deposited between transcriptionally active versus repressed loci of all three TE regulators (Cdx2, Gata3, and Tead4), our functional analyses with mESCs and mTSCs indicated that only Cdx2 and Gata3 transcription is critically regulated by deposition of the H3K27Me3 mark at their chromatin domains. In mTSCs, ectopic expression of EED and depletion of KDM6B induce deposition of the H3K27Me3 mark at the Cdx2 and Gata3 loci and abrogate their transcription. Similar to the findings for mTSCs, ectopic expression of EED and depletion of KDM6B in preimplantation mouse embryos deplete expression of GATA3 and CDX2 from the TE lineage. Furthermore, the ectopic EED-expressing/KDM6B-depleted embryos either do not develop to the blastocyst stage or, if they develop to the blastocyst stage, are unable to implant. Our results unfold how molecular components modulating a specific histone modification regulate the transcriptional outcome from specific chromatin domains for proper progression of preimplantation development and ensure embryo implantation.
MATERIALS AND METHODS
Cell culture and reagents.
E14tg2a (E14) mESCs were cultured without a feeder layer in mESC culture medium with 100 IU/ml mouse leukemia-inhibiting factor (mLIF) (ESGRO; Invitrogen, Carlsbad, CA) following procedures described earlier (20). Undifferentiated ESCs from Eed−/− mice (a kind gift from Terry Magnuson) were maintained on irradiated mouse embryonic fibroblasts (MEFs) with the mESC medium with twice the concentration of mLIF (i.e., 200 IU/ml).
mTSCs were derived following previously described procedures (21). Blastocysts from 6-week-old female 129S2/SvPasCrl (129/Sv) strain mice (Charles River Laboratory, Wilmington, MA) were isolated at 3.5 days postcoitum and transferred on irradiated MEFs to mTSC culture medium consisting of RPMI 1640 (Sigma) supplemented with 20% fetal bovine serum (FBS; Sigma, St. Louis, MO), 100 μM β-mercaptoethanol, 1 mM sodium pyruvate (Cellgro; Mediatech Inc., Herndon, VA), 2 mM l-glutamine, penicillin-streptomycin (100 IU/ml and 100 mg/ml, respectively) in the presence of 25 ng/ml fibroblast growth factor 4 (FGF4; Invitrogen), and heparin (1 μg/ml; Sigma). Blastocyst outgrowths were disaggregated after 5 to 7 days, and a female TSC line, confirmed by a lack of sex-determining region Y (Sry) expression (data not shown), was selected for subsequent experiments. For experimental purposes, mTSCs were expanded in the proliferative state without MEF feeders by culturing in the presence of MEF-conditioned medium and normal mTSC medium (as described above) in a ratio of 7:3 (22).
Human embryonic kidney 293T (HEK293T) cells were cultured in Dulbecco modified Eagle medium supplemented with 10% FBS and 1% penicillin-streptomycin. Transfection of HEK293T cells was done in a similar medium devoid of antibiotics.
Isolation and in vitro culture of preimplantation embryos from mouse, rat, and monkey.
All procedures to isolate preimplantation embryos from mouse, rat, and monkey were performed after obtaining IACUC approvals at the University of Kansas Medical Center and the University of Wisconsin—Madison.
Human preimplantation embryos were collected at Stanford University, obeying all the institutional rules: a two-stage consent process (23) along with the approval from institutional committees at both Stanford University and the University of Kansas Medical Center. Deidentified and discarded human blastocysts were fixed in 4% paraformaldehyde solutions and analyzed for the H3K27Me3 mark by immunostaining. Embryos isolated from other species are described below.
Mouse embryos.
Adult (age, 3 to 4 weeks) CD-1 female mice were superovulated by intraperitoneal injection of 5 IU of P.G. 600 (Intervet, Millsboro, DE), followed by 5 IU of human chorionic gonadotropin (hCG; Sigma) 48 h later. Females were then mated with C57BL/6 males and euthanized either on the following morning to collect one-cell embryos (day 0.5), 2 days later (day 2.5) to collect morulae, or 3 days later (day 3.5) to collect matured blastocysts. Oviducts were removed by surgery; the embryos were harvested in M2 medium (Millipore, Billerica, MA) and cultured in KSOM (Millipore) at 37°C in a 5% CO2 incubator with a relative humidity of 95%. Embryos were visualized daily, photographed, and sampled for RNA preparation at the morula or blastocyst stage (9).
Rat embryos.
Female dark agouti rats (age, 6 weeks; DA/OlaHsd strain; Harlan Laboratories) were mated overnight with dark agouti males (age, 7 weeks) and then euthanized by cervical dislocation on day 4.5 postcoitum. Uteri were collected from the females after euthanasia and flushed with M2 medium for the collection of blastocysts.
Monkey embryos.
Colonies of rhesus monkey were maintained at the Wisconsin National Primate Research Center (WNPRC; Madison, WI). Monkey oocytes were obtained from the donor monkey after treatment with 30 or 45 IU of recombinant human follicle-stimulating hormone administered twice daily for 7 days, followed by one injection of 1,000 IU of recombinant hCG hormone. Oocytes were retrieved from visible follicles by laparoscopic surgery. These were matured in vitro, followed by in vitro fertilization (IVF) with sperms collected on the same day. IVF-derived zygotes were cultured for 7 days until they formed matured blastocysts. All the surgical procedures were performed in accordance with the NIH guidelines for the care and use of laboratory animals (24) with the approval of the University of Wisconsin Graduate School Animal Care and Use Committee.
Isolation of ICMs and TEs from blastocysts.
ICMs were isolated by embryo immunosurgery according to the method described earlier (1). Zonae pellucidae were removed using 0.5% pronase (Sigma) for 5 min. The zona-free embryos were then treated with rabbit anti-mouse serum (Sigma) at a 1:100 dilution for 30 min, washed with phosphate-buffered saline (PBS), and incubated with guinea pig complement (Innovative Research, Novi, MI) at 37°C in 5% CO2 for 30 min. After washing, ICM cells were collected for RNA preparation or chromatin immunoprecipitation (ChIP)–whole-genome amplification (WGA) analyses.
TE samples were acquired under an inverted microscope (a Nikon Ti-U Eclipse microscope equipped with Hoffman modulation contrast optics) by using Leica manual manipulators after microdissection and a customized ultrasharp splitting blade (ESE020; Bioniche Animal Health USA, Inc.). Purified TE samples were further processed for generating samples for either ChIP-WGA or extraction of RNA.
qRT-PCR.
Total RNA was extracted from cells with the TRIzol reagent (Invitrogen) and analyzed for quantitative reverse transcription-PCR (qRT-PCR) following procedures described earlier (22, 25). For analysis of expression in preimplantation embryos, total RNA was isolated using a PicoPure RNA isolation kit (MDS Analytical Technology, Sunnyvale, CA) and processed as described earlier (9). The oligonucleotides used for qRT-PCR are provided in Table S1 in the supplemental material.
Quantitative ChIP and ChIP-WGA analyses.
Real-time PCR-based quantitative ChIP analysis was performed following published protocols (9, 22, 26). In brief, cells were trypsinized; protein-DNA cross-linking was then conducted by treating cells with 1% formaldehyde (Sigma) for 10 min at room temperature with gentle agitation. Glycine (125 mM; Sigma) was added to quench the reaction. Sonicated, cross-linked chromatin fragments were immunoprecipitated with different antibodies (listed in Table S3 in the supplemental material). Mouse IgG1 (Sigma) was used as a negative control of antibody binding in all sets of experiments. Immunoprecipitated chromatins were reverse cross-linked, digested with proteinase K (Sigma), and purified. Quantification of the precipitated DNA was performed using quantitative PCR (qPCR) amplification. The amount of DNA precipitated by each antibody was normalized against 1/5, 1/25, 1/125, and 1/625 of the starting input DNA samples. A list of the primers used for ChIP analyses is provided in Table S2 in the supplemental material.
Quantitative ChIP-WGA analyses with purified ICM and TE samples from preimplantation mouse embryo (≥300) were performed in the same manner described above, with a few modifications. Column-purified immunoprecipitated DNA fragments were amplified using a whole-genome amplification kit (WGA4; Sigma) prior to qPCR analysis.
Western blot analysis.
For Western blot analyses, whole-cell lysates were extracted with a lysis buffer described earlier (1) and electrophoresed in an 8 to 12% polyacrylamide gel (Sigma) using Tris-glycine running buffer (pH 8.3). Proteins were then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Nonspecific binding was blocked with 5% nonfat dry milk (Difco, Becton, Sparks, MD) and 0.05% Tween 20 (Sigma) in 20 mM Tris-HCl, pH 7.6 (Tris-buffered saline with Tween [TBS-T]). After incubation with appropriate primary antibodies overnight at 4°C, membranes were washed in TBS-T and reincubated with appropriate secondary antibodies conjugated with horseradish peroxidase. Enhanced chemiluminescence (ECL) signals were developed in films using an ECL-plus reagent (GE Healthcare UK Limited, Little Chalfont, Buckinghamshire, United Kingdom). The primary antibodies along with the dilutions used are listed in Table S3 in the supplemental material.
Immunofluorescence study.
Preimplantation embryos were washed with ice-cold PBS and fixed with freshly prepared 4% paraformaldehyde (Sigma) in PBS for 30 min at room temperature. Cells were permeabilized in 0.25% Triton X-100 in PBS and blocked with 1% bovine serum albumin (BSA) for 1 h at room temperature. Appropriate primary antibody, diluted in wash buffer (containing 0.1% BSA and 0.05% Tween 20 in PBS), was incubated overnight at 4°C. Primary antibodies along with the dilutions used are listed in Table S3 in the supplemental material. Washed embryos were labeled with appropriate secondary antibody and conjugated with either Alexa Fluor 488 (green) or Alexa Fluor 568 (red) in a dilution of 1:400 for 1 h in the dark at room temperature. After the labeled embryos were counterstained with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen), they were observed under either a fluorescence microscope (Leica) or a laser scanning confocal microscope (LSM 5; Carl Zeiss Microimaging, Thornwood, NY). Images were acquired using a ×40 water-immersion objective on a confocal microscope (LSM 5 laser scanning microscope; Carl Zeiss Microimaging, Thornwood, NY) operated by LSM 5 Pa software. Alexa Fluor 488, Alexa Fluor 568 fluorophores, and DAPI were excited individually with 488-nm, 543-nm, and 405-nm lasers, respectively, with appropriate filter sets. Uncompressed images were processed with the Zeiss LSM image browser. Where indicated, images were taken as z stacks and rendered as three-dimensional projections for detailed visualization. All immunofluorescence images for a specific set of experiment were captured with an identical set up, including the exposure time.
RNA interference (RNAi) and overexpression via lentiviral transduction.
Short hairpin RNAs (shRNAs) _targeting mouse Kdm6b were cloned in lentiviral vector pLKO.1 (10878; Addgene). Lentiviral supernatants were prepared in HEK293T cells as described earlier (9, 22, 26). Three different shRNA constructs with Kdm6B _target sequences, 5′-CCTGTATATGTCTCTTGTTTA-3′ (shRNA1), 5′-CCTCTGTTCTTGAGGGACAAA-3′ (shRNA2), and 5′-GCTGGATGAATCCATTCGGAA-3′ (shRNA3), were used. Among them, two shRNAs, shRNA1 and shRNA3, were found to be the most effective for successful knockdown. For ectopic EED expression, cDNA for mouse Eed along with a ribosome-binding site was cloned under the control of the human U6 promoter in the pLKO.3G (14748; Addgene) lentiviral vector. The vector also expressed an enhanced green fluorescent protein (EGFP) reporter.
Lentiviral particles were transduced in undifferentiated mTSCs, and cells were selected with puromycin under the undifferentiated TSC culture condition in the presence of FGF4. After 3 to 4 days of lentiviral transduction, experimental analyses were performed. To transduce one-cell preimplantation embryos, lentiviral soups were concentrated using an ultracentrifuge (35,000 rpm for 3 h), and the concentrates were dissolved in EmbryoMax injection buffer (Millipore). About 150 one-cell fertilized embryos in each set were then subjected to subzonal injection. Transducted embryos were grown in KSOM at 37°C and 95% relative humidity in a 5% CO2 chamber for 3 to 4 days. Embryos were visualized and photographed daily under fluorescence and phase-contrast microscopes. Unmanipulated embryos and embryos infected with lentiviral particles containing pLKO.3G were used as untreated and negative-control experimental sets, respectively.
To transduce TE, early-blastocyst-stage (about embryonic day 2.5 [E2.5] to E3.0) mouse embryos were subjected to subzonal microinjection with lentiviral particles containing either pLKO.3G (control vector) or vectors expressing EED with an EGFP reporter and shRNAs for KDM6B. Transduced embryos were grown in KSOM at 37°C with 95% relative humidity in a 5% CO2 chamber for 36 h before further analyses.
Analyses of blastocyst implantation efficiency.
Blastocysts were surgically transferred into the uterine horns of E2.5 foster pseudopregnant, 6- to 8-week-old FVB female mice (Charles River) that were previously mated with a vasectomized male. Blastocysts were transferred into a single uterine horn. After 5 days (at E7.5), Chicago sky blue (27) was injected into the tail vein and mice were sacrificed to monitor the sites of embryo implantation.
RESULTS
During preimplantation development, locus-specific mechanisms regulate differential deposition of the repressive H3K27Me3 mark at the chromatin domains of TE regulators.
To understand the importance of H3K27Me3 modification during preimplantation mammalian development, we initiated our study by testing the distribution of the global H3K27Me3 mark in preimplantation mouse embryos at different developmental stages beginning from the 2-cell stage. Similar to an earlier report (28), we found that the H3K27Me3 mark exists in the nuclei of all blastomeres at all developmental stages (Fig. 1A). However, at the blastocyst stage, the H3K27Me3 modification is enriched in ICM nuclei compared to TE nuclei (Fig. 1B). To further understand the importance of the H3K27Me3 modification, we tested whether enrichment of the global H3K27Me3 content in the ICM of a blastocyst is a common event for other mammalian species. We analyzed rat, monkey, and human blastocysts. We found that, unlike in a mouse blastocyst, the global levels of the H3K27Me3 mark are very similar among the nuclei of TE and ICM lineage cells in rat, monkey, and human blastocysts (Fig. 1B). Thus, reduced global levels of the H3K27Me3 modification in the TE nuclei compared to those in the ICM nuclei are not a common event for all mammalian embryos.
Fig 1.

Global H3K27Me3 levels in preimplantation mammalian embryos. (A) Preimplantation mouse embryos at different developmental stages were analyzed by confocal microscopy for the presence of the H3K27Me3 modification (green) with respect to nuclei (blue). Bar, 50 μm. (B) Immunofluorescence analyses showing the H3K27Me3 modification (green) with respect to nuclei (blue) in mouse, rat, monkey, and human blastocysts observed by confocal microscopy. Compared to TE, the ICM of mouse blastocysts has higher global levels of the H3K27Me3 modification. However, TEs and ICMs of rat, monkey, and human blastocysts have similar levels of the global H3K27Me3 modification.
Although global H3K27Me3 levels vary between the TE and ICM of a mouse embryo and remain similar in rat, monkey, and human embryos, interestingly, expression of TE regulators like CDX2 is suppressed in the ICM of all these species (1). Thus, we next tested whether the H3K27Me3 mark is differentially incorporated between active versus repressed chromatin domains of key TE regulators in the TE and ICM lineages, respectively. To make a comparative analysis in two different mammalian species, we obtained both mouse and rat blastocysts, isolated TEs (Fig. 2A) and ICMs (9), confirmed TE- and ICM-specific gene expression (see Fig. S1A in the supplemental material), and performed ChIP-WGA analyses (as described in references 9 and 29) to determine H3K27Me3 levels at the chromatin domains of key TE regulators. Our analyses showed that the Cdx2, Gata3, and Tead4 promoters as well as the intron 1 region of Cdx2, an enhancer element for Cdx2 transcription (9), harbor low levels of the H3K27Me3 modification in early-stage embryos (8- to 16-cell stage) and the incorporation of the H3K27Me3 mark is highly induced within the ICM at the blastocyst stage, confirming an earlier observation in relation to the mouse Cdx2 promoter (17). However, the incorporation of the H3K27Me3 mark is very low at the Cdx2, Gata3, and Tead4 loci in the TEs of both mouse and rat blastocysts (Fig. 2B). Furthermore, analysis of H3K27 acetylation (H3K27Ac), a chromatin mark that is indicated to have an antagonistic switch with the H3K27Me3 modification and is often associated with transcriptionally active genes (30), showed that the Cdx2, Gata3, and Tead4 loci within the TE harbor increased levels of the H3K27Ac mark (see Fig. S1B in the supplemental material). These results indicated that, in the TE versus the ICM, the differential H3K27Me3 incorporation at the chromatin domains of TE regulators is not dependent upon global levels of H3K27Me3 modification. Interestingly, in contrast to the TE regulators, the chromatin domains of two X-linked genes, cysteine-rich hydrophobic domain 1 (Chic1) and alpha-thalassemia/mental retardation syndrome X linked (Atrx), contain high levels of the H3K27Me3 mark in the TE (Fig. 2C). These results indicate that the machinery essential to incorporate the H3K27Me3 mark at a chromatin domain is functional within the TE lineage. However, that machinery is unable to incorporate the H3K27Me3 mark at the Cdx2, Gata3, and Tead4 loci. Collectively, our results indicate that chromatin domain-specific mechanisms rather than global levels of H3K27Me3 ensure reduced incorporation of the H3K27Me3 mark at the Cdx2, Gata3, and Tead4 loci in the TE lineage of a mammalian embryo.
Fig 2.

H3K27Me3 incorporation at the chromatin domains of TE regulators is not correlated with global H3K27Me3 levels. (A) Steps in isolating TE. (a) A blastocyst was captured with a micropipette near the ICM/polar TE. (b) Cuts were made at the indicated (arrowheads) regions. (c) ICM/polar TE was separated from the mural TE with the micropipette. Bars, 50 μm. (B) Quantitative ChIP-WGA analyses (mean ± SE, n = 3) showing H3K27Me3 incorporation at the Cdx2, Gata3, and Tead4 loci in mouse (top) and rat (bottom) preimplantation embryos at different developmental stages. The dotted lines show the average ChIP-WGA signals that were obtained using nonspecific mouse IgG1 (IgG) as the negative-control antibody. *, significant (P < 0.05) difference. (C) Quantitative ChIP-WGA analyses (mean ± SE, n = 3; P < 0.05) show H3K27Me3 incorporation at the promoter regions of X-linked genes Chic1 and Atrx in TEs and ICMs isolated from mouse (top) and rat (bottom) blastocysts. The promoter region of the large subunit of RNA polymerase II (Rpl27), a constitutively active gene, was analyzed as the negative-control region for H3K27Me3 incorporation.
Similar to the expression patterns in the TE and ICM lineages, CDX2 and GATA3 are expressed only in the mouse TE-derived mTSCs but are repressed in the mouse ICM-derived mESCs (see Fig. S2A in the supplemental material) (9, 10). Similarly, expression of mRNA of TEAD4, another master regulator of TE fate specification, is severalfold higher in mTSCs than mESCs (1, 13). Thus, mESCs and mTSCs can be exploited for mechanistic studies to understand the transcriptional mechanisms that activate and repress the Cdx2, Gata3, and Tead4 loci in the TE and the ICM, respectively, so we compared the global levels of the H3K27Me3 mark and its incorporation at the chromatin domains of TE regulators in mESCs versus mTSCs. We found that, similar to mouse TE and mouse ICM, the global H3K27Me3 level is low in mTSCs compared to that in mESCs (see Fig. S2A in the supplemental material). Our ChIP analyses also confirmed that activated Cdx2, Gata3, and Tead4 loci in mTSCs contain reduced levels of the H3K27Me3 modification (see Fig. S2B in the supplemental material). However, similar to mouse TE, the chromatin domains of X-linked genes contain high levels of the H3K27Me3 modification in mTSCs compared to those in mESCs (see Fig. S2B in the supplemental material). These results indicated that similar mechanisms might be involved in both mouse TE and mTSCs to reduce the H3K27Me3 mark at the Cdx2, Gata3, and Tead4 loci. Thus, in the next series of experiments, using mESCs and mTSCs as model systems, we asked what molecular mechanism regulates H3K27Me3 deposition at the chromatin domains of TE regulators.
Mammalian blastocysts have reduced EED expression in the TE lineage and high levels of KDM6B expression in both the TE and ICM lineages.
Methylation at the H3K27 residue is mediated by the PRC2 complex (31), and demethylation is mediated by histone demethylases KDM6A (also known as UTX) and KDM6B (also known as JMJD3) (32). A recent study showed that KDM6B is involved in regulating global H3K27Me3 levels in bovine preimplantation embryos (33). In addition, studies in mESCs showed that JARID2 (also known as Jumonji) orchestrates PRC2 function and fine-tunes deposition of the H3K27Me3 mark at chromatin domains (34–36). Therefore, we tested which of these mechanisms reduces the H3K27Me3 mark at the chromatin domains of TE regulators.
We compared expression of PRC2 core components EZH2, SUZ12, and EED in mTSCs versus mESCs. We found that SUZ12 and EZH2 are expressed at very similar levels in mTSCs and mESCs (Fig. 3A). However, EED expression is significantly lower in the mTSCs (Fig. 3A), an observation also reported by an earlier study (19). Next, we compared the expression patterns of histone demethylase KDM6A and KDM6B along with JARID2 in mTSCs versus mESCs. We found that, compared to mESCs, mTSCs express very high levels of KDM6B (Fig. 3A). On the contrary, using two different antibodies, we could not detect KDM6A by Western blot analysis in either mESCs or mTSCs (data not shown). Thus, we concluded that KDM6B is the major H3K27 demethylase present in the mTSCs. Furthermore, compared to JARID2 protein expression in mESCs, JARID2 protein expression was almost undetectable in mTSCs (Fig. 3A). Thus, we also concluded that JARID2 does not modulate PRC2 function in mTSCs.
Fig 3.

EED expression is repressed in the TE lineage of mammalian blastocysts. (A) Western blot analyses of undifferentiated mESCs and mTSCs showing relative levels of expression of molecular components that regulate the H3K27Me3 modification. Representative blots are shown. Compared to mESCs, mTSCs express reduced levels of EED and higher levels of KDM6B, whereas the JARID2 protein was undetectable in mTSCs under our experimental condition. (B) qRT-PCR analyses (mean ± SE, n = 3) showing the relative levels of mRNA expression of PRC2 core components and Kdm6B in ICMs and TEs isolated from mouse blastocysts (RT, reverse transcriptase). The plot shows that only Eed mRNA expression in the TE is significantly (P < 0.01) lower. (C) Confocal images showing expression of PRC2 core components and KDM6B in mouse blastocysts. Noticeably, the KDM6B protein is detected in both the cytoplasm and nuclei of TE and ICM cells. Bars, 50 μm. (D) qRT-PCR analyses showing the relative levels of mRNA expression of PRC2 core components and Kdm6B in ICMs and TEs isolated from rat blastocysts (mean ± SE, n = 3).
Our expression analyses in mTSCs versus mESCs revealed two major differences, reduced EED expression and elevated KDM6B expression, that could contribute to the reduced H3K27Me3 mark at the chromatin domains of key TE regulators. Therefore, we next tested expression of PRC2 components along with KDM6B in the TE and the ICM of mouse blastocysts. Our analyses showed that, although Suz12 and Ezh2 mRNAs and proteins are expressed at similar levels in both TE and ICM (Fig. 3B and C), expression of EED is much lower in the mouse TE (Fig. 3B and C). These results indicated that the patterns of expression of core PRC2 components in the TE and ICM are maintained in the stem cell population derived from those lineages. Next, we tested expression of KDM6B in mouse blastocysts and found that, unlike KDM6B expression in the stem cells, KDM6B is abundantly expressed in both the TE and ICM of a mouse embryo (Fig. 3B and C), confirming an earlier observation that showed that Kdm6B expression is repressed during derivation of ESCs from the ICM (37). Interestingly, we also noticed that KDM6B is present in both the cytoplasm and nuclei of TE and ICM cells, with a relatively greater abundance within the cytoplasm.
To test whether reduced expression of EED in the TE is a common event for other mammalian species, we isolated TE and ICM from rat blastocysts and tested mRNA expression. We found that, similar to the mouse TE, Eed mRNA expression in the rat TE is significantly lower than that in the ICM (Fig. 3D) and Kdm6B mRNA expression is similar in both rat TE and rat ICM (Fig. 3D). Unfortunately, we were unable to obtain specific antibodies to study the EED and KDM6B proteins in rat embryos. Nonetheless, as we observed reduced Eed mRNA expression and high levels of Kdm6B mRNA expression in both mouse and rat TE as well as in mTSCs, we next asked whether reduced levels of EED and the function of KDM6B contribute to reduce the H3K27Me3 mark at the chromatin domains of key TE regulators in TE-derived mTSCs, thereby facilitating their transcription.
Reduced levels of EED lead to loss of PRC2 recruitment at the chromatin domains of TE regulators in mTSCs.
EED is implicated in _targeting the PRC2 complex to chromatin through its interaction with the histone H3 tail (38, 39). So, we tested whether reduced levels of EED result in reduced PRC2 recruitment at the Cdx2, Gata3, and Tead4 chromatin domains in mTSCs. Our ChIP analyses revealed that, compared to mESCs (Fig. 4A), PRC2 recruitment is significantly lower at those chromatin domains in mTSCs. Therefore, we hypothesized that reduced expression of EED contributes to reduced PRC2 recruitment and H3K27Me3 incorporation at the chromatin. To test this hypothesis, we ectopically expressed EED in mTSCs (Fig. 4B and C) and asked whether ectopic expression of EED in mTSCs represses TE regulators. We found that, although ectopic expression of EED did not increase the total content of the H3K27Me3 mark in mTSCs (Fig. 4C), the levels of CDX2 and GATA3 mRNA and protein expression were strongly reduced (Fig. 4B and C). Furthermore, ChIP analyses revealed that transcriptional repression of Cdx2 and Gata3 was associated with PRC2 recruitment and enrichment of the H3K27Me3 mark at their chromatin domains (Fig. 4D). Thus, from gain-in-function analyses, we concluded that, in mTSCs, PRC2 recruitment and H3K27Me3 deposition at the Cdx2 and Gata3 loci are dependent upon EED protein levels and reduced levels of EED ensure impairment of PRC2 recruitment and H3K27Me3 deposition at those loci, thereby activating their transcription. Interestingly, despite increased PRC2 recruitment and H3K27Me3 incorporation at the Tead4 chromatin domain (Fig. 4D), TEAD4 expression was not significantly reduced in ectopic EED-expressing mTSCs (Fig. 4B and C). Therefore, we also concluded that, unlike transcription of Cdx2 and Gata3, transcription of Tead4 in mTSCs is not critically dependent upon the EED/PRC2-mediated H3K27Me3 modification.
Fig 4.

EED regulates PRC2 recruitment at the Cdx2 and Gata3 loci. (A) Quantitative ChIP analyses (mean ± SE, n = 3; P < 0.05) of SUZ12 occupancy (as an indicator of PRC2 recruitment) at the Cdx2, Gata3, and Tead4 loci in mESCs and mTSCs. The promoter region of the constitutive Rpl27 gene was analyzed as the negative-control region for PRC2 recruitment. (B) mTSCs were infected with lentiviral vectors with or without expressing ectopic EED, and qRT-PCR analyses were performed (mean ± SE, n = 3) to determine mRNA expression. The plot shows that ectopic expression of EED strongly represses Cdx2 and Gata3 mRNA expression in mTSCs, whereas Tead4 mRNA expression was marginally repressed and Suz12 mRNA expression was unchanged. (C) Western blot analyses confirming ectopic EED expression and repression of CDX2 and GATA3 protein levels in samples, analyzed as described for panel B. These representative blots also show that global H3K27Me3 levels were unchanged in ectopic EED-expressing mTSCs. (D) Quantitative ChIP analyses (mean ± SE, n = 3) showing induction of PRC2 recruitment (left) and incorporation of the H3K27Me3 mark (right) at the chromatin domains of TE regulators in ectopic EED-expressing (clone 1) mTSCs.
The EED function in mESCs represses CDX2 and GATA3 expression, thereby contributing to suppression of the trophoblast fate.
EED is expressed in the mouse ICM (40) or ICM-derived mESCs (19). However, EED is dispensable for the development of the ICM lineage (41), and mESCs can be derived from Eed−/− mouse blastocysts (42). Therefore, the question arises, what is the importance of EED in the ICM or ICM-derived mESCs? We found that, in mTSCs, ectopic induction of EED expression leads to the repression of key trophoblast regulators, CDX2 and GATA3, in mTSCs. Thus, we hypothesized that an EED function might be important to suppress induction of the trophoblast fate in ICM or ICM-derived mESCs by repressing expression of TE regulators. To test this, we took a loss-of-function approach using Eed−/− mESCs (42). We cultured wild-type E14tg2a (E14) and Eed−/− mESCs under both mESC and FGF4-containing mTSC culture conditions (22) and determined the expression of TE regulators. We found that the expression of CDX2 and GATA3 was not induced in E14 cells, when cultured under the mTSC culture condition (Fig. 5A and B). We also noticed that expression of TEAD4 was marginally induced in both E14 and Eed−/− mESCs under the mTSC culture condition. However, expression of both CDX2 and GATA3 was robustly induced in Eed−/− mESCs, when they were cultured under the mTSC culture condition (Fig. 5A and B). Intriguingly, the induction of CDX2 and GATA3 was also associated with a gradual morphological change of Eed−/− mESC colonies toward mTSC-like colonies (Fig. 5C). These results indicated that, even though EED is dispensable for development of the mouse ICM or derivation of mESCs, an EED function could be important to repress CDX2 and GATA3 expression and to prevent induction of the trophoblast fate under conditions that facilitate TE/TSC development.
Fig 5.

EED contributes to the suppression of the trophoblast fate in mESCs. (A) E14 and Eed−/− mESCs were cultured under the mESC and mTSC culture conditions (Cond) for 5 days, and qRT-PCR analyses (mean ± SE, n = 3; P < 0.05) were performed to determine Cdx2, Gata3, and Tead4 mRNA expression. (B) Western blot analyses of samples described in panel A showing strong induction of CDX2 and GATA3 protein expression in Eed−/− mESCs under the mTSC culture condition. (C) Micrographs showing representative E14 (top) and Eed−/− (bottom) mESC colonies when cultured under the mESC or mTSC culture conditions for different numbers of days. The Eed−/− mESC colonies maintained the typical mESC colony morphology when cultured under the mESC culture condition (bottom left micrograph). However, unlike E14 mESCs (top, middle and right micrographs), the colony morphology of the Eed−/− mESCs gradually changed to the mTSC-like colony morphology (indicated by the red border in the bottom middle micrograph and the colony morphology of the bottom right micrograph) under the mTSC culture condition. (Inset, bottom right) Example of an mTSC colony. Bars, 100 μm.
The KDM6B function ensures reduced levels of H3K27Me3 incorporation and maximal transcription at the Cdx2 and Gata3 loci in mTSCs.
As KDM6B is highly expressed in the TE lineage and TE-derived mTSCs, we next asked whether the KDM6B function contributes to reduction of the H3K27Me3 mark and transcription of TE regulators in mTSCs. To that end, we tested whether KDM6B physically occupies the Cdx2 Gata3, and Tead4 chromatin domains in mTSCs. Our ChIP analyses detected KDM6B recruitment at the Cdx2 and Gata3 chromatin domains. However, KDM6B recruitment was not observed at the Tead4 promoter or the promoter region of the X-linked gene Atrx (Fig. 6A). To test whether KDM6B is functionally important to regulation of Cdx2 and Gata3 transcription in mTSCs, we knocked down KDM6B expression via RNAi (Fig. 6B and C). We found that depletion of KDM6B increases the total H3K27Me3 mark in mTSCs (Fig. 6C) and strongly represses expression of Cdx2 and Gata3 (Fig. 6B and C) but does not affect TEAD4 and SUZ12 expression. ChIP analyses in KDM6B-depleted mTSCs showed that loss of KDM6B recruitment at the Cdx2 and Gata3 chromatin domains is associated with increased deposition of the H3K27Me3 mark (Fig. 6D). Moreover, a combination of KDM6B depletion and ectopic expression of EED in mTSCs abrogated expression of CDX2 and GATA3 (Fig. 6E). Analyses of the Cdx2 and Gata3 chromatin domains indicated that combinatorial ectopic EED expression/KDM6B depletion further increased H3K27Me3 deposition, strongly reduced H3K27Ac deposition (see Fig. S3A in the supplemental material), and inhibited RNA polymerase II recruitment (see Fig. S3B). We also found that loss of CDX2 and GATA3 expression upon combinatorial EED induction/KDM6B depletion is associated with induced expression of differentiated trophoblast markers (Fig. 6F) like placental lactogen I (PL-I) and PL-II (markers for trophoblast giant cells), trophoblast-specific protein alpha (Tpbpa; spongiotrophoblast marker), and glial cell missing 1 (Gcm1; syncytiotrophoblast marker). These results indicate that mTSC self-renewal can be negatively affected by altered KDM6B/EED expression.
Fig 6.

KDM6B function in mTSCs reduces H3K27Me3 incorporation at the Cdx2 and Gata3 chromatin domains. (A) Quantitative ChIP analyses (mean ± SE, n = 3; P < 0.05) in mTSCs showing KDM6B occupancy at the Cdx2 and Gata3 loci but not at the Tead4 and X-linked gene Atrx loci. (B) mTSCs were infected either with empty lentiviral vectors or with vectors expressing Kdm6B shRNAs to knock down Kdm6B expression. Three different shRNA molecules (shRNA1, shRNA2, and shRNA3) with different knockdown efficiencies were studied, and qRT-PCR analyses were performed (mean ± SE, n = 3; P < 0.05) to determine mRNA expression of the indicated genes. The plot shows that depletion of Kdm6B strongly represses Cdx2 and Gata3 mRNA expression in mTSCs, whereas Tead4 and Suz12 mRNA expression was unaffected. (C) Western blot analyses confirming KDM6B knockdown and repression of CDX2 and GATA3 protein levels in samples analyzed for panel B. Representative blots also show that global H3K27Me3 levels were increased in KDM6B-depleted mTSCs. (D) Quantitative ChIP analyses (mean ± SE, n = 3; P < 0.05) showing induction of H3K27Me3 incorporation at the Gata3 and Cdx2 chromatin domains in KDM6B-depleted mTSCs. (E) mTSCs were infected with lentiviral particles expressing empty vectors or with vectors expressing ectopic EED (clone 1 and clone 2) and KDM6B shRNAs (shRNA1 and shRNA3) in combination. Protein samples were analyzed by Western blotting. Representative blots show the nearly complete loss of CDX2 and GATA3 protein expression in ectopic EED-expressing/KDM6B-depleted mTSCs. (F) qRT-PCR analyses showing induction of differentiated trophoblast cell markers in EED-expressing/KDM6B-depleted mTSCs (mean ± SE, n = 3; P < 0.05).
Ectopic EED expression along with KDM6B depletion in preimplantation embryos inhibits CDX2 and GATA3 expression in the TE lineage.
Our analyses in mTSCs confirmed that two distinct mechanisms, reduced PRC2 recruitment due to repressed expression of EED and the function of KDM6B, prevent H3K27Me3 deposition at the Cdx2 and Gata3 chromatin domains and activate their transcription. Therefore, we next analyzed whether coordinated regulation of EED and KDM6B is important to ensure CDX2 and GATA3 expression and proper development of the TE lineage in a preimplantation mouse embryo. First, we tested whether expressions of EED and KDM6B is dynamically regulated during the course of mouse preimplantation development. Earlier studies (43, 44) indicated that EED mRNA could be detected in preimplantation mouse embryos and the EED protein can be detected within the nuclei of mouse TE and ICM lineages (40, 45), with a greater abundance found within the ICM (46). However, none of these prior studies performed any quantitative analyses of EED expression between the TE and ICM lineages. Therefore, for measuring EED and KDM6B expression, we performed quantitative RT-PCR analyses with embryos isolated at different developmental stages (from 4-cell to blastocyst stages).
We found that Eed and Kdm6B mRNAs are present at all developmental stages. Interestingly, Eed mRNA expression was strongly induced within the ICM lineage during the morula-to-blastocyst transition (Fig. 7A), whereas KDM6B induction was a common event in both TE and ICM (Fig. 7B). A similar pattern for Kdm6B expression was also observed during bovine preimplantation development (33). As expected, we noticed that the induction of EED expression during the morula-to-blastocyst transition occurs solely in the ICM lineage (Fig. 7A). We also compared PRC2 recruitment at the Cdx2 and Gata3 loci and found that, compared to cells in morula-stage embryos, cells in the ICM lineage have high levels of the PRC2 complex at the Cdx2 and Gata3 chromatin domains (Fig. 7C), whereas PRC2 recruitment at those loci within TE cells is reduced. Collectively, these results indicate that the relative concentrations of EED and KDM6B are altered in the nascent TE and ICM lineages and relatively less EED expression correlates with the loss of PRC2 recruitment and H3K27Me3 incorporation at the Cdx2 and Gata3 loci in the nascent TE lineage. Thus, we next tested whether alteration of EED and KDM6B expression alters CDX2 and GATA3 expression within the developing TE lineage and affects proper preimplantation mouse development.
Fig 7.
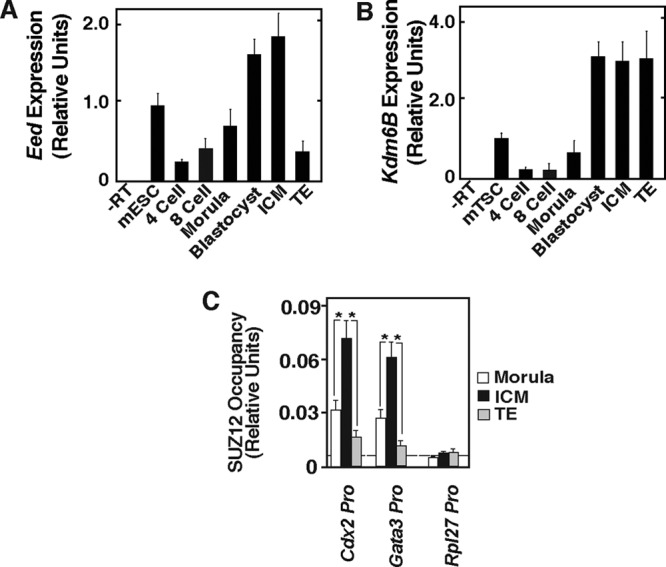
Relative expression of EED and KDM6B is altered during TE and ICM lineage specification and correlates with PRC2 recruitment at the Cdx2 and Gata3 loci. (A and B) Quantitative RT-PCR analyses (mean ± SE, n = 3) showing the levels of Eed and Kdm6B mRNA, respectively, in mouse preimplantation embryos at different developmental stages. The plots show relative expression levels compared to those in undifferentiated mESCs (for Eed expression) and mTSCs (for Kdm6B expression). During transition from the morula to blastocyst stage, Eed is induced within the ICM and is repressed in the TE, whereas Kdm6B is induced in both the ICM and the TE. (C) ChIP-WGA analyses of SUZ12 recruitment (as an indicator of PRC2 recruitment). The data show that in comparison to morula-stage embryos, the ICM cells have significantly (P ≤ 0.05) higher levels of PRC2 recruitment at the Cdx2 and Gata3 loci. The plot also shows that PRC2 recruitment is reduced at those loci within TE-lineage cells.
We infected one-cell mouse embryos with lentiviral vectors expressing ectopic EED or shRNAs against KDM6B individually or in combination. We monitored preimplantation development by culturing infected embryos. We used lentiviral vectors expressing only EGFP as a control. We found that the presence of EGFP-expressing lentiviral vectors does not significantly inhibit development of preimplantation mouse embryos and they form matured blastocysts when cultured for 96 h (Fig. 8A and B). When infected with ectopic EED- and KDM6B shRNA-expressing vectors individually, we saw a partial but significant reduction in blastocyst maturation and found that 65 to 70% of the embryos developed to the blastocyst stage (Fig. 8B; see Fig. S4A in the supplemental material). However, when we infected embryos with lentiviral vectors expressing Kdm6B shRNAs along with ectopic EED-expressing vectors, we noticed stronger inhibition in blastocyst development and that ∼45% of the embryos did not develop to the blastocyst stage (Fig. 8A and B), and we found that expression of both the CDX2 and GATA3 proteins was strongly repressed in those development-arrested embryos (Fig. 8C).
Fig 8.
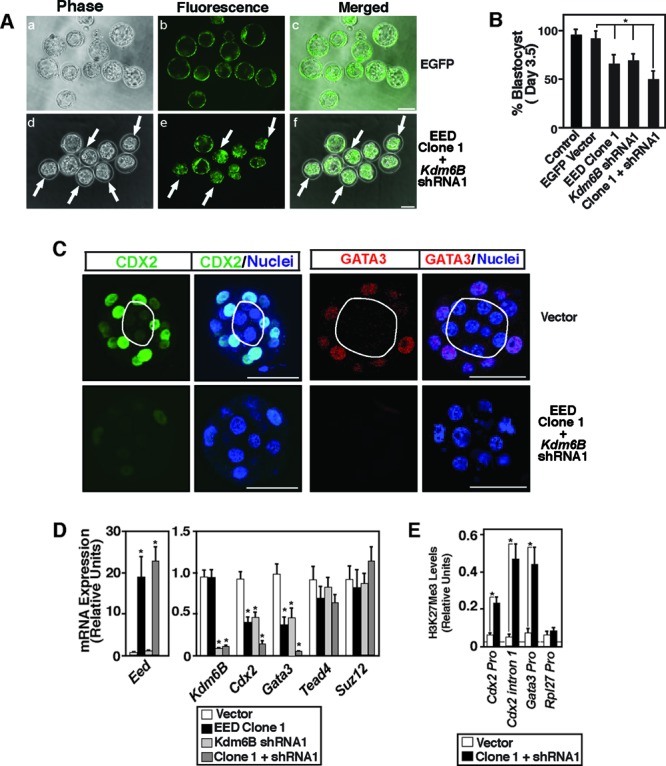
EED and KDM6B coordinate CDX2 and GATA3 expression in preimplantation mouse embryos by regulating the H3K27Me3 modification at their chromatin domains. (A) One-cell mouse embryos were infected with the lentiviral particles with vectors expressing either EGFP (top) or both ectopic Eed (clone 1) and Kdm6B shRNA (shRNA1) (bottom) and were cultured for 96 h. The vectors expressing ectopic EED also expressed an EGFP reporter. Phase-contrast and fluorescence micrographs show that several of the ectopic EED-expressing/KDM6B-depleted embryos (arrows) did not mature to the blastocyst stage. Bar, 50 μm. (B) Percentage of embryos maturing to the blastocyst stage after 96 h of culture under different experimental conditions (*, P < 0.05). (C) Confocal images show the loss of CDX2 (green) and GATA3 (red) expression in the nuclei (blue) of ectopic EED-expressing/KDM6B-depleted morula-stage (16-cell) embryos. Bars, 50 μm. (D) TEs were isolated from ectopic EED-expressing and/or KDM6B-depleted embryos that were matured to the blastocyst stage, and qRT-PCR analyses were performed to compare mRNA expression. TEs isolated from control vector-infected blastocysts were used as a control (*, P < 0.01). (E) ChIP-WGA analyses showing strong induction of H3K27Me3 incorporation at the Cdx2 and Gata3 chromatin domains in TEs isolated from ectopic EED-expressing/KDM6B-depleted blastocysts (*, P < 0.01).
Next, we isolated TEs from the ectopic EED-expressing/KDM6B-depleted embryos which matured to the blastocyst stage. We compared mRNA expression with that of TEs from control embryos and also performed ChIP-WGA to determine H3K27Me3 deposition at the chromatin domains of the Cdx2 and Gata3 loci. We found that Gata3 and Cdx2 mRNA expression was strongly repressed in TEs that were isolated from ectopic EED-expressing/KDM6b-depleted blastocysts (Fig. 8D) and transcriptional repression was associated with increased H3K27Me3 deposition at their chromatin domains (Fig. 8E). Interestingly, similar to our observation in mTSCs, we did not notice a significant reduction in Tead4 or Suz12 mRNA expression in ectopic EED-expressing/KDM6B-depleted TEs.
Blastocysts with altered expression of EED and KDM6B fail to implant.
Both CDX2 and GATA3 are implicated in proper TE function (9, 11), and earlier studies (10, 47–49) showed that the absence of CDX2 in a mouse blastocyst results in ultrastructural abnormalities within the TE, leading to the failure of embryo implantation to the uterus. We found that, although several of the ectopic EED-expressing/KDM6B-depleted embryos formed blastocysts, they showed defective hatching compared to the control embryos expressing only vectors (see Fig. S4B in the supplemental material). Therefore, we next tested whether the ectopic EED-expressing/KDM6B-depleted blastocysts show reduced implantation efficiency. We transferred blastocysts expressing ectopic EED or shRNAs against KDM6B individually or in combination into the uterine horns of E2.5 pseudopregnant female mice to test their implantation efficiency. We noticed a partial defect in the implantation efficiency of blastocysts with ectopic EED expression as well as KDM6B depletion (Fig. 9A and B). However, a complete failure of implantation was observed in blastocysts with both ectopic EED expression and KDM6B depletion (Fig. 9A and B).
Fig 9.
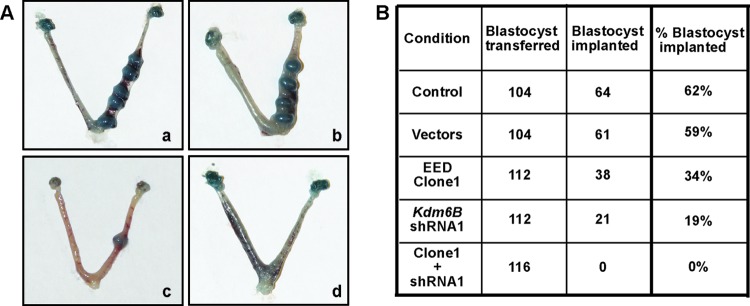
Ectopic EED-expressing/KDM6B-depleted blastocysts fail to implant into the uterus. (A) Blastocysts expressing either an empty vector (a), ectopic Eed (b), or Kdm6B shRNA (c) or both ectopic Eed and Kdm6B shRNA (d) were transferred into the uterine horns of E2.5 pseudopregnant mice. Mice were sacrificed at E7.5, and the uterine horns were isolated to compare implantation efficiencies. Images of representative uterine horns are shown. (B) Total number of blastocysts under different experimental conditions that were transferred into the uterine horns of E2.5 pseudopregnant mice and percentage of implanted embryos that were observed at E7.5.
To further define the importance of TE-specific regulation of EED and KDM6B, we tested whether TE-specific induction of EED and depletion of KDM6B result in the loss of CDX2 and GATA3 expression and negatively affect implantation efficiency. We took early blastocyst-stage mouse embryos and performed subzonal injection to specifically transduce TE (Fig. 10A) with lentiviral particles to ectopically induce EED expression and deplete KDM6B expression (Fig. 10B). We found that TE-specific alteration of EED and KDM6B expression resulted in a significant loss of Cdx2 and Gata3 mRNA expression (Fig. 10B). Immunofluorescence analyses also indicated strong inhibition of GATA3 and CDX2 protein expression in several blastocysts (Fig. 10C). Furthermore, the loss of CDX2 and GATA3 expression was associated with a reduced efficiency of blastocyst hatching (Fig. 10D and E) and implantation in surrogate females (Fig. 10F). Collectively, from our results, we concluded that coordinated functions of EED and KDM6B regulate the spatial expression patterns of key TE regulators, thereby leading to proper development and function of the TE, ensuring successful implantation.
Fig 10.

Trophectoderm-specific functions of EED and KDM6B coordinate optimal CDX2 and GATA3 expression and implantation of embryo. (A) Early-blastocyst-stage mouse embryos were subjected to subzonal injection with the lentiviral particles with either vectors or both ectopic Eed (clone 1) and Kdm6B shRNA (shRNA1) and were cultured for 36 h. Phase-contrast and fluorescence micrographs show TE-specific expression of the EGFP reporter gene from the ectopic EED-expressing vector. Bar, 50 μm. (B) TEs expressing ectopic Eed and Kdm6B shRNA were isolated, and qRT-PCR analyses were performed to compare mRNA expression. TEs expressing control vector were used as a control (*, P < 0.05). (C) Confocal images show representative blastocysts with loss of CDX2 (green) and GATA3 (red) expression in the TE nuclei (blue) upon TE-specific ectopic EED expression and KDM6B depletion. Bars, 50 μm. (D) Micrographs show defective hatching of blastocysts upon ectopic EED expression and KDM6B depletion within the TE. White arrows, hatched or hatching blastocysts; black arrows, empty zonae pellucidae. (E) Quantitation of blastocyst hatching described in panel D. The plot is shown in percent (n = 3; P < 0.05). (F) Total number of blastocysts under different experimental conditions that were transferred into the uterine horns of E2.5 pseudopregnant mice and percentage of implanted embryos that were observed at E7.5.
DISCUSSION
The spatial distribution of key transcription factors in the outer versus inner cells of a developing preimplantation embryo specifies TE and ICM lineages (5, 50, 51). Proper regulation of this process is crucial for embryo implantation and subsequent development. In recent years, several studies characterized histone modification patterns at the chromatin domains in preimplantation embryos as well as in mESCs and mTSCs (19, 28, 30, 52–54). However, prior to this study, the molecular mechanisms that regulate deposition of a histone mark at the chromatin domains of key TE regulators and their functional importance in TE lineage development had not been evaluated. In this study, we demonstrated that EED- and KDM6B-mediated regulation of H3K27Me3 deposition at the chromatin domains of key TE regulators Cdx2 and Gata3 is essential for their expression in the TE and suppression in the ICM, ensuring proper preimplantation development and implantation of the embryo.
_targeted knockout studies of EZH2, the catalyst for H3K27Me3 modification, and EED in mice revealed the importance of the H3K27Me3 modification during mammalian development (41, 55). However, preimplantation mouse development is not impaired in either Ezh2−/− (45, 55) or Eed−/− (41, 56) embryos. Interestingly, EZH1, a homolog of EZH2, could complement the EZH2 function in certain contexts (57, 58). So, the EZH1 function might compensate for the loss of EZH2 during preimplantation development. Similarly, preimplantation embryos contain maternal EED (44, 45). Thus, it is possible that maternal EED protein or expression of EED from maternal mRNA could compensate for the loss of zygotic EED in preimplantation embryos.
Our findings in this study indicate that one of the key regulatory components for differential deposition of the H3K27Me3 mark within the TE and ICM lineages is altered expression of EED. Cells in the ICM lineage express higher levels of EED. In contrast, cells in the TE lineage and TE-derived mTSCs have reduced levels of EED expression. Furthermore, ectopic expression of EED in those cells increases PRC2 recruitment and H3K27Me3 modification at the Gata3 and Cdx2 chromatin domains and represses their expression. The correlation between suppression of EED expression and induction of key TE/TSC regulators like CDX2 and GATA3 supports earlier findings that EED function is dispensable for mTSC self-renewal (56). On the other hand, induction of CDX2 and GATA3 and a TSC-like fate in Eed−/− mESCs under TSC culture conditions indicate that the EED function could be important to suppress the key TE/TSC-specific genes in the mESCs and in the ICM, the source of mESCs.
Interestingly, during preimplantation mouse development, CDX2 and GATA3 mRNA expression is ubiquitously induced in the totipotent blastomeres of 4- to 8-cell-stage embryos (9, 14). Thus, the question arises, how do EED/PRC2 recruitment and H3K27Me3 incorporation at the Cdx2 and Gata3 chromatin domains occur only in ICM-lineage cells at the blastocyst stage? Earlier studies indicated that the prior presence of the seed H3K27Me3 mark at a chromatin domain further facilitates EED-mediated PRC2 recruitment (39, 59). Our analyses with 8- to 16-cell mouse embryos detected low levels of the H3K27Me3 mark at both the Cdx2 and Gata3 loci. Thus, it is attractive to propose a model (Fig. 11) in which induction of EED expression in the nascent ICM lineage facilitates PRC2 recruitment at the Cdx2 and Gata3 loci due to the presence of the seed H3K27Me3 modification. The recruitment of PRC2 further increases the H3K27Me3 mark, leading to strong repression of transcription in the ICM.
Fig 11.

EED- and KDM6B-regulated spatial expression of CDX2 and GATA3 during preimplantation mammalian development. The model predicts that in the blastomeres of early-stage (8-cell) embryos, the Cdx2 and Gata3 chromatin domains harbor low levels of H3K27Me3 along with the H3K27Ac modification. Thus, those loci are ubiquitously but weakly (+) transcribed in those blastomeres. We showed that EED expression is induced in the nascent ICM lineage and is repressed in the nascent TE lineage, whereas KDM6B is induced in both the nascent ICM and nascent TE lineages. The model predicts that differential relative concentrations of EED and KDM6B lead to EED and PRC2 recruitment at the Cdx2 and Gata3 chromatin domains in the nascent ICM lineage or KDM6B recruitment within the TE lineage, thereby inducing or reducing incorporation of the H3K27Me3 modification, respectively. The induction of the H3K27Me3 modification leads to the abrogation of transcription in the ICM lineage. On the contrary, KDM6B recruitment and loss of the H3K27Me3 modification recruit histone acetyl transferases (HATs), leading to incorporation of more of the H3K27Ac mark, RNA polymerase II (Pol II) recruitment, and strong (+++) transcriptional activation within the TE lineage.
Our analyses with mouse embryos indicated that, along with Eed, Kdm6B expression is also induced during the morula-to-blastocyst transition, a finding also observed during bovine preimplantation development (33). Furthermore, mechanistic analyses in mTSCs indicated that KDM6B could regulate Cdx2 and Gata3 transcription via two different mechanisms: (i) it reduces global H3K27Me3 levels (Fig. 6C), and (ii) it could act via direct demethylation of the H3K27 residue by occupying the Cdx2 and Gata3 chromatin domains (Fig. 6A). Our analyses did not indicate which of these two mechanisms is more important to regulate Cdx2 and Gata3 transcription. However, global levels of the H3K27Me3 modification do not correlate with the spatial expression patterns of Cdx2 in rat, monkey, and human blastocysts (Fig. 1) (9). Thus, direct demethylation of the H3K27 residues at the Cdx2 and Gata3 chromatin domains is probably the predominant mechanism by which KDM6B contributes to the transcriptional upregulation of those genes in the TE lineage. Along this line, we predict that the recruitment of KDM6B at the Cdx2 and Gata3 chromatin domains in the nascent TE lineage eliminates the seed H3K27Me3 mark and thereby prevents further recruitment of the PRC2 complex, ensuring their progressive transcriptional activation (Fig. 11). In this aspect, it is also attractive to hypothesize that the relative concentration of EED and KDM6B is the key factor to shift the equilibrium either toward PRC2 recruitment or toward KDM6B recruitment at the Cdx2 and Gata3 loci in nascent ICM and TE lineages, respectively (Fig. 11). However, it is possible that alteration of global H3K27Me3 levels by KDM6B is a key regulatory mechanism to attenuate expression of other genes during preimplantation development.
Our findings in this study raise several questions. (i) How does EED expression become selectively repressed in the TE and induced in the ICM? (ii) How does KDM6B expression become induced during blastocyst formation? (iii) Why does reduced expression of EED result in the loss of PRC2 recruitment only at the selective loci like Cdx2 and Gata3 within the TE lineage? Studies in mESCs showed that JARID2 could modulate PRC2 recruitment at a chromatin domain (34–36). However, the JARID2 protein is almost undetectable in the mTSCs, indicating that a different mechanism might be involved in fine-tuning PRC2 recruitment at distinct chromatin domains in the cells of the TE lineage.
Despite low levels of EED expression within the TE, earlier studies showed that establishment of H3K27Me3 marks on inactive X chromosomes within the cells of the nascent TE lineage is mediated by transient recruitment of EED (45, 46). We predict that the lack of H3K27Me3 incorporation at the Cdx2 and Gata3 loci within the nascent TE is prevented by a TEAD4-dependent mechanism. We and other groups have implicated TEAD4 as the major upstream factor that directly regulates Cdx2 and Gata3 transcription during TE development (1, 11–13). Thus, it is possible that in the TE lineage, TEAD4 occupancy at the Cdx2 and Gata3 chromatin facilitates KDM6B recruitment, thereby eliminating the seed H3K27Me3 modification and preventing EED/PRC2 recruitment. However, as other histone modifications, like H3K27Ac, could modulate PRC2 recruitment (60), it is also possible that selective incorporation of the H3K27Ac mark prevents further PRC2 recruitment at the Cdx2 and Gata3 loci in the nascent TE lineage. Future studies capturing the detailed dynamics of chromatin domains during preimplantation development will address these questions.
Interestingly, unlike Cdx2 and Gata3 repression, Tead4 repression is not critically dependent on H3K27Me3 modification during preimplantation development. The insensitivity of Tead4 expression to altered H3K27Me3 modification indicates that, in preimplantation embryos, a specific histone modification could be crucial for regulation of some selective genes without significantly affecting expression of others. This selectivity in gene expression is encouraging, considering that preimplantation development could be modulated by regulating expression of a specific set of genes through alterations in histone modification patterns. Thus, we predict that _targeting histone modification could be an important therapeutic approach to alter specific genes in preimplantation embryos to enhance their quality and implantation efficiency.
Supplementary Material
ACKNOWLEDGMENTS
We thank Terry Magnuson for providing the Eed−/− mESCs, Thaddeus G. Golos for providing monkey blastocysts, Michael J. Soares for important suggestions, and Partho Chattoraj for experimental help.
The work is supported by NIH grants HD062546, HL104322, and HL106311 to S. Paul. B. Saha is supported by a Biomedical Research Training Program postdoctoral fellowship, and P. Home is supported by an American Heart Association postdoctoral fellowship.
All work with human embryos was done with private funding and without any support (including support for instruments, facilities, reagents, or personnel) from federal funding.
Footnotes
Published ahead of print 13 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00069-13.
REFERENCES
- 1.Home P, Saha B, Ray S, Dutta D, Gunewardena S, Yoo B, Pal A, Vivian JL, Larson M, Petroff M, Gallagher PG, Schulz VP, White KL, Golos TG, Behr B, Paul S. 2012. Altered subcellular localization of transcription factor TEAD4 regulates first mammalian cell lineage commitment. Proc. Natl. Acad. Sci. U. S. A. 109:7362–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunath T, Strumpf D, Rossant J. 2004. Early trophoblast determination and stem cell maintenance in the mouse—a review. Placenta 25(Suppl A):S32–S38 [DOI] [PubMed] [Google Scholar]
- 3.Rossant J. 2004. Lineage development and polar asymmetries in the peri-implantation mouse blastocyst. Semin. Cell Dev. Biol. 15:573–581 [DOI] [PubMed] [Google Scholar]
- 4.Zernicka-Goetz M. 2005. Cleavage pattern and emerging asymmetry of the mouse embryo. Nat. Rev. 6:919–928 [DOI] [PubMed] [Google Scholar]
- 5.Cockburn K, Rossant J. 2010. Making the blastocyst: lessons from the mouse. J. Clin. Invest. 120:995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfeffer PL, Pearton DJ. 2012. Trophoblast development. Reproduction 143:231–246 [DOI] [PubMed] [Google Scholar]
- 7.Roberts RM, Fisher SJ. 2011. Trophoblast stem cells. Biol. Reprod. 84:412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossant J, Cross JC. 2001. Placental development: lessons from mouse mutants. Nat. Rev. Genet. 2:538–548 [DOI] [PubMed] [Google Scholar]
- 9.Home P, Ray S, Dutta D, Bronshteyn I, Larson M, Paul S. 2009. GATA3 is selectively expressed in the trophectoderm of peri-implantation embryo and directly regulates Cdx2 gene expression. J. Biol. Chem. 284:28729–28737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, Rossant J. 2005. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell 123:917–929 [DOI] [PubMed] [Google Scholar]
- 11.Ralston A, Cox BJ, Nishioka N, Sasaki H, Chea E, Rugg-Gunn P, Guo G, Robson P, Draper JS, Rossant J. 2010. Gata3 regulates trophoblast development downstream of Tead4 and in parallel to Cdx2. Development 137:395–403 [DOI] [PubMed] [Google Scholar]
- 12.Nishioka N, Yamamoto S, Kiyonari H, Sato H, Sawada A, Ota M, Nakao K, Sasaki H. 2008. Tead4 is required for specification of trophectoderm in pre-implantation mouse embryos. Mech. Dev. 125:270–283 [DOI] [PubMed] [Google Scholar]
- 13.Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML, Buonanno A. 2007. Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 134:3827–3836 [DOI] [PubMed] [Google Scholar]
- 14.Ralston A, Rossant J. 2008. Cdx2 acts downstream of cell polarization to cell-autonomously promote trophectoderm fate in the early mouse embryo. Dev. Biol. 313:614–629 [DOI] [PubMed] [Google Scholar]
- 15.Workman JL, Kingston RE. 1998. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 67:545–579 [DOI] [PubMed] [Google Scholar]
- 16.Farthing CR, Ficz G, Ng RK, Chan CF, Andrews S, Dean W, Hemberger M, Reik W. 2008. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 4:e1000116. 10.1371/journal.pgen.1000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alder O, Lavial F, Helness A, Brookes E, Pinho S, Chandrashekran A, Arnaud P, Pombo A, O'Neill L, Azuara V. 2010. Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment. Development 137:2483–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl JA, Reiner AH, Klungland A, Wakayama T, Collas P. 2010. Histone H3 lysine 27 methylation asymmetry on developmentally-regulated promoters distinguish the first two lineages in mouse preimplantation embryos. PLoS One 5:e9150. 10.1371/journal.pone.0009150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rugg-Gunn PJ, Cox BJ, Ralston A, Rossant J. 2010. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. U. S. A. 107:10783–10790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutta D, Ray S, Home P, Larson M, Wolfe MW, Paul S. 2011. Self-renewal versus lineage commitment of embryonic stem cells: protein kinase C signaling shifts the balance. Stem Cells 29:618–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. 1998. Promotion of trophoblast stem cell proliferation by FGF4. Science 282:2072–2075 [DOI] [PubMed] [Google Scholar]
- 22.Ray S, Dutta D, Rumi MA, Kent LN, Soares MJ, Paul S. 2009. Context-dependent function of regulatory elements and a switch in chromatin occupancy between GATA3 and GATA2 regulate Gata2 transcription during trophoblast differentiation. J. Biol. Chem. 284:4978–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalista T, Freeman HA, Behr B, Pera RR, Scott CT. 2011. Donation of embryos for human development and stem cell research. Cell Stem Cell 8:360–362 [DOI] [PubMed] [Google Scholar]
- 24.National Institutes of Health 2000. Public Health Service policy on humane care and use of laboratory animals. Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, MD. [Google Scholar]
- 25.Dutta D, Ray S, Vivian JL, Paul S. 2008. Activation of the VEGFR1 chromatin domain: an angiogenic signal-ETS1/HIF-2alpha regulatory axis. J. Biol. Chem. 283:25404–25413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutta D, Ray S, Home P, Saha B, Wang S, Sheibani N, Tawfik O, Cheng N, Paul S. 2010. Regulation of angiogenesis by histone chaperone HIRA-mediated incorporation of lysine 56-acetylated histone H3.3 at chromatin domains of endothelial genes. J. Biol. Chem. 285:41567–41577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dey SK. 2006. Visualizing early embryo implantation sites by dye injection. CSH Protoc. 2006(2): pii=pdb.prot4361. 10.1101/pdb.prot4361 [DOI] [PubMed] [Google Scholar]
- 28.Wongtawan T, Taylor JE, Lawson KA, Wilmut I, Pennings S. 2011. Histone H4K20me3 and HP1alpha are late heterochromatin markers in development, but present in undifferentiated embryonic stem cells. J. Cell Sci. 124:1878–1890 [DOI] [PubMed] [Google Scholar]
- 29.O'Geen H, Nicolet CM, Blahnik K, Green R, Farnham PJ. 2006. Comparison of sample preparation methods for ChIP-chip assays. Biotechniques 41:577–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, Helin K. 2010. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of polycomb group _target genes. Nucleic Acids Res. 38:4958–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margueron R, Reinberg D. 2011. The polycomb complex PRC2 and its mark in life. Nature 469:343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swigut T, Wysocka J. 2007. H3K27 demethylases, at long last. Cell 131:29–32 [DOI] [PubMed] [Google Scholar]
- 33.Canovas S, Cibelli JB, Ross PJ. 2012. Jumonji domain-containing protein 3 regulates histone 3 lysine 27 methylation during bovine preimplantation development. Proc. Natl. Acad. Sci. U. S. A. 109:2400–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. 2010. JARID2 regulates binding of the polycomb repressive complex 2 to _target genes in ES cells. Nature 464:306–310 [DOI] [PubMed] [Google Scholar]
- 35.Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J. 2009. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and _target gene occupancy in pluripotent cells. Cell 139:1290–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH. 2009. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 139:1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang F, Barbacioru C, Bao S, Lee C, Nordman E, Wang X, Lao K, Surani MA. 2010. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell 6:468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tie F, Stratton CA, Kurzhals RL, Harte PJ. 2007. The N terminus of Drosophila ESC binds directly to histone H3 and is required for E(Z)-dependent trimethylation of H3 lysine 27. Mol. Cell. Biol. 27:2014–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu C, Bian C, Yang W, Galka M, Ouyang H, Chen C, Qiu W, Liu H, Jones AE, MacKenzie F, Pan P, Li SS, Wang H, Min J. 2010. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc. Natl. Acad. Sci. U. S. A. 107:19266–19271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mak W, Nesterova TB, de Napoles M, Appanah R, Yamanaka S, Otte AP, Brockdorff N. 2004. Reactivation of the paternal X chromosome in early mouse embryos. Science 303:666–669 [DOI] [PubMed] [Google Scholar]
- 41.Faust C, Lawson KA, Schork NJ, Thiel B, Magnuson T. 1998. The Polycomb-group gene eed is required for normal morphogenetic movements during gastrulation in the mouse embryo. Development 125:4495–4506 [DOI] [PubMed] [Google Scholar]
- 42.Chamberlain SJ, Yee D, Magnuson T. 2008. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 26:1496–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puschendorf M, Terranova R, Boutsma E, Mao X, Isono K, Brykczynska U, Kolb C, Otte AP, Koseki H, Orkin SH, van Lohuizen M, Peters AH. 2008. PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 40:411–420 [DOI] [PubMed] [Google Scholar]
- 44.Schumacher A, Faust C, Magnuson T. 1996. Positional cloning of a global regulator of anterior-posterior patterning in mice. Nature 384:648. [DOI] [PubMed] [Google Scholar]
- 45.Erhardt S, Su IH, Schneider R, Barton S, Bannister AJ, Perez-Burgos L, Jenuwein T, Kouzarides T, Tarakhovsky A, Surani MA. 2003. Consequences of the depletion of zygotic and embryonic enhancer of zeste 2 during preimplantation mouse development. Development 130:4235–4248 [DOI] [PubMed] [Google Scholar]
- 46.Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N. 2003. Establishment of histone H3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 4:481–495 [DOI] [PubMed] [Google Scholar]
- 47.Chawengsaksophak K, de Graaff W, Rossant J, Deschamps J, Beck F. 2004. Cdx2 is essential for axial elongation in mouse development. Proc. Natl. Acad. Sci. U. S. A. 101:7641–7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, Beck F, Rossant J. 2005. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development 132:2093–2102 [DOI] [PubMed] [Google Scholar]
- 49.Wu G, Gentile L, Fuchikami T, Sutter J, Psathaki K, Esteves TC, Arauzo-Bravo MJ, Ortmeier C, Verberk G, Abe K, Scholer HR. 2010. Initiation of trophectoderm lineage specification in mouse embryos is independent of Cdx2. Development 137:4159–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruce AW, Zernicka-Goetz M. 2010. Developmental control of the early mammalian embryo: competition among heterogeneous cells that biases cell fate. Curr. Opin. Genet. Dev. 20:485–491 [DOI] [PubMed] [Google Scholar]
- 51.Takaoka K, Hamada H. 2012. Cell fate decisions and axis determination in the early mouse embryo. Development 139:3–14 [DOI] [PubMed] [Google Scholar]
- 52.Abell AN, Jordan NV, Huang W, Prat A, Midland AA, Johnson NL, Granger DA, Mieczkowski PA, Perou CM, Gomez SM, Li L, Johnson GL. 2011. MAP3K4/CBP-regulated H2B acetylation controls epithelial-mesenchymal transition in trophoblast stem cells. Cell Stem Cell 8:525–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kidder BL, Palmer S. 2010. Examination of transcriptional networks reveals an important role for TCFAP2C, SMARCA4, and EOMES in trophoblast stem cell maintenance. Genome Res. 20:458–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kidder BL, Palmer S. 2012. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 40:2925–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. 2001. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 21:4330–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalantry S, Mills KC, Yee D, Otte AP, Panning B, Magnuson T. 2006. The polycomb group protein Eed protects the inactive X-chromosome from differentiation-induced reactivation. Nat. Cell Biol. 8:195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. 2008. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32:503–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. 2008. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 32:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10:1291–1300 [DOI] [PubMed] [Google Scholar]
- 60.Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, Behrens A, Bertone P, Hendrich B. 2012. NuRD-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 31:593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.