

Abstract
Amyloid imaging is currently introduced to the market for clinical use. We will review the evidence demonstrating that the different amyloid PET ligands that are currently available are valid biomarkers for Alzheimer-related β amyloidosis. Based on recent findings from cross-sectional and longitudinal imaging studies using different modalities, we will incorporate amyloid imaging into a multidimensional model of Alzheimer's disease. Aside from the critical role in improving clinical trial design for amyloid-lowering drugs, we will also propose a tentative algorithm for when it may be useful in a memory clinic environment. Gaps in our evidence-based knowledge of the added value of amyloid imaging in a clinical context will be identified and will need to be addressed by dedicated studies of clinical utility.
Keywords: Flutemetamol, Florbetapir, Florbetaben, PIB, Clinic, Diagnostic
Highlights
-
•
Amyloid imaging has been validated as a biomarker for cerebral β amyloidosis.
-
•
Biomarkers are essential for progress in therapy for Alzheimer's disease.
-
•
They may have a role in clinical diagnosis under specific conditions.
-
•
An evidence-based medicine approach will have to prove their added value.
-
•
We position amyloid imaging within a multidimensional model of Alzheimer's disease.
1. Introduction
Cognitive and behavioral decline due to neurodegenerative disease is highly prevalent across the world. Apart from the pervasive personal and familial impact, the public health burden may become unsurmountable for healthcare systems over the next decades (Brookmeyer et al., 2007; Hebert et al., 2001) except if more efficacious interventions to prevent, halt or slow down Alzheimer's disease (AD) (Brookmeyer et al., 1998) are discovered. The ability to characterize directly in humans the underlying pathophysiological processes is fundamental to progress in AD research and therapy. After more than a decade of therapeutic stagnation, the time may have come to re-think the clinical concept of Alzheimer's disease and to re-draw the clinical development path for testing novel candidate AD drugs. We will evaluate to which degree the availability of amyloid imaging, amid other techniques, could alter our clinical-diagnostic approach to AD and how this may enhance progress towards more efficacious therapy.
2. Some basic AD principles relevant to amyloid imaging
The initial abnormalities in AD probably occur at the functional level, involving synaptic and neuronal dysfunction, possibly initiated by abnormalities in soluble Aβ42. The exact temporal relationship between the diverse structural alterations that follow is still a matter of active neuropathological and in vivo research (see below). In the revised National Institute on Aging-Alzheimer's Association (NIA-AA) neuropathological criteria (Hyman et al., 2012), three different classification schemes have been adopted in parallel, depending on the phase of the spread of amyloid plaques (Thal et al., 2002) and neurofibrillary tangles (Braak and Braak, 1991) and the amount of neuritic plaques as defined by the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria.
The principal structural changes are:
-
•
Loss of synaptic density (Davis et al., 1999; DeKosky and Scheff, 1990; Scheff et al., 2006; Terry et al., 1991), starting in the dentate gyrus and correlated to episodic memory scores (Scheff et al., 2006)
-
•
Neuronal loss, starting in entorhinal cortex (Gómez-Isla et al., 1996), and correlated to cognitive scores (Giannakopoulos et al., 2003)
-
•
Neurofibrillary tangles (NFT) and neuropil threads (Hyman et al., 2012). Initially, neurofibrillary tangles are mainly present in entorhinal and perirhinal cortex spreading to the hippocampus (stage I/II). They subsequently spread to inferior temporal and lateral temporal cortex (stage III/IV) and then become widely distributed over neocortical association zones (stages V/VI) (Braak and Braak, 1991; Hyman et al., 2012; Tiraboschi et al., 2004). These NFT stages correlate relatively well with neuronal loss and with the severity of clinical symptoms (Arriagada et al., 1992; Bancher et al., 1993; Giannakopoulos et al., 1998, 2003; Guillozet et al., 2003; Mesulam, 1999; Mesulam et al., 2004).
-
•
Aβ amyloid aggregates, which may take different forms (Hyman et al., 2012): According to some authors, diffuse plaques should not be considered as pathological since they are not associated with synapse loss or neuronal loss, key features of Alzheimer's disease (Masliah et al., 1990). Others have suggested that diffuse amyloid plaques are related to the presymptomatic stage of AD (Morris et al., 1996). In contrast to diffuse plaques, neuritic plaques stain with thioflavine S or Bielschowsky, indicative for the presence of tau pathology invading or surrounding the plaque. A time sequence analogous to that described for NFTs has been developed to describe the propagation of amyloid, starting in neocortical areas (phase 1) and then spreading to allocortical regions including, among other regions, entorhinal cortex, CA1, anterior and posterior cingulate (phase 2), basal forebrain nuclei, diencephalic nuclei and striatum (phase 3), brain stem nuclei (phase 4) and further into the molecular layer of the cerebellum (phase 5) (Thal et al., 2002). At a certain stage the increase in β amyloid aggregates may level off, also referred to as a ‘growth arrest’ of the amyloid plaques (Christie et al., 2001; Hyman et al., 1993).
The clinical phenotype and severity in AD is determined principally by the topographic distribution and density of neuronal loss and NFTs, starting in the hippocampal formation and extending into inferior and lateral temporal cortex and beyond (Arriagada et al., 1992; Bancher et al., 1993; Giannakopoulos et al., 1998, 2003; Guillozet et al., 2003; Mesulam, 1999; Mesulam et al., 2004). In atypical variants of AD, such as posterior cortical atrophy (Hof et al., 1993, 1997; von Gunten et al., 2006) or the logopenic variant of primary progressive aphasia (Gefen et al., 2012), the unusual clinical phenotype at initial presentation is mirrored by an unusual distribution of neuronal loss and NFTs. Conversely, when other proteinopathies, such as Tar DNA Binding Protein (TDP)43-proteinopathies (Pao et al., 2011) or tauopathies (Hornberger et al., 2012) have an atypical distribution and predominantly affect medial temporal cortex, they may mimic the amnestic presentation of AD.
As age increases, the classical hallmark lesions of AD (NFT and neuritic plaques) allow one to discriminate less and less reliably between subjects with versus without dementia (Davis et al., 1999; Prohovnik et al., 2006). In a prospective population-based series of 456 autopsy cases between 70 and 100 years of age (Savva et al., 2009), a strong relationship between dementia and the presence of NFT and neuritic plaques was seen until 75 years of age (Savva et al., 2009). This relationship became weaker above the age of 80, especially for neuritic plaques. Diffuse plaques were only weakly associated with dementia, even at the age of 75 (Savva et al., 2009). In contrast, neuropathological measures of volume loss retained their strong relationship with the presence or absence of dementia over the entire age range (Savva et al., 2009). Prospective community-based neuropathological studies have highlighted the contribution of other lesions to cognitive decline, such as cerebrovascular lesions or Lewy bodies, independently (Schneider et al., 2012) or interactively (Snowdon et al., 1997) with Alzheimer pathology. Cognitive decline therefore is the integral of a combination of different, at least partly independent factors, leading to the concept of ‘multifactorial AD’.
In line with this concept, NIA-AA guidelines for the neuropathologic assessment of AD put emphasis on the systematic evaluation of concomitant abnormalities, e.g. Lewy bodies, TDP43 inclusions and vascular changes (Hyman et al., 2012). In the same vein, the novel NIA-AA criteria for clinical AD separate out a category of possible AD with ‘etiologically mixed’ presentation (McKhann et al., 2011). This subgroup includes subjects who have concomitant neuropathological findings of cerebrovascular disease, TDP43-proteinopathy or Lewy bodies. This subgroup also includes cases where medical comorbidity and use of drugs contribute to the cognitive dysfunction.
3. Comparisons between amyloid ligands
The amyloid ligands that are available for research use are the thioflavin T derived 11C-Pittsburgh Compound B (PIB) (Klunk et al., 2004), its 18F-labeled analog 18F-flutemetamol (Koole et al., 2009; Nelissen et al., 2009), the benzofuranes 11C-AZD-2184 (Nyberg et al., 2009) and 18F-AZD-4694 (Cselényi et al., 2012), the benzoxazole 11C-BF-227 (Furukawa et al., 2010), the stilbene compounds 11C-SB (Verhoeff et al., 2004), 18F-florbetaben (Barthel et al., 2011; Rowe et al., 2008) and 18F-florbetapir (Wong et al., 2010), and the naphthol 18F-FDDNP (Small et al., 2006). Of these, at the time of writing, only 18F-florbetapir is approved by the Food and Drugs Administration (FDA) and European Medicines Agency (EMA) for the clinical evaluation of cognitive deficits in patients. For review of the development and biochemical characteristics of these compounds as well as methodological nuclear imaging issues we refer to Herholz and Ebmeier (2011), Kadir and Nordberg (2010), and Mathis et al. (2012). Here we will focus on direct comparative studies between different amyloid ligands within the same subjects (Tables 1, 2).
Table 1.
Cohen's d effect size for discriminating between AD and controls (difference between the means of the two study populations divided by the pooled standard deviation). These values are for indicative purposes and, as indicated, may substantially vary between studies depending on the exact processing and analysis method used. For the sake of comparison, the effect size obtained for FDG-PET for discriminating AD from controls in a comparative study of FDG-PET and 11C-PIB-PET was 1.53 (Ng et al., 2007).
| Cohen's d effect size | Reference | |
|---|---|---|
| 11C-PIB | 2.23–3.8 | Verhoeff et al. (2004), Ng et al. (2007) and Villemagne et al. (2012) |
| 11C-SB | 1.22 | Verhoeff et al. (2004) |
| 18F-flutemetamol | 2.7 | Vandenberghe et al. (2010) |
| 18F-florbetapir | 2.3 | Wong et al. (2010) |
| 18F-florbetaben | 1.37–3 | Barthel et al. (2011) and Villemagne et al. (2011, 2012) |
Table 2.
Within-subject correlations between 11C-PIB and 18F-labeled ligands (Tolboom et al., 2009a; Vandenberghe et al., 2010; Villemagne et al., 2012; Wolk et al., 2012). m: region-wise linear slope. r: Pearson correlation coefficient. SUVR: Standardized Uptake Value Ratio; BP: Binding Potential. Dash: not reported.
| SUVR |
Global BP |
|||||
|---|---|---|---|---|---|---|
|
18F-flutemetamol |
18F-florbetapir |
18F-florbetaben |
18F-FDDNP |
|||
| r | m | r | m | r | r | |
| Composite | 0.91 | 0.99 | 0.78 | 0.33 | 0.97 | 0.45 |
| Frontal | 0.92 | 1.00 | 0.81 | – | 0.94–0.96 | – |
| Parietal | 0.92 | 1.01 | 0.58 | – | 0.94 | – |
| Lateral temporal | 0.91 | 0.99 | 0.68 | – | 0.96 | – |
| Posterior cingulate | 0.91 | 1.01 | 0.79 | – | 0.96 | – |
| Anterior cingulate | 0.88 | 0.91 | 0.81 | – | 0.94 | – |
| Medial temporal | 0.83 | 0.74 | – | – | 0.82 | – |
| Occipital | 0.89 | 1.03 | – | – | 0.92 | – |
| Striatum | 0.84 | 0.88 | – | – | 0.95 | – |
| Subcortical white matter | 0.22 | 0.36 | – | – | 0.63 | – |
| Pons | 0.63 | 0.50 | 0.38 | – | 0.50 | – |
Verhoeff et al. (2004) compared the stilbene 11C-SB and 11C-PIB (Verhoeff et al., 2004) (Table 1). Overall, the discriminative power was higher for the latter compound (Table 1). Other studies compared 18F-labeled amyloid ligands to 11C-PIB (Table 2). In 20 AD and 20 amnestic MCI patients (Vandenberghe et al., 2010), volume-of-interest based Standardized Uptake Value Ratios (SUVR) with cerebellar gray matter as reference region were compared between 11C-PIB and 18F-flutemetamol by means of linear regression (Table 2) (Vandenberghe et al., 2010). There was a close match of SUVR levels between the two compounds in neocortical volumes of interest (Table 2). The correlation was much weaker in subcortical white matter and pons where retention of the 18F amyloid ligand was higher than that of 11C-PIB (Vandenberghe et al., 2010).
Wolk et al. (2012) compared 18F-florbetapir and 11C-PIB in 14 AD and 15 healthy controls. The separation between controls and AD was wider for 11C-PIB, both in absolute terms and in terms of S.D. (healthy controls (HC) mean SUVR composite cortical VOI (SUVRcomp) = 1.07 (SD 0.23), AD mean SUVRcomp 1.94 (SD 0.36)) than for 18F-florbetapir (HC mean SUVRcomp 1.06 (SD 0.17), AD mean SUVRcomp 1.38 (SD 0.15)).
11C-PIB has also been directly compared with 18F-FDDNP in 14 patients with AD, 11 patients with amnestic MCI and 13 controls. The correlation of binding potentials between the two tracers was only 0.45 (Table 2). Values in hippocampus were higher for 18F-FDDNP than for 11C-PIB (Shin et al., 2010; Tolboom et al., 2009a). This was also true in inferior temporal gyrus and secondary visual association cortex (Shin et al., 2010), probably related to the predilection of NFTs for these regions (Giannakopoulos et al., 1998). In other neocortical association areas, binding potential values in the AD group were more than 9-fold lower for 18F-FDDNP than for 11C-PIB, reflecting lower affinity of 18F-FDDNP for neuritic plaques. The differences between groups (AD, MCI and controls) were more pronounced for 11C-PIB than for 18F-FDDNP (Tolboom et al., 2009a).
It is often assumed that 18F-amyloid ligands perform more or less equivalently for separating AD versus controls. Only direct comparisons between the 18F-labeled amyloid ligands within the same subjects can resolve this important question, and head-to-head comparisons are currently underway. As several competing ligands will enter the commercial arena, such direct comparisons will be of great clinical interest.
4. Comparisons between amyloid ligands and neuropathology
3H-PIB colocalizes with neuritic plaques and also with diffuse amyloid plaques and Aβ in the blood vessel wall (Lockhart et al., 2007), and much less so with tangles (Lockhart et al., 2007) or other β sheets aggregates such as Lewy bodies (Lockhart et al., 2007). 6-CN-PIB, a highly fluorescent derivative of 11C-PIB, has affinity for plaques (Ikonomovic et al., 2008), more so for neuritic than for diffuse plaques (Ikonomovic et al., 2012), as well as affinity for β amyloid in the vessel walls (Bacskai et al., 2007) and for striatal plaques (Ikonomovic et al., 2008). There is no affinity for cerebellar plaques and only partial affinity for extracellular ‘ghost tangles’, probably due to adjacent neuritic plaques (Ikonomovic et al., 2008). 11C-PIB retention values correlate with Enzyme Linked ImmunoSorbent Assay (ELISA) measurements of Aβ42 more so than Aβ40 (Ikonomovic et al., 2012).
The level of exposure of the different types of aggregates to the tracer may drastically differ between the brain slices and the in vivo imaging situation (Lockhart et al., 2007). For that reason, quantitative correlational studies between in vivo measures of amyloid ligand retention and quantitative neuropathological measures of the presence of Aβ are an essential step for biomarker validation.
4.1. Large-scale prospective postmortem validation studies
In the clinicopathological 18F-florbetapir phase 3 study (Clark et al., 2011, 2012), 59 cases (mean age 79 years) received a 10-min florbetapir PET and a neuropathological assessment within 1–2 years following the PET (Clark et al., 2012). Twelve had no cognitive impairment, 29 had clinical AD, and 13 had a clinical diagnosis of some other form of dementia (Lewy body dementia (LDB), Parkinson's Disease with Dementia (PDD), frontotemporal lobar degeneration (FTLD), unspecified, mixed). When binary scan reads were compared to binarized CERAD neuritic amyloid plaque scores (moderate/frequent vs sparse/none), specificity was 100% and sensitivity around 92–96%. 18F-florbetapir retention levels also correlated with β amyloid area as determined by 4G8 amyloid immunostaining (antibody against Aβ residues 17–24). The phase 3 clinicopathological studies with the other 18F-ligands have not been published at the time of writing.
A series of biopsy studies correlated β amyloid 4G8 immunostaining and 18F-flutemetamol retention in normal pressure hydrocephalus. These studies confirmed the correlation between ligand retention levels and β amyloid plaque surface area (Leinonen et al., 2013; Rinne et al., 2012; Wong et al., 2013).
4.2. Other clinicopathological studies
The large-scale clinicopathological studies have been pivotal in demonstrating the relationship between ligand retention and amyloid plaque surface area. Smaller case series from academic investigator-driven studies provide complimentary information to what can be obtained from larger-scale industry-sponsored trials. A number of cases exhibited a dissociation between amyloid positivity/negativity and the presence/absence of Alzheimer's disease according to consensus CERAD or The National Institute on Aging and Reagan Institute (NIA-RI) neuropathological criteria. It is important to note that the terms ‘false-positive’ and ‘false-negative’ depend on the comparator used. If the amyloid plaque area is used as comparator and both neuritic and diffuse plaques are counted, false-positives have not been described until now and false-negatives only exceptionally (Cairns et al., 2009). If conventional neuropathological criteria are used as comparator, mismatches will inevitably occur as these criteria also integrate other AD-related features such as NFTs. Nevertheless, in a clinical review, when assessing clinicopathological correlations, it is useful to compare with such well-established nosological criteria. Conceivably, in the future biomarker research may lead to novel taxonomic classifications that are based on one specific lesion type (such as a diagnostic category of β amyloidosis), among the different abnormalities that currently define Alzheimer's disease, but such novel classifications would first have to be validated and prove their utility in drug development and clinical practice.
4.2.1. False-positive cases with CERAD and NIA-RI criteria as comparator
In the Baltimore Longitudinal Study of Aging (BLSA), a community-recruited cohort is followed longitudinally with cognitive assessments until close to time of death and brain autopsy (Driscoll et al., 2012; Sojkova et al., 2011). In the 5 nondemented and 1 demented case reported, 11C-PIB retention values had an almost linear relationship with Aβ immunostaining using 6E10 antibody (which is directed at aminoacids 1–17 of the Aβ protein). One 11C-PIB positive nondemented case had mainly diffuse plaques, sparse neuritic plaques, and NFT Braak stage IV. According to NIA-Reagan Institute (RI) criteria (Hyman and Trojanowski, 1997), the likelihood of AD was low and according to CERAD criteria for neuropathologically definite AD, the case was normal.
Five other false-positive cases have been reported, all with a diagnosis of Lewy-body disease during life (Bacskai et al., 2007; Burack et al., 2010; Kantarci et al., 2012). For instance, Burack et al. (2010) reported 3 cases with Parkinson disease dementia, two of whom had a positive 11C-PIB scan within 15 months of death. These two positive cases had abundant diffuse plaques and only sparse neuritic plaques and intermediate NFT pathology (Burack et al., 2010). Neuropathologically, the diagnosis was Braak and Braak Lewy body disease stage 6, possible AD according to CERAD criteria and low-probability AD according to NIA-RI criteria (Burack et al., 2010).
Taken together, these single case reports and small case series provide evidence that despite the higher affinity of 11C-PIB for neuritic compared to diffuse plaques (Ikonomovic et al., 2012), cases with abundant diffuse plaques and no or sparse neuritic plaques may be associated with a positive amyloid scan, although such cases would not meet CERAD or NIA-RI neuropathological criteria for AD (Bacskai et al., 2007; Burack et al., 2010; Kantarci et al., 2012; Sojkova et al., 2011).
4.2.2. False-negative cases with CERAD and NIA-RI criteria as comparator
Another subject of the BLSA series (Driscoll et al., 2012; Sojkova et al., 2011) had a moderate amount of neuritic plaques according to CERAD classification while Distribution Volume Ratio (DVR) for 11C-PIB was only around the cut-off of 1.2. This would be considered a false-negative scan if conventional neuropathological criteria for AD diagnosis are used as comparator. Another case with clinically probable Lewy body disease met CERAD criteria for definite AD due to focal deposition of abundant neuritic plaques limited to a prefrontal region but was 11C-PIB-negative (Ikonomovic et al., 2012). In a biopsy study of cases with Normal-Pressure Hydrocephalus (NPH) (Leinonen et al., 2008), one 11C-PIB-negative case met neuropathological criteria for definite AD. Cairns et al. (2009) described a case above the age of 85 who had progressive cognitive decline, a negative 11C-PIB scan, abundant diffuse plaques and a neuropathological diagnosis of possible AD according to CERAD criteria and low-probability of AD according to NIA-RI criteria (Cairns et al., 2009). This case is not a false-negative when conventional criteria are used as comparator but could be regarded as such if the diffuse plaque count is used as comparator, as is the case in the Katchaturian criteria (Cairns et al., 2009).
Part of the mismatch between the amyloid scan and the neuropathological diagnosis can be attributed to the way neuropathologically definite AD is defined: The regions sampled in the CERAD protocol do not strictly match the regions of predilection of amyloid ligand retention. Furthermore, in the NIA-RI criteria NFTs play an important role which are obviously not detected by amyloid imaging. Some neuropathologically definite PIB-negative AD cases however have neuritic plaques that seem ‘11C-PIB refractory’ (Rosen et al., 2010). For yet unknown reasons, 11C-PIB may have a low affinity for specific and rare types of neuritic plaques (Leinonen et al., 2008). Other such examples are the plaques occurring in the Arctic mutation carriers (Schll et al., 2012).
4.3. Affinity for white matter lesions
White matter lesions have a highly heterogeneous neuropathological basis. White matter lesions due to cerebral amyloid angiopathy (CAA) would be expected to bind amyloid ligands given the affinity of 11C-PIB for CAA (Bacskai et al., 2007; Dhollander et al., 2011; Dierksen et al., 2010; Johnson et al., 2007; Lockhart et al., 2007). PIB also binds to β sheet structures contained in myelin, e.g. myelin basic protein. This can lead to a focal white matter signal decrease in demyelinating lesions in multiple sclerosis (Stankoff et al., 2011). In such inflammatory conditions it may be important to model changes in blood–brain-barrier permeability that are associated with the disease as this may affect tracer kinetics. The same could possibly be true for white matter lesions due to vascular disease.
4.4. Summary
Continuous measures of ‘amyloid plaque area’ highly correlate with amyloid ligand retention values (Clark et al., 2011, 2012; Driscoll et al., 2012; Sojkova et al., 2011). The amyloid ligands bind both neuritic and diffuse plaques, although with higher affinity for neuritic than for diffuse plaques (Ikonomovic et al., 2012). The three main explanations for mismatches between binary results from amyloid ligand scans and neuropathological CERAD or NIA-RI classification are:
-
1.
Binding of the amyloid ligand to diffuse plaques (Lockhart et al., 2007) in cases where diffuse plaques are abundant and neuritic plaques sparse or absent.
-
2.
‘PIB refractory cases’ where affinity of the amyloid ligand for neuritic plaques appears to be much lower for as yet poorly understood reasons.
-
3.
A sampling bias that is inherent to the CERAD criteria and where the regions selected for neuropathological diagnosis do not match those that typically show high ligand retention in vivo (such as posterior and anterior cingulate and precuneus) (Driscoll et al., 2012).
Overall, the mismatches may be of high scientific interest but among the total group of amyloid PET scans performed, they probably represent only a minority of cases as evidenced by the prospective phase 3 neuropathological studies (Clark et al., 2011, 2012).
5. The AD puzzle: putting the pieces together by means of multimodal imaging
Let us turn to the relationship between amyloid imaging and other in vivo measures of AD pathology that are in clinical use (including cognitive assessment, structural and functional MRI or FDG PET), cross-sectionally and longitudinally. If correlations are calculated between ligand retention levels and cognitive scores across diagnostic groups (patients and cognitively intact controls), correlations will be almost certainly found but such correlations may be indirect: For example, as AD patients have lower cognitive scores than controls and also higher Aβ load, a correlation of cognitive scores with Aβ load across groups will be easily found. For a discussion of related issues in subjects with subjective cognitive impairment (SCI) (see Chételat et al., 2013).
5.1. Topography
Different brain regions have intrinsically different susceptibilities to different pathological expressions of AD (Jack et al., 2008; La Joie et al., 2012). A relatively consistent spatiotemporal pattern of volume loss has been described using structural MRI (Baron et al., 2001; Becker et al., 2011; Rombouts et al., 2000; Whitwell et al., 2008). According to this ‘AD signature’ (Dickerson et al., 2009), most severe cortical thinning occurs in rostral medial temporal and anterior temporal cortex extending posteriorly along the middle temporalgyrus, inferior parietal cortex, temporoparietal junction (TPJ), ventral premotor cortex and the precuneus and posterior cingulate (Dickerson et al., 2009). The typical topography of hypometabolism in early-stage Alzheimer's disease versus normal controls encompasses posterior cingulate and precuneus, temporoparietal association cortex, prefrontal and ventromedial frontal cortex, with sparing of primary sensory and motor cortex, basal ganglia and thalami (Herholz et al., 2002; Mosconi et al., 2008).
The regional distribution of 11C-PIB retention only partly overlaps with these well-known patterns of brain volume loss and hypometabolism. The overlap between 11C-PIB increase and volume loss mainly occurs in precuneus and lateral temporoparietal association cortex in controls, amnestic MCI and AD (Jack et al., 2008). In these regions, there is also concordance between Aβ deposition and hypometabolism (La Joie et al., 2012; Shin et al., 2010). In hippocampus, however, there is a discordance between volume loss and amyloid ligand retention: volume loss is pronounced with relatively little Aβ deposition and limited hypometabolism. A third pattern is seen in dorsolateral prefrontal cortex, with high ligand retention and limited volume loss or hypometabolism (Jack et al., 2008; La Joie et al., 2012).
Within the MCI or AD group, global 11PIB-retention levels do not correlate with structural volume loss (Bourgeat et al., 2010; Chételat et al., 2010). At the single-case level, several cases show dissociations between the presence of cortical 11C-PIB retention and hippocampal volume loss, in both directions (Jack et al., 2008; Vandenberghe et al., 2013).
5.2. Relationship with disease severity
In AD, a structural MRI-based linear model of medial temporal, inferior temporal and inferior frontal regions best predicts clinical severity, as measured by the Clinical Dementia Rating scale—Sum of Boxes (Dickerson et al., 2009). The pattern of hypometabolism also correlates with the type (Engler et al., 2006) and severity (Herholz et al., 2002; Jagust et al., 2009; Rabinovici et al., 2010) of clinical deficits in AD. In contrast, the correlation between PET measures of Aβ deposition and cognitive scores is weak or absent within the group of clinically probable AD subjects (Engler et al., 2006; Jagust et al., 2009; Rabinovici et al., 2010). Within the MCI group, episodic memory scores are lower in the amyloid-positive than the − negative MCI cases (Rowe et al., 2010; Wolk et al., 2009). If both hippocampal volume and 11C-PIB retention values are entered into a regression model in 39 11C-PIB-positive MCI cases and 17 healthy controls, only hippocampal volume make an independent contribution to episodic memory scores (Mormino et al., 2009).
5.3. Longitudinal change
In one of the first multimodal imaging studies with longitudinal amyloid imaging in controls, MCI and AD (Jack et al., 2009), ventricular expansion rate measured with MRI differed between groups (controls 1.3 cm3/yr, MCI 2.5 cm3/yr, AD: 7.7 cm3/yr). This correlated with clinical decline (Jack et al., 2009). In contrast, in controls, MCI and AD, amyloid ligand retention only showed a small increase over time in the absence of between-group differences (Jack et al., 2009) and without any relationship to cognitive decline (Fouquet et al., 2009; Furst et al., 2012; Landau et al., 2011). In MCI in particular, there was a wide variability in the amount of increase in amyloid ligand retention and in some cases the rate of change was higher than in any of the two other groups (Jack et al., 2009). Longitudinal studies based on FDG-PET have demonstrated widespread metabolic changes in MCI and AD, affecting, among other regions, posterior cingulate, precuneus, and medial temporal cortex (Chen et al., 2010; Fouquet et al., 2009). These changes correlate with longitudinal cognitive decline (Fouquet et al., 2009; Furst et al., 2012; Landau et al., 2011).
Other studies have confirmed that the longitudinal change in Aβ deposition in AD and MCI is relatively small. For instance, in both AD and controls, a 3–4% increase in 11C-PIB uptake over a two-year period was restricted to medial frontal cortex (Scheinin et al., 2009). In a study with 3–5 years follow-up, 11C-PIB uptake increased in MCI but remained stationary in the AD group, be it with considerable inter-individual heterogeneity (Kadir and Nordberg, 2010). One of the largest longitudinal studies reported so far included 103 controls, 49 MCI patients and 32 AD patients (Villain et al., 2012). There was a small but noticeable increase in amyloid ligand retention over time, even in the clinical stage. Higher rates of increase in Aβ deposition were seen with higher baseline levels, regardless of the group, leading to the novel concept of 11C-PIB accumulators and non-accumulators (Villain et al., 2012). This resonates with the basic neurobiological idea of self-propagation of Aβ deposition (Lee et al., 2010). The amount of increase in amyloid ligand retention was regionally dependent: At baseline, precuneus and temporoparietal junction showed the highest levels while the rate of change was most pronounced in posterior temporal cortex (see also Chételat et al., 2013). Between-subject variability, however, was high, which is also apparent from visual inspection of the data (Villain et al., 2012). Besides Aβ load at baseline, other baseline factors that have been associated with a higher increase in Aβ load over time in AD are APOE ϵ4 status (Grimmer et al., 2010) and vascular white matter lesions (Grimmer et al., 2012). The latter effect has been attributed to reduced Aβ clearance (Grimmer et al., 2012).
Driscoll et al. (2011) evaluated whether longitudinal changes in structural MRI over the preceding 10 years differed between amyloid-positive versus amyloid-negative non-demented cases. No such correlation was seen at the global or the regional level, except for a trend in the precuneus (Driscoll et al., 2011). This paper therefore shows a within-subject dissociation between MRI structural volume loss estimated over a 10-year period and amyloid ligand retention levels. In MCI, Tosun et al. (2011) performed a parallel independent component analysis of baseline amyloid scans and brain atrophy rates over the subsequent year. The main components consisted of increased amyloid levels in medial parietal regions together with higher medial temporal atrophy rate (Tosun et al., 2011).
5.4. Comparison of diagnostic performance
From a clinical perspective, diagnostic and prognostic performance at the individual level is the critical parameter. For instance, while group comparisons robustly show more pronounced hippocampal volume loss in AD patients versus controls, at the individual level there is considerable overlap in hippocampal volumes between AD and healthy controls (Jack and Petersen, 2000).
When the diagnostic performance of 18F-flutemetamol PET and structural MRI is compared at the individual level in AD, MCI and controls using machine learning techniques, overall sensitivity was comparable between the two diagnostic techniques when the clinical diagnosis was used as comparator (85.2%) (Vandenberghe et al., 2013). Specificity however of the 18F-flutemetamol based classifier (92%) was higher than that of the gray matter based classifier (68%) (Vandenberghe et al., 2013). The gray matter based classifier classified more scans from healthy controls as pathological than the 18F-flutemetamol based classifier (Vandenberghe et al., 2013). This could be partly explained by the overlap in hippocampal volumes between the AD group and the controls (Duara et al., 2012; Thurfjell et al., 2012). One of the regions that best discriminated between Alzheimer's disease and normal controls for 18F-flutemetamol according to the classifier's feature weights was the striatum, mainly in its anterior portion (Vandenberghe et al., 2013). Striatum is involved in AD-related β amyloidosis from Thal stage 3 onwards (see above) (Thal et al., 2002). Early neuropathological studies have revealed the presence of amyloid plaques in striatum (Brilliant et al., 1997; Gearing et al., 1993; Rudelli et al., 1984), in particular the ventral striatum (Suenaga et al., 1990). The plaques in dorsal and ventral striatum are immunochemically distinct from cortical plaques and also differ between dorsal and ventral striatum, with relatively more diffuse plaques in dorsal striatum and more dystrophic neurites in association with the plaques in ventral striatum (Suenaga et al., 1990). Possibly, the latter pattern may relate to the connections of ventral striatum with mesolimbic and allocortical regions and with orbitofrontal cortex (Aggleton et al., 1987; Cavada et al., 2000; Haber et al., 1990), structures that are affected early in the AD course by neuronal loss and tau pathology.
It is not common clinical practice in radiology to provide binary classifications of MRI scans in relation to AD. Binary reads, however, of FDG-PET scans are performed by nuclear medicine physicians on a daily basis and it is therefore of clinical relevance to compare the diagnostic accuracy of binary classification of FDG-PET with that of amyloid PET. When clinical diagnosis (AD versus healthy controls) is used as comparator, visual readings as well as Receiver Operating Characterics (ROC) analysis show higher accuracy for 11C-PIB than for FDG-PET (90% vs 70% for the reads, 95% and 83% for the ROC) (Ng et al., 2007). Higher sensitivity and specificity of 11C-PIB than FDG-PET was also seen in a study by Devanand et al. (2010). Values were compared at a global level and also for the regions that showed highest diagnostic accuracy per technique (precuneus for 11C-PIB and parietal cortex for FDG-PET). Sensitivity and specificity of global 11C-PIB retention levels were 94% while sensitivity of mean regional cerebral metabolic glucose rate (CMRGlu) was 81% and specificity 71%. In precuneus, sensitivity and specificity of 11C-PIB was 94%, while sensitivity and specificity of rCMRGlu in parietal cortex was 87% and 88%, respectively (Devanand et al., 2010). In MCI, discordances between 11C-PIB PET and FDG-PET were more marked and 11C-PIB PET had higher diagnostic accuracy for the distinction between healthy controls and MCI than FDG-PET (Devanand et al., 2010).
When clinically probable AD patients (n = 62) were compared with FTLD patients (n = 45, including behavioral variant frontotemporal dementia and nonfluent and semantic variants of primary progressive aphasia (PPA)) and the clinical diagnosis was used as standard-of-truth, sensitivity of visual reads of 11C-PIB scans for AD (89%) was higher than that of FDG-PET (73%), with similar specificity (83–84%) (Rabinovici et al., 2007, 2008, 2011). Interrater agreement of the visual reads was higher for 11C-PIB (Fleiss κ 0.96) than for FDG-PET (Fleiss κ 0.72) (Rabinovici et al., 2011). Accordingly, when neuropathology is used as gold standard, unanimity in the overall interpretation of FDG-PET scans between readers for discriminating AD from FTLD was relatively low, in particular for the scans from FTLD cases (unanimity in 7 out of 14 cases, compared to 27 out of 31 AD cases) (Womack et al., 2011). With quantitative analysis, PiB had a higher sensitivity (89% vs 73%) but FDG a higher specificity (83% vs 98%) (Rabinovici et al., 2011). The higher specificity probably relates to the occurrence of positive amyloid scans even in cognitively intact older adults. High interrater agreement (Rabinovici et al., 2011) together with higher sensitivity (Rabinovici et al., 2011) may be important clinical advantages that amyloid PET would bring compared to FDG PET in the differential diagnosis between AD and FTLD, although further studies with neuropathological diagnosis as comparator will be required.
5.5. Task-related fMRI versus amyloid PET
Task-related fMRI can reveal adaptive changes in cognitive brain circuits during cognitive processing which may provide resilience against the functional impact of amyloid pathology. Brain resilience and functional reorganization may gain importance in the field as AD therapies may increasingly _target multiple facets of AD. To the best of our knowledge, two studies combined amyloid PET and task-related fMRI within the same subjects in MCI or AD.
The first such study focused on changes in the language circuit in early-stage clinically probable AD and how this related to amyloid ligand retention (Nelissen et al., 2007). This study built on a preceding study in amnestic MCI: in amnestic MCI, the earliest changes in the language network occur in the posterior part of the left superior temporal sulcus and these changes correlate with subclinical changes in written word identification speed, in line with the critical role of this region in lexical-semantic retrieval (Vandenbulcke et al., 2007). In early-stage AD, hypoactivity is also seen in left posterior temporal cortex during associative-semantic versus visuoperceptual processing. These fMRI changes in the left posterior STS in AD correlate with offline measures of confrontation naming: clinically probable AD subjects with hypoactivity in this region are impaired on confrontation naming (Nelissen et al., 2007). There is no correlation between 11C-PIB levels in this region and the clinical deficit (Nelissen et al., 2007). fMRI activity levels to the homotopical right side show increased activity in those patients who had preserved naming on the confrontation naming, suggestive of a compensatory increase (Nelissen et al., 2007).
In the episodic memory domain, a combined 11C-PIB-fMRI study in subjects with a Clinical Dementia Rating Scale of 0–0.5 revealed mainly differential activity decreases as a function of Aβ deposition. During encoding of associations between faces and names, the precuneus and temporoparietal cortex, partially overlapping with the posterior nodes of the default mode network, were less deactivated in amyloid-positive subjects compared to amyloid-negative subjects (Sperling et al., 2009). Deactivation in PCC is inversely related to hippocampal activation (Celone et al., 2006).
5.6. Summary
Imaging techniques such as structural MRI and FDG PET reveal the topography of structurally and functionally affected regions at an anatomical level. This correlates with the type and degree of clinical deficit, cross-sectionally and longitudinally. Uniquely, amyloid imaging provides direct information about lesion type, yielding a superior specificity in terms of disease diagnosis. Within the AD group, however, the extent and topographical distribution of Aβ deposition correlates relatively poorly with cognitive profile, severity or progression of the clinical deficit.
6. Amyloid imaging as a road sign in the clinical development path
6.1. Requirements
In clinical trial research, the issues related to evaluating longitudinal change are similar for amyloid imaging as for any other method (e.g. longitudinal neuropsychological assessment):
-
1.
The change over time in the normal population and the normal variance needs to be quantified. This is necessary to determine if any change observed in an individual, either positive or negative, falls within the normal range or not.
-
2.
Test–retest variability of the technique at a short interval must be known: Test–retest variability of 11C-PIB SUVR in AD is 8.0% (SD 7) (Tolboom et al., 2009b), but can be improved to 3% by using dynamic scans with reference tissue methods (Tolboom et al., 2009b). The test–retest variability of the 18F-labeled amyloid ligand is provided in Table 3 (Cselényi et al., 2012; Joshi et al., 2012; Vandenberghe et al., 2010; Villemagne et al., 2011). To detect a drug-induced difference, test–retest variability within the same center needs to be kept as low as possible: this will depend not only on the analysis method (Tolboom et al., 2009b) but also on changes in structural brain volume over time and partial volume effect, motion during scanning etc.
-
3.
Between-center variability should be measured: differences in scanner, reconstruction algorithm etc. may affect retention levels. This is an active area of investigation which is highly relevant for multicenter trials. A European multicenter study of 11C-PIB using different scanners and reconstruction algorithm suggested that the range of values in AD, MCI and controls are relatively comparable between centers (Nordberg et al., 2013).
-
4.
The study drug should not affect the metrics of the technique directly. Both in Rinne et al. (2010) and Ostrowitzki et al. (2012), competition assays did not reveal any alteration of the tracer affinity of amyloid plaques due to the passive immunization.
Table 3.
Test–retest variability of SUVR for 18F-labeled amyloid ligands with cerebellum as reference region in AD patients scanned with an interval less than one month (Cselényi et al., 2012; Joshi et al., 2012; Vandenberghe et al., 2010; Villemagne et al., 2011). Expressed in %. The exact definition of volumes of interest, e.g. the composite cortical VOI, may differ between the studies and the current comparison should therefore be considered an indirect comparison. Dash: not reported.
|
18F-flutemetamol |
18F-florbetapir |
18F-florbetaben |
18F-AZD4694 |
|||||
|---|---|---|---|---|---|---|---|---|
| AD cases | 5 |
10 |
8 |
3 |
||||
| Mean | SD | Mean | SD | Mean | Range | Mean | SD | |
| Composite | 1.5 | 0.7 | 2.40 | 1.41 | 6.2 | 0.6–12.2 | 7.5 | 6.5 |
| Frontal | 1.4 | 0.4 | – | – | – | – | – | – |
| Parietal | 2.1 | 1.8 | – | – | – | – | – | – |
| Lateral temporal | 1.8 | 0.8 | – | – | – | – | – | – |
| Posterior cingulate | 1.2 | 0.5 | – | – | – | – | 7.8 | 5.3 |
| Anterior cingulate | 2.0 | 0.9 | – | – | – | – | – | – |
| Medial temporal | 3.8 | 2.4 | – | – | – | – | – | – |
| Occipital | 0.9 | 0.5 | – | – | – | – | – | – |
| Striatum | 0.9 | 0.5 | – | – | – | – | – | – |
| Subcortical white matter | 3.2 | 2.1 | – | – | – | – | – | – |
| Pons | 3.1 | 2.7 | – | – | – | – | – | – |
The more fundamental question concerns the biological significance of a change in amyloid ligand retention and how this translates into clinical benefit given the fact that the relationship between amyloid ligand retention and clinical expression is limited (see above). In our view, the critical contribution of amyloid imaging to amyloid lowering drug development lies in the selection of study patients and the demonstration of drug _target engagement.
6.2. Patient selection
In most mono- and multicentric diagnostic trials of early-stage clinically probable AD from academic memory clinics, the proportion of AD cases with a negative amyloid PET scan is approximately 10% (Devanand et al., 2010; Nelissen et al., 2007, 2009; Vandenberghe et al., 2010). In other multicenter diagnostic trials these percentages have been as high as 32% (Doraiswamy et al., 2012). In the PET substudy of the bapineuzumab phase 2 therapeutic trial, 8 of the 53 patients screened for inclusion did not meet 11C-PIB criteria (Rinne et al., 2010). It is likely that similar or higher proportions of amyloid-negative cases have entered amyloid-lowering drug trials as these trials most often did not verify amyloid status at inclusion, possibly leading to dismissal of potentially useful drugs. The proportion of false-positive clinical AD diagnoses may further increase as one moves towards the predementia stage, reducing the chance of a positive study outcome. Regardless of the exact mechanism of action of the drug tested, improved patient selection may be one of the principal advantages that amyloid imaging at inclusion will bring. This may restrict the applicability of the drug once it is licensed. Such considerations however lose their validity in the face of the current crisis in AD therapeutic development.
6.3. _target engagement
For drugs that have as a purpose to lower amyloid levels, amyloid imaging also allows one to evaluate _target engagement. If a drug engages its _target but still does not exert a beneficial effect, this is highly informative from a scientific viewpoint, while a negative trial in the absence of any knowledge about _target engagement is open to many more interpretations. Rinne et al. (2010) and Ostrowitzki et al. (2012) reported longitudinal changes during passive immunization with bapineuzumab (a humanized monoclonal antibody) and gantenerumab (a fully human monoclonal antibody), respectively (Table 5). In both studies, the relative small sample size led to imbalances in the 11C-PIB baseline values between groups. In the Ostrowitzki et al. (2012) study, the highest initial SUVR value in the placebo group was 2.3 while in the actively treated groups at least 5 subjects had values above 3, and in two of them between 3.5 and 4 (Ostrowitzki et al., 2012). This mismatch in baseline value makes interpretation of differences in change between groups difficult, in particular since initial 11C-PIB level affects the subsequent rate of increase in 11C-PIB retention (Villain et al., 2012). Furthermore, in the Ostrowitzki et al. (2012) study, the number of infusions received also differed between groups: two of the placebo received all 7 infusions, 6 of the 60 mg group and only 1 of the 200 mg group (1 case only two, 2 cases only 3 infusions, 2 four infusions) (Table 5). From the report it is unclear whether the number of infusions received affected the interval between first and end-of-treatment scan so that there could be a mismatch in duration between cases. The confidence intervals for change to baseline also included zero in all treatment groups (Ostrowitzki et al., 2012). For these reasons one has to be cautious in drawing strong conclusions from these initial studies using 11C-PIB to provide proof of _target engagement by amyloid-lowering drugs. Based on these proof-of-concept studies, one could wonder whether prior stratification based on the biomarker value would help prevent initial mismatches at baseline.
Table 5.
Numerical results from longitudinal trials with passive immunization using 11C-PIB (Ostrowitzki et al., 2012; Rinne et al., 2010).
| Number of cases | Initial SUVR | Observed mean difference | |
|---|---|---|---|
| Bapineuzumab | |||
| Placebo | 9 | 1.89 (0.19) | + 0.15 |
| 0.5, 1 or 2 mg | 19 | 2.06 (0.20) | − 0.09 |
| Ganteneurumab | |||
| Placebo | 4 | 2.18 (0.17) | + 0.23 |
| 60 mg | 6 | 2.86 (0.67) | + 0.02 |
| 200 mg | 6 | 2.86 (0.63) | − 0.27 |
7. Amyloid imaging in clinical practice
While the validation of amyloid PET as a biomarker is relatively advanced, there have been very few studies that directly tested its utility (added value) and cost-effectiveness in diagnosis and patient management in a clinical context.
7.1. Amyloid imaging and the ideal biomarker
So far, clinical development of amyloid imaging markers has mainly focused on validating the imaging measure as a biomarker. Ideally, AD biomarkers have to fulfill a number of requirements (The Ronald and Nancy Reagan Research Institute of the Alzheimer Association, 1998). The biomarker should:
-
1.
Detect a fundamental feature of pathophysiological processes active in AD. There is general consensus that β amyloidosis is a fundamental feature to AD (Hardy and Selkoe, 2002). There however remains debate about the pathological nature of diffuse as opposed to neuritic plaques (Masliah et al., 1990; Morris et al., 1996). The selectivity of amyloid ligands for neuritic as opposed to diffuse plaques is discussed above.
-
2.
Be validated in neuropathologically confirmed cases. As described above, the correlational studies with neuropathology have shown a sensitivity of 90% and specificity of 100% (Clark et al., 2011, 2012). It remains to be seen to which degree this can be extrapolated to the clinical populations in which the test would be applied. These patients would generally be in a much earlier disease stage, when, for instance, the relative proportion of diffuse versus neuritic plaques might be different. Conversely, in advanced cases the scans may be relatively more difficult to classify due to advance cortical atrophy.
-
3.
Be precise, i.e. should detect AD early in its course and distinguish it from other dementias. For instance, in the phase 2 18F-flutemetamol study, in which the prevalence of amyloid-positive healthy controls was relatively low, sensitivity and specificity to discriminate AD from controls with the clinical diagnosis as comparator was 93–96% (Vandenberghe et al., 2010). These values vary markedly between studies (Doraiswamy et al., 2012). This can be partly accounted for by differences in the prevalence of positive scans in the healthy control groups and differences in the prevalence of negative scans in the AD group. High sensitivity and specificity values have also been obtained in the phase 3 clinicopathological studies, which included a variety of non-AD dementias, each in low number (Clark et al., 2011, 2012). A growing number of papers have evaluated diagnostic performance in AD compared to other disease groups, such as Lewy body disease (Edison et al., 2008; Rowe et al., 2007; Villemagne et al., 2011), cerebral amyloid angiopathy (Dhollander et al., 2011; Dierksen et al., 2010; Johnson et al., 2007) and frontotemporal degeneration (Rabinovici et al., 2007, 2008, 2011; Rowe et al., 2007; Villemagne et al., 2011) and determined sensitivity and specificity with the clinical diagnosis as standard-of-truth (Villemagne et al., 2011). The added value compared to clinical diagnosis will only become clear once an independent standard-of-truth becomes available, e.g. through clinicopathological examination.
-
4.
Should be reliable: Test–retest variability is reported in Table 3 and interrater reliability κ in Table 4. For the sake of comparison, the interrater reliability of visual reads of FDG-PET in 20 healthy controls and 15 AD patients was 0.56 according to a comparative study of 11C-PIB and FDG-PET (Ng et al., 2007), but this may improve when statistical maps are used based on the contrast between an individual's scan and a normal database rather than visual reads of native images (Foster et al., 2007; Herholz et al., 2002; Mosconi et al., 2008).
-
5.
Simple to perform and inexpensive: For a health system to be sustainable given the demographic evolution, the cost of a diagnostic technique should be outweighed by the benefit the technique brings in terms of patient's well-being. This benefit does not purely relate to therapeutic benefit but also in terms of communication of a correct diagnosis.
-
6.
Recommended steps to establish a biomarker include confirmation by at least two independent studies from different groups of researchers.
Table 4.
Inter-reader consistency of binary reads (Fleiss' κ). Dash: not reported.
| AD |
MCI |
Controls |
κ | Reference | |
|---|---|---|---|---|---|
| (n) | (n) | (n) | |||
| 11C-PIB | 15 | 25 | 0.90 | Ng et al. (2007) | |
| 18F-flutemetamol | 27 | 20 | 25 | 0.96 | Vandenberghe et al. (2010) |
| 18F-florbetapir | 10 | 10 | 0.76 | Joshi et al. (2012) | |
| 31 | 51 | 69 | 0.58 | Doraiswamy et al. (2012) | |
| 18F-florbetaben | 81 | – | 69 | 0.60 | Barthel et al. (2011) |
7.2. Clinical utility of amyloid imaging
Both amyloid imaging and CSF biomarkers directly reflect one of the underlying neuropathological hallmark lesions of AD. The place that we will define for these biomarkers in our tentative diagnostic algorithm will be generally applicable to in vivo measures of AD-related amyloidosis, be it by means of PET or CSF (Fig. 1). The difference in utility between imaging and biochemical biomarkers will lie in the metrics of the tests, i.e. between-center and within-center replicability, the prevalence of intermediate values, and the between-reader variability in diagnostic interpretation of the values for amyloid imaging and CSF, respectively. It has been shown that binary assignment of cases based on 11C-PIB uptake into positive and negative cases closely corresponds to values of Aβ42 in cerebrospinal fluid but not with any of the other CSF variables such as total tau or Aβ40 (Fagan et al., 2006). From the viewpoint of implementation in a clinical environment, CSF biomarkers still have to go through the standardization steps that other diagnostic laboratory tests, e.g. for measuring protein levels in blood, have gone through (Bartlett et al., 2012; Mattsson et al., 2012). The values measured for a same sample vary between centers and the interpretation of a given CSF profile may also be center- and examiner-dependent. The factors that contribute to the between-center variability of CSF measurements are relatively poorly understood. In this sense, the strength of amyloid imaging compared to CSF at the time of writing seems to lie in its performance in terms of replicability between and within centers, between-reader replicability and standardization (see above).
Fig. 1.
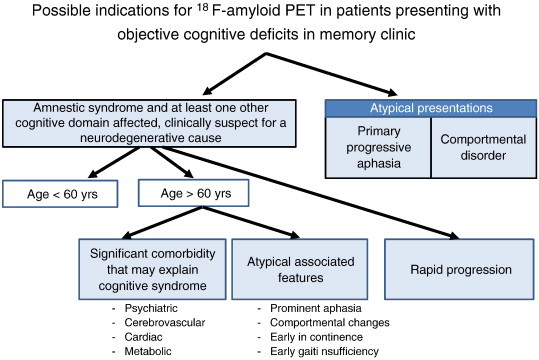
Tentative clinical algorithm for the utility of amyloid imaging in patients presenting at a memory clinic with a cognitive deficit that is confirmed by clinical assessment of cognitive functions or by neuropsychological evaluation. This algorithm is tentative and subject to further empirical evaluation.
The occurrence of AD neuropathology in cognitively intact individuals (see side-to-side review by Chételat et al. (2013)) necessarily puts an upper limit to the maximal specificity the technique can reach in comparison with healthy controls. The occurrence of a positive scan in a patient who presents at the memory clinic with cognitive complaints may reflect the underlying cause of cognitive dysfunction or, alternatively, may constitute a coincidental finding given the high prevalence of positive scans even in cognitively intact older adults in the higher age range (see Chételat et al. (2013)).
A second clinically relevant issue is the prevalence of ‘intermediate values’ (see also side-to-side review by Chételat et al. (2013)). This is a classical problem in diagnostic tests, which also exists for CSF and for FDG-PET. The number of cases within the gray zone will depend on the population tested and may differ substantially between modalities.
The added value of biomarkers for cerebral amyloidosis depends on:
-
1.
The clinical context in which a diagnosis is made: primary care, neurology/psychiatry/geriatrics practice, academic memory clinic, … Different clinical contexts are associated with differences in a priori probability of Alzheimer's disease and non-AD dementias, age differences, differences in a priori probability of comorbidity or reversible cognitive causes, etc. These differences may affect the accuracy of a clinical AD diagnosis and the clinical impact of an additional biomarker measurement.
-
2.
The healthcare system in which amyloid imaging is implemented. Factors that play a role and that are outside the scope of the current review are the degree of equality of access to healthcare that a society wishes to guarantee and budgetary restraints that have to be put in place to be able to provide this guarantee. Budgetary restraints will determine the cost-benefit ratio that is required for granting reimbursement of the biomarker.
-
3.
The societal perception of Alzheimer's disease. The latter determines the emotional impact of an AD diagnosis on the individual's wellbeing in the different stages of the disease course.
Paradoxically, the added value of a biomarker for β amyloidosis may be higher in a clinical context where clinical expertise is relatively limited. Under such circumstances, the added value and cost-benefit of amyloid imaging would have to be compared with corrective measures directed at enhancing knowledge and clinical skills in this domain at different healthcare levels. Nevertheless, even in centers of excellence false-positive diagnoses of AD in the early disease stage may be more frequent than previously assumed. For instance, awareness is increasing that FTLD may present initially with an amnestic syndrome that is hardly distinguishable from what is seen in AD (Hornberger et al., 2012; Pao et al., 2011).
The role of clinical context is also reflected by the NIA-AA criteria (Albert et al., 2011; McKhann et al., 2011). One set of clinical criteria is intended for use in all clinical settings. A separate set of criteria refers to ‘probable or possible AD dementia or mild cognitive impairment due to AD with low, high or intermediate level of evidence of the AD pathophysiological process’. A high level is defined as a concordance between markers for Aβ and markers of neuronal injury. The latter set is for use in three circumstances: investigational use, clinical trials (Section 5), and as optional clinical tools for use where available and when deemed appropriate by the clinician (McKhann et al., 2011). The topic of the remainder of this review is to delineate a number of circumstances where the use of biomarkers may be appropriate in clinical practice in our opinion.
The main clinical situations that we discern are predictive value in MCI and differential diagnosis of AD under specific clinical circumstances.
7.3. Predictive value in MCI
Approximately 50% of MCI patients have a positive amyloid scan (Okello et al., 2009; Pike et al., 2007; Vandenberghe et al., 2010; Wolk et al., 2009), with highest proportions (89%) in the multidomain amnestic MCI subtype (Wolk et al., 2009). It has been suggested that in MCI the amyloid ligand retention values have a bimodal distribution, but currently, to the best of our knowledge, there is no statistical proof of such a bimodal distribution (Mormino et al., 2009). A positive amyloid PET has a predictive value for future conversion to Alzheimer's disease according to early studies with follow-up periods up to 3 years (Forsberg et al., 2008; Nordberg et al., 2013; Okello et al., 2009). In such studies, the clinician who decides about conversion to clinically probable AD ought to be blinded to the result of the initial PET scan. The interval over which MCI patients may convert to AD can be relatively long and a positive scan in a subject who has not converted after a 3 or 5 year interval is not necessarily a false-positive. It is also of clinical importance to note that a negative scan in a case with amnestic MCI does not preclude progression. Other diseases not associated with brain amyloidosis may cause an amnestic syndrome, such as frontotemporal degeneration, hippocampal sclerosis, argyrophilic grain disease or tangle-only AD. The risk for ‘conversion’ in such cases is probably as high as for MCI due to AD. A final point relates to the clinical utility of prediction in MCI patients in the absence of efficacious therapy. Ethically, it is important to avoid situations where the patient knows more than what he or she would consider desirable or beneficial. Such situations could for instance be patients with a diagnosis of amnestic MCI in whom acetylcholinesterase inhibitors are not of proven benefit and who would receive an earlier AD diagnosis due to the diagnostic advances.
In the phase 2 18F-florbetapir study (Doraiswamy et al., 2012), 51 MCI patients participated along with 69 healthy controls and 31 AD patients. 37% of MCI patients were amyloid positive (Doraiswamy et al., 2012). Forty-six of the MCI patients were followed for 18 months. This is one of the few studies that explicitly mentions blinding of the evaluator. Of the amyloid positive MCI subjects (n = 17) 5 progressed in an 18-months follow-up period. Of those who were amyloid negative (n = 29), 3 progressed. This study however suffers from a relatively low sample size, short follow-up time, and a relatively poor interrater reliability of the visual reads. Furthermore, the low prevalence of amyloid positivity in the AD group raises concerns about the clinical diagnostic accuracy prior to inclusion also in the MCI group.
7.4. Differential diagnosis
Accessibility of amyloid imaging in clinical practice will probably precede the evidence for clinical utility based on strict evidence-based medicine criteria. In our generic algorithm for use of biomarkers in an academic memory clinic, the indications for additional biomarkers are based on the a priori probability of Alzheimer's disease in a given individual and the level of diagnostic certainty that one wishes to obtain.
We discern four situations where biomarker evidence may contribute to the differential diagnostic process in a way that is relevant for the patient and the caregivers concerned:
-
1.
Early-onset dementia (onset before 60 years of age), in the absence of a known genetic mutation in the family: in early-onset dementia, the a priori probability of AD as a cause of cognitive decline is equal to that of FTLD. Furthermore, given the impact of the diagnosis that is communicated, a high level of diagnostic certainty is desirable (Hodges, 2001). The impact of a correct and early diagnosis in young-onset dementia is situated at the personal and familial emotional and relational level, and also at the level of third-party payers who have to make decisions on the strength of evidence for an underlying neurological cause.
-
2.
Atypical presentations such as PPA or predominant comportmental disorder. Although detailed neurolinguistic phenotyping allows to estimate the probability of AD as an underlying cause of PPA (Gorno-Tempini et al., 2011), biomarker evidence may be very helpful. In an early stage of the disease, subtyping of individual cases may sometimes be difficult and may require a high level of neurolinguistic expertise that may not be widely available. Even with adequate clinical subtyping, the relationship between clinical phenotype and underlying pathophysiological substrate remains probabilistic, meaning that inferring the underlying lesion type based on the clinical phenotype will always be liable to a substantial degree of uncertainty (Mesulam, 2000). Furthermore, when the distribution of the AD pathology is atypical, imaging measures that reflect topography will provide less diagnostic information than evidence based on biomarkers measuring the underlying lesion type.
-
3.Cases where concomitant pathology could explain the cognitive decline but a diagnosis of possible AD is also entertained.
-
•A high vascular load on structural MRI does not exclude the concomitant presence of AD. In a study of 45 cases with subcortical vascular disease in the absence of large-vessel stroke or macrohemorrhage, approximately one third of cases were 11C-PIB positive (Lee et al., 2011). The amount of white matter hyperintensities correlates with 11C-PIB based measures of Aβ load in patients with clinically probable CAA but not in AD (Gurol et al., 2012).
-
•Cases with a long-standing psychiatric history of depression may develop Alzheimer's disease and it may be impossible to make a diagnosis of clinically probable AD under such conditions. A false-negative diagnosis will lead to poor caregiver comprehension of the drastic changes that occur over the more recent period of time and it is important to disentangle the psychiatric origin from any acquired neurodegenerative contribution. A false-positive diagnosis would also have serious consequences in this vulnerable population.
-
•Medical comorbidity such as chronic renal insufficiency, heart failure or obstructive pulmonary disease, to the degree that the presence of AD would impact on patient management in such cases (e.g. candidates for transplantation surgery).
-
•Normal-pressure hydrocephalus: Among 433 patients with a clinical diagnosis of NPH based on neurological and radiological criteria, half of whom received a ventriculoperitoneal shunt, 20% showed cognitive decline over the following years. The presence of Aβ alone or in combination with hyperphosphorylated tau at the time of shunting was a strong predictor for subsequent cognitive decline. In such cases amyloid imaging may increase preoperative diagnostic accuracy and contribute to the decision making with regards to a surgical intervention (Leinonen et al., 2010).
-
•
-
4.
Rapidly progressive cognitive deterioration: There is no strict definition of ‘rapidly progressive AD’: an interval from clinical onset to death of less than two years would be a reasonable definition but is essentially retrospective. Besides rapidly progressive AD, the differential diagnosis of rapidly progressive dementia includes conditions such as Lewy body dementia and Creutzfeldt Jakob but also potentially reversible causes such as paraneoplastic or non-paraneoplastic autoimmune encephalitis, central nervous system lymphoma or angiitis. To reach a high level of confidence that reversible causes of rapid progression are excluded, hospitalization and costly diagnostic investigations will often be required. Evidence for the presence or absence of cerebral amyloidosis may help direct the investigations. Of course, under such dramatic conditions, biomarkers for cerebral amyloidosis will not replace other tests such as diffusion-weighted MRI or cerebrospinal fluid assays. The added value of biomarkers for cerebral amyloidosis in rapidly progressive dementia is an important topic for further investigation which will probably require a multicenter approach given the relative rarity of this syndrome.
Under such conditions the clinical utility of amyloid imaging has to be weighed against the clinical utility of other tools such as FDG-PET and CSF biomarkers. Structural MRI will likely retain its place in the diagnostic work-up, for exclusion of neoplastic lesions (basal forebrain tumors, non-Hodgkin lymphoma, low-grade glioma of medial temporal cortex…) and for assessment of the vascular lesion load. A high vascular load may direct diagnostics and therapeutics into the vascular risk prevention strategies. Also, if disease-modifying drugs are introduced into the clinic, MRI Gradient Echo sequence may allow to detect cerebral amyloid angiopathy which is associated with a higher risk for Amyloid-Related Imaging Abnormalities (Sperling et al., 2011), as seen in most passive immunization trials.
Under the conditions outlined above, the benefit of amyloid imaging is a benefit in terms of diagnostic clarity. This should not be under-estimated as under the abovementioned conditions, accurate diagnosis determines patient management. It is also one of the principal questions by patients and caregivers to the physician. It is hard to quantify this benefit by monetary values and the benefit of accurate diagnosis early in the disease course is a complex issue. Furthermore, with most other diagnostics (e.g. 123I β-CIT SPECT in Parkinson's disease), there is a clear cost-effectiveness based on appropriate therapy choices, which is difficult for dementia when therapeutic options are still limited.
8. The multidimensional space of Alzheimer's disease
According to one of the most influential current AD models (Jack et al., 2010, 2013), CSF alterations in Aβ42, amyloid aggregation, MRI volume loss and cognitive decline follow an orderly temporal sequence, initiated by changes in amyloid and culminating in the clinical expression of cognitive symptoms. This model has a high heuristic value and has engendered a number of testable hypotheses from which a clearer picture of the time-dependent trajectories of AD biomarkers relative to clinical disease stage and to each other will be derived (Jack et al., 2010). This model could be viewed as a translation of the amyloid cascade hypothesis (Hardy and Selkoe, 2002) into a sequence of changes that are measurable in humans in vivo. Inherent to the amyloid cascade hypothesis is a linear sequence of events that is initiated by an amyloid related change.
Alternatives exist to such linear sequential models. For instance, the different in vivo imaging modalities may reflect partly independent events. According to that scenario, AD would have to be depicted in a multidimensional space where different measures co-determine cognitive outcome rather than as a linear sequence from Aβ42 to cognitive symptoms. The term ‘multidimensional space’ emphasizes the partial independence of the different in vivo measures of Alzheimer's disease. It refers to the multidimensional scaling of sets of parameters that is often used when one wants to discern separate clusters within complex datasets. The multidimensional space of Alzheimer's disease relates to the concept of multifactorial AD but remains more at the level of description of the complex relationship between different features while the term multifactorial refers more to the factors that contribute to AD, such as neuropathological core lesions or concomitant Lewy bodies (see above).
A multidimensional view fits with recent genetic evidence in sporadic AD (Lambert et al., 2009; Sleegers et al., 2010): AD can be regarded as a network of multiple pathways with multiple hubs that may separately and interactively result in a given behavioral phenotype. The multidimensional model incorporates the partial independence between processes that we can measure in vivo by means of the different techniques, in particular between amyloid ligand retention levels and volume loss (Driscoll et al., 2012; La Joie et al., 2012). There is also neuropathological evidence for partial independence: Synapse loss in dentate gyrus does not correlate with the amount of NFT (Scheff et al., 2006) and volume loss does not correlate with amyloid load or NFT (Driscoll et al., 2012). Such multidimensional models resonate with the multifactorial model of AD (Savva et al., 2009) and are distinct from an amyloidocentric linear sequential model. The way in which different measures combine may give rise to clusters or patterns that characterize a specific subtype of patients. The multidimensional space therefore not only allows for partial independence between measures but also for inter-individual heterogeneity.
The distinction between a linear sequential model and a multidimensional model hinges on the relative independence of the different processes that can be imaged with current techniques. At the group level the evidence for a probabilistic relationship between brain volume loss, presence of amyloid aggregates, and zones of hypometabolism is strong. At the regional and at the individual level, the interdependence between these measures may be more variable. The distinction between a multidimensional and parallel model of AD versus a linear model bears resemblance to the distinction between serial box-and-arrow models of cognitive function and parallel distributed models. As is the case in such neuropsychological models, linear models have a strong heuristic advantage as they engender clearly defined hypotheses that can be empirically tested. Such models have contributed a lot to our understanding of how cognitive processes are related to each other. Multidimensional models however may allow one to capture the complexity of the brain's operations in a more truthful manner. Empirical data obtained from multimodal longitudinal studies will have to determine which of the two types of models is correct.
9. Conclusion
Amyloid imaging provides a direct window on one of the components of AD. This component stands in a relatively complex relationship to a diversity of other component processes. Only some of these processes can be imaged and there is an ongoing search for techniques to detect and quantify in vivo some of the other pathogenetic mechanisms, such as glial cell involvement (Cagnin et al., 2001) or tau hyperphosphorylation (Fodero-Tavoletti et al., 2011; Okamura et al., 2005). Potential key early aspects of AD pathophysiology, such as Aβ oligomers, remain below the radar of current biomarker detection capabilities (Jack et al., 2013). Amyloid imaging provides hope for progress as it allows for direct measurement of one component contributing to AD-related cognitive decline. This can enhance the chance of success of trials by allowing to restrict inclusion to those patients who have the _target of interest and by showing whether the drug engages the _target or not. While amyloid imaging is already being introduced to the market, many gaps can be identified in our evidence-based medical knowledge of its role in clinical practice. These gaps will have to be filled over the years to come by studies of clinical utility and added value.
Acknowledgments
Supported by FWO grants G.0076.02 (R.V.), KU Leuven Research grants OT/08/056 and OT/12/097 (R.V.), Federaal Wetenschapsbeleid belspo Inter-University Attraction Pole P6/29 and P7/11, and Stichting Alzheimer Onderzoek grant 11020, R.V. and K.V.L. are senior clinical investigators of the Fund for Scientific Research, Flanders, Belgium (FWO), and K.A. is a doctoral research fellow of the Fund for Scientific Research, Flanders, Belgium (FWO).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- Aggleton J.P., Friedman D.P., Mishkin M. A comparison between the connections of the amygdala and hippocampus with the basal forebrain in the macaque. Experimental Brain Research. 1987;67:556–568. doi: 10.1007/BF00247288. [DOI] [PubMed] [Google Scholar]
- Albert M.S., DeKosky S.T., Dickson D., Dubois B., Feldman H.H., Fox N.C., Gamst A., Holtzman D.M., Jagust W.J., Petersen R.C., Snyder P.J., Carrillo M.C., Thies B., Phelps C.H. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the national institute on aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers & Dementia. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bacskai B.J., Frosch M.P., Freeman S.H., Raymond S.B., Augustinack J.C., Johnson K.A., Irizarry M.C., Klunk W.E., Mathis C.A., DeKosky S.T., Greenberg S.M., Hyman B.T., Growdon J.H. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Archives of Neurology. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- Bancher C., Braak H., Fischer P., Jellinger K.A. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neuroscience Letters. 1993;162:179–182. doi: 10.1016/0304-3940(93)90590-h. [DOI] [PubMed] [Google Scholar]
- Baron J.C., Chételat G., Desgranges B., Perchey G., Landeau B., de la Sayette V., Eustache F. In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer's disease. NeuroImage. 2001;14:298–309. doi: 10.1006/nimg.2001.0848. [DOI] [PubMed] [Google Scholar]
- Barthel H., Gertz H.-J., Dresel S., Peters O., Bartenstein P., Buerger K., Hiemeyer F., Wittemer-Rump S.M., Seibyl J., Reininger C., Sabri O. Cerebral amyloid-β PET with florbetaben (18f) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurology. 2011;10:424–435. doi: 10.1016/S1474-4422(11)70077-1. [DOI] [PubMed] [Google Scholar]
- Bartlett J.W., Frost C., Mattsson N., Skillbäck T., Blennow K., Zetterberg H., Schott J.M. Determining cut-points for Alzheimer's disease biomarkers: statistical issues, methods and challenges. Biomarkers in Medicine. 2012;6:391–400. doi: 10.2217/bmm.12.49. [DOI] [PubMed] [Google Scholar]
- Becker J.A., Hedden T., Carmasin J., Maye J., Rentz D.M., Putcha D., Fischl B., Greve D.N., Marshall G.A., Salloway S., Marks D., Buckner R.L., Sperling R.A., Johnson K.A. Amyloid-β associated cortical thinning in clinically normal elderly. Annals of Neurology. 2011;69:1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeat P., Chételat G., Villemagne V.L., Fripp J., Raniga P., Pike K., Acosta O., Szoeke C., Ourselin S., Ames D., Ellis K.A., Martins R.N., Masters C.L., Rowe C.C., Salvado O., and the AIBL Research Group Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology. 2010;74:121–127. doi: 10.1212/WNL.0b013e3181c918b5. [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brilliant M.J., Elble R.J., Ghobrial M., Struble R.G. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer's disease. Neuropathology and Applied Neurobiology. 1997;23:322–325. [PubMed] [Google Scholar]
- Brookmeyer R., Gray S., Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. American Journal of Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookmeyer R., Johnson E., Ziegler-Graham K., Arrighi H.M. Forecasting the global burden of Alzheimer's disease. Alzheimers & Dementia. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- Burack M.A., Hartlein J., Flores H.P., Taylor-Reinwald L., Perlmutter J.S., Cairns N.J. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology. 2010;74:77–84. doi: 10.1212/WNL.0b013e3181c7da8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin A., Brooks D.J., Kennedy A.M., Gunn R.N., Myers R., Turkheimer F.E., Jones T., Banati R.B. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Cairns N.J., Ikonomovic M.D., Benzinger T., Storandt M., Fagan A.M., Shah A.R., Reinwald L.T., Carter D., Felton A., Holtzman D.M., Mintun M.A., Klunk W.E., Morris J.C. Absence of Pittsburgh Compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Archives of Neurology. 2009;66:1557–1562. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavada C., Compay T., Tejedor J., Cruz-Rizzolo R.J., Reinoso-Surez F. The anatomical connections of the macaque monkey orbitofrontal cortex. a review. Cerebral Cortex. 2000;10:220–242. doi: 10.1093/cercor/10.3.220. [DOI] [PubMed] [Google Scholar]
- Celone K.A., Calhoun V.D., Dickerson B.C., Atri A., Chua E.F., Miller S.L., DePeau K., Rentz D.M., Selkoe D.J., Blacker D., Albert M.S., Sperling R.A. Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. Journal of Neuroscience. 2006;26:10222–10231. doi: 10.1523/JNEUROSCI.2250-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Langbaum J.B.S., Fleisher A.S., Ayutyanont N., Reschke C., Lee W., Liu X., Bandy D., Alexander G.E., Thompson P.M., Foster N.L., Harvey D.J., de Leon M.J., Koeppe R.A., Jagust W.J., Weiner M.W., Reiman E.M., A.D.N.I. Twelve-month metabolic declines in probable Alzheimer's disease and amnestic mild cognitive impairment assessed using an empirically pre-defined statistical region-of-interest: findings from the Alzheimer's disease neuroimaging initiative. NeuroImage. 2010;51:654–664. doi: 10.1016/j.neuroimage.2010.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G., Joie R.L., Villain N., Perrotin A., de La Sayette V., Eustache F., Vandenberghe R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disea. Neuroimage Clinical. 2013;2:356–365. doi: 10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie R.H., Bacskai B.J., Zipfel W.R., Williams R.M., Kajdasz S.T., Webb W.W., Hyman B.T. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. Journal of Neuroscience. 2001;21:858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G., Villemagne V.L., Bourgeat P., Pike K.E., Jones G., Ames D., Ellis K.A., Szoeke C., Martins R.N., O'Keefe G.J., Salvado O., Masters C.L., Rowe C.C., AIBL Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Annals of Neurology. 2010;67:317–324. doi: 10.1002/ana.21955. [DOI] [PubMed] [Google Scholar]
- Clark C.M., Schneider J.A., Bedell B.J., Beach T.G., Bilker W.B., Mintun M.A., Pontecorvo M.J., Hefti F., Carpenter A.P., Flitter M.L., Krautkramer M.J., Kung H.F., Coleman R.E., Doraiswamy P.M., Fleisher A.S., Sabbagh M.N., Sadowsky C.H., Reiman E.P., Reiman P.E.M., Zehntner S.P., Skovronsky D.M. Use of florbetapir-PET for imaging beta-amyloid pathology. Journal of the American Medical Association. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark C.M., Pontecorvo M.J., Beach T.G., Bedell B.J., Coleman R.E., Doraiswamy P.M., Fleisher A.S., Reiman E.M., Sabbagh M.N., Sadowsky C.H., Schneider J.A., Arora A., Carpenter A.P., Flitter M.L., Joshi A.D., Krautkramer M.J., Lu M., Mintun M.A., Skovronsky D.M., for the AV45-A07 group Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic Amyloid-β plaques: a prospective cohort study. Lancet Neurology. 2012;11:669–678. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- Cselényi Z., Jönhagen M.E., Forsberg A., Halldin C., Julin P., Schou M., Johnström P., Varnäs K., Svensson S., Farde L. Clinical validation of 18F-AZD4694, an Amyloid-β-specific PET radioligand. Journal of Nuclear Medicine. 2012:415–424. doi: 10.2967/jnumed.111.094029. [DOI] [PubMed] [Google Scholar]
- Davis D.G., Schmitt F.A., Wekstein D.R., Markesbery W.R. Alzheimer neuropathologic alterations in aged cognitively normal subjects. Journal of Neuropathology and Experimental Neurology. 1999;58:376–388. doi: 10.1097/00005072-199904000-00008. [DOI] [PubMed] [Google Scholar]
- DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Annals of Neurology. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Devanand D.P., Mikhno A., Pelton G.H., Cuasay K., Pradhaban G., Dileep Kumar J.S., Upton N., Lai R., Gunn R.N., Libri V., Liu X., van Heertum R., Mann J.J., Parsey R.V. Pittsburgh Compound B (11C-PIB) and fluorodeoxyglucose (18F-FDG) PET in patients with Alzheimer disease, mild cognitive impairment, and healthy controls. Journal of Geriatric Psychiatry and Neurology. 2010;23:185–198. doi: 10.1177/0891988710363715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhollander I., Nelissen N., Van Laere K., Peeters D., Demaerel P., Van Paesschen W., Thijs V., Vandenberghe R. In vivo amyloid imaging in cortical superficial siderosis. Journal of Neurology, Neurosurgery, and Psychiatry. 2011;82:469–471. doi: 10.1136/jnnp.2009.194480. [DOI] [PubMed] [Google Scholar]
- Dickerson B.C., Bakkour A., Salat D.H., Feczko E., Pacheco J., Greve D.N., Grodstein F., Wright C.I., Blacker D., Rosas H.D., Sperling R.A., Atri A., Growdon J.H., Hyman B.T., Morris J.C., Fischl B., Buckner R.L. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild ad dementia and is detectable in asymptomatic amyloid-positive individuals. Cerebral Cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierksen G.A., Skehan M.E., Khan M.A., Jeng J., Nandigam R.N.K., Becker J.A., Kumar A., Neal K.L., Betensky R.A., Frosch M.P., Rosand J., Johnson K.A., Viswanathan A., Salat D.H., Greenberg S.M. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Annals of Neurology. 2010;68:545–548. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doraiswamy P.M., Sperling R.A., Coleman R.E., Johnson K.A., Reiman E.M., Davis M.D., Grundman M., Sabbagh M.N., Sadowsky C.H., Fleisher A.S., Carpenter A., Clark C.M., Joshi A.D., Mintun M.A., Skovronsky D.M., Pontecorvo M.J., for the AV45-A11 group Amyloid-β assessed by florbetapir 18F PET and 18-month cognitive decline: a multicenter study. Neurology. 2012;79:1636–1644. doi: 10.1212/WNL.0b013e3182661f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I., Zhou Y., An Y., Sojkova J., Davatzikos C., Kraut M.A., Ye W., Ferrucci L., Mathis C.A., Klunk W.E., Wong D.F., Resnick S.M. Lack of association between 11C-PIB and longitudinal brain atrophy in non-demented older individuals. Neurobiology of Aging. 2011;32:2123–2130. doi: 10.1016/j.neurobiolaging.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I., Troncoso J.C., Rudow G., Sojkova J., Pletnikova O., Zhou Y., Kraut M.A., Ferrucci L., Mathis C.A., Klunk W.E., O'Brien R.J., Davatzikos C., Wong D.F., Resnick S.M. Correspondence between in vivo 11C-PIB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathologica. 2012;124:823–831. doi: 10.1007/s00401-012-1025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duara R., Loewenstein D.A., Shen Q., Barker W., Potter E., Varon D., Heurlin K., Vandenberghe R., Buckley C. Amyloid positron emission tomography with 18F-flutemetamol and structural magnetic resonance imaging in the classification of mild cognitive impairment and Alzheimer's disease. Alzheimers & Dementia. 2012 doi: 10.1016/j.jalz.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P., Rowe C.C., Rinne J.O., Ng S., Ahmed I., Kemppainen N., Villemagne V.L., O'Keefe G., Nagren K., Chaudhury K.R., Masters C.L., Brooks D.J. Amyloid load in Parkinson's disease dementia and lewy body dementia measured with 11C-PIB positron emission tomography. Journal of Neurology, Neurosurgery, and Psychiatry. 2008;79:1331–1338. doi: 10.1136/jnnp.2007.127878. [DOI] [PubMed] [Google Scholar]
- Engler H., Forsberg A., Almkvist O., Blomquist G., Larsson E., Savitcheva I., Wall A., Ringheim A., Langström B., Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- Fagan A.M., Mintun M.A., Mach R.H., Lee S.-Y., Dence C.S., Shah A.R., LaRossa G.N., Spinner M.L., Klunk W.E., Mathis C.A., DeKosky S.T., Morris J.C., Holtzman D.M. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid abeta42 in humans. Annals of Neurology. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti M.T., Okamura N., Furumoto S., Mulligan R.S., Connor A.R., McLean C.A., Cao D., Rigopoulos A., Cartwright G.A., O'Keefe G., Gong S., Adlard P.A., Barnham K.J., Rowe C.C., Masters C.L., Kudo Y., Cappai R., Yanai K., Villemagne V.L. 18F-thk523: a novel in vivo tau imaging ligand for Alzheimer's disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- Forsberg A., Engler H., Almkvist O., Blomquist G., Hagman G., Wall A., Ringheim A., Langström B., Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiology of Aging. 2008;29:1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Foster N.L., Heidebrink J.L., Clark C.M., Jagust W.J., Arnold S.E., Barbas N.R., DeCarli C.S., Turner R.S., Koeppe R.A., Higdon R., Minoshima S. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer's disease. Brain. 2007;130:2616–2635. doi: 10.1093/brain/awm177. [DOI] [PubMed] [Google Scholar]
- Fouquet M., Desgranges B., Landeau B., Duchesnay E., Mzenge F., de la Sayette V., Viader F., Baron J.-C., Eustache F., Chételat G. Longitudinal brain metabolic changes from amnestic mild cognitive impairment to Alzheimer's disease. Brain. 2009;132:2058–2067. doi: 10.1093/brain/awp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst A.J., Rabinovici G.D., Rostomian A.H., Steed T., Alkalay A., Racine C., Miller B.L., Jagust W.J. Cognition, glucose metabolism and amyloid burden in Alzheimer's disease. Neurobiology of Aging. 2012;33:215–225. doi: 10.1016/j.neurobiolaging.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K., Okamura N., Tashiro M., Waragai M., Furumoto S., Iwata R., Yanai K., Kudo Y., Arai H. Amyloid PET in mild cognitive impairment and Alzheimer's disease with BF-227: comparison to FDG-PET. Journal of Neurology. 2010;257:721–727. doi: 10.1007/s00415-009-5396-8. [DOI] [PubMed] [Google Scholar]
- Gearing M., Wilson R.W., Unger E.R., Shelton E.R., Chan H.W., Masters C.L., Beyreuther K., Mirra S.S. Amyloid precursor protein (APP) in the striatum in Alzheimer's disease: an immunohistochemical study. Journal of Neuropathology and Experimental Neurology. 1993;52:22–30. doi: 10.1097/00005072-199301000-00004. [DOI] [PubMed] [Google Scholar]
- Gefen T., Gasho K., Rademaker A., Lalehzari M., Weintraub S., Rogalski E., Wieneke C., Bigio E., Geula C., Mesulam M.-M. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain. 2012;135:1554–1565. doi: 10.1093/brain/aws076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P., Hof P.R., Bouras C. Selective vulnerability of neocortical association areas in Alzheimer's disease. Microscopic Research Techniques. 1998;43:16–23. doi: 10.1002/(SICI)1097-0029(19981001)43:1<16::AID-JEMT3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P., Herrmann F.R., Bussire T., Bouras C., Kvari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T., Price J.L., McKeel D., Jr., Morris J.C., Growdon J.H., Hyman B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. Journal of Neuroscience. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini M.L., Hillis A.E., Weintraub S., Kertesz A., Mendez M., Cappa S.F., Ogar J.M., Rohrer J.D., Black S., Boeve B.F., Manes F., Dronkers N.F., Vandenberghe R., Rascovsky K., Patterson K., Miller B.L., Knopman D.S., Hodges J.R., Mesulam M.M., Grossman M. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T., Tholen S., Yousefi B.H., Alexopoulos P., Förschler A., Förstl H., Henriksen G., Klunk W.E., Mathis C.A., Perneczky R., Sorg C., Kurz A., Drzezga A. Progression of cerebral amyloid load is associated with the apolipoprotein e ϵ4 genotype in Alzheimer's disease. Biological Psychiatry. 2010;68:879–884. doi: 10.1016/j.biopsych.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T., Faust M., Auer F., Alexopoulos P., Förstl H., Henriksen G., Perneczky R., Sorg C., Yousefi B.H., Drzezga A., Kurz A. White matter hyperintensities predict amyloid increase in Alzheimer's disease. Neurobiology of Aging. 2012;33:2766–2773. doi: 10.1016/j.neurobiolaging.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Guillozet A.L., Weintraub S., Mash D.C., Mesulam M.M. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Archives of Neurology. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- Gurol M.E., Viswanathan A., Gidicsin C., Hedden T., Martinez-Ramirez S., Dumas A., Vashkevich A., Ayres A.M., Auriel E., van Etten E., Becker A., Carmasin J., Schwab K., Rosand J., Johnson K.A., Greenberg S.M. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Annals of Neurology. 2012 doi: 10.1002/ana.23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber S.N., Lynd E., Klein C., Groenewegen H.J. Topographic organization of the ventral striatal efferent projections in the rhesus monkey: an anterograde tracing study. The Journal of Comparative Neurology. 1990;293:282–298. doi: 10.1002/cne.902930210. [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hebert L.E., Beckett L.A., Scherr P.A., Evans D.A. Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer's Disease and Associated Disorders. 2001;15:169–173. doi: 10.1097/00002093-200110000-00002. [DOI] [PubMed] [Google Scholar]
- Herholz K., Ebmeier K. Clinical amyloid imaging in Alzheimer's disease. Lancet Neurology. 2011;10:667–670. doi: 10.1016/S1474-4422(11)70123-5. [DOI] [PubMed] [Google Scholar]
- Herholz K., Salmon E., Perani D., Baron J.C., Holthoff V., Frölich L., Schönknecht P., Ito K., Mielke R., Kalbe E., Zündorf G., Delbeuck X., Pelati O., Anchisi D., Fazio F., Kerrouche N., Desgranges B., Eustache F., Beuthien-Baumann B., Menzel C., Schröder J., Kato T., Arahata Y., Henze M., Heiss W.D. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. NeuroImage. 2002;17:302–316. doi: 10.1006/nimg.2002.1208. [DOI] [PubMed] [Google Scholar]
- Hodges J., editor. Early-onset Dementia. A Multidisciplinary Approach. Oxford University Press; Oxford: 2001. [Google Scholar]
- Hof P.R., Archin N., Osmand A.P., Dougherty J.H., Wells C., Bouras C., Morrison J.H. Posterior cortical atrophy in Alzheimer's disease: analysis of a new case and re-evaluation of a historical report. Acta Neuropathologica. 1993;86:215–223. doi: 10.1007/BF00304135. [DOI] [PubMed] [Google Scholar]
- Hof P.R., Vogt B.A., Bouras C., Morrison J.H. Atypical form of Alzheimer's disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Research. 1997;37:3609–3625. doi: 10.1016/S0042-6989(96)00240-4. [DOI] [PubMed] [Google Scholar]
- Hornberger M., Wong S., Tan R., Irish M., Piquet O., Kril J., Hodges J., Halliday G. In vivo and post-mortem memory circuit integrity in frontotemporal dementia and Alzheimer's disease. Brain. 2012;135:3015–3025. doi: 10.1093/brain/aws239. [DOI] [PubMed] [Google Scholar]
- Hyman B.T., Trojanowski J.Q. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute working group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- Hyman B.T., Marzloff K., Arriagada P.V. The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. Journal of Neuropathology and Experimental Neurology. 1993;52:594–600. doi: 10.1097/00005072-199311000-00006. [DOI] [PubMed] [Google Scholar]
- Hyman B.T., Phelps C.H., Beach T.G., Bigio E.H., Cairns N.J., Carrillo M.C., Dickson D.W., Duyckaerts C., Frosch M.P., Masliah E., Mirra S.S., Nelson P.T., Schneider J.A., Thal D.R., Thies B., Trojanowski J.Q., Vinters H.V., Montine T.J. National institute on aging-Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers & Dementia. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic M.D., Klunk W.E., Abrahamson E.E., Mathis C.A., Price J.C., Tsopelas N.D., Lopresti B.J., Ziolko S., Bi W., Paljug W.R., Debnath M.L., Hope C.E., Isanski B.A., Hamilton R.L., DeKosky S.T. Post-mortem correlates of in vivo PIB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic M.D., Abrahamson E.E., Price J.C., Hamilton R.L., Mathis C.A., Paljug W.R., Debnath M.L., Cohen A.D., Mizukami K., DeKosky S.T., Lopez O.L., Klunk W.E. Early AD pathology in a 11C-PIB-negative case: a PIB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathologica. 2012;123:433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C., Petersen R. Humana Press; 2000. Early Diagnosis of Alzheimer's Disease, Chapter Structural Imaging Approaches to AD. [Google Scholar]
- Jack C.R., Jr., Lowe V.J., Senjem M.L., Weigand S.D., Kemp B.J., Shiung M.M., Knopman D.S., Boeve B.F., Klunk W.E., Mathis C.A., PETersen R.C. 11C-PIB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Jr., Lowe V.J., Weigand S.D., Wiste H.J., Senjem M.L., Knopman D.S., Shiung M.M., Gunter J.L., Boeve B.F., Kemp B.J., Weiner M., Petersen R.C., and the Alzheimer's Disease Neuroimaging Initiative Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Jr., Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., Petersen R.C., Trojanowski J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurology. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Jr., Knopman D.S., Jagust W.J., Petersen R.C., Weiner M.W., Aisen P.S., Shaw L.M., Vemuri P., Wiste H.J., Weigand S.D., Lesnick T.G., Pankratz V.S., Donohue M.C., Trojanowski J.Q. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurology. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W.J., Landau S.M., Shaw L.M., Trojanowski J.Q., Koeppe R.A., Reiman E.M., Foster N.L., Petersen R.C., Weiner M.W., Price J.C., Mathis C.A., and the Alzheimer's Disease Neuroimaging Initiative Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–1199. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.A., Gregas M., Becker J.A., Kinnecom C., Salat D.H., Moran E.K., Smith E.E., Rosand J., Rentz D.M., Klunk W.E., Mathis C.A., Price J.C., DeKosky S.T., Fischman A.J., Greenberg S.M. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Annals of Neurology. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Joshi A.D., Pontecorvo M.J., Clark C.M., Carpenter A.P., Jennings D.L., Sadowsky C.H., Adler L.P., Kovnat K.D., Seibyl J.P., Arora A., Saha K., Burns J.D., Lowrey M.J., Mintun M.A., Skovronsky D.M., and the Florbetapir F18 Study Investigators Performance characteristics of amyloid PET with florbetapir f 18 in patients with Alzheimer's disease and cognitively normal subjects. Journal of Nuclear Medicine. 2012;53:378–384. doi: 10.2967/jnumed.111.090340. [DOI] [PubMed] [Google Scholar]
- Kadir A., Nordberg A. _target-specific PET probes for neurodegenerative disorders related to dementia. Journal of Nuclear Medicine. 2010;51:1418–1430. doi: 10.2967/jnumed.110.077164. [DOI] [PubMed] [Google Scholar]
- Kantarci K., Yang C., Schneider J.A., Senjem M.L., Reyes D.A., Lowe V.J., Barnes L.L., Aggarwal N.T., Bennett D.A., Smith G.E., PETersen R.C., Jack C.R., Jr., Boeve B.F. Ante mortem amyloid imaging and β-amyloid pathology in a case with dementia with lewy bodies. Neurobiology of Aging. 2012;33:878–885. doi: 10.1016/j.neurobiolaging.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W.E., Engler H., Nordberg A., Wang Y., Blomqvist G., Holt D.P., Bergstrm M., Savitcheva I., Huang G.-f., Estrada S., Ausn B., Debnath M.L., Barletta J., Price J.C., Sandell J., Lopresti B.J., Wall A., Koivisto P., Antoni G., Mathis C.A., Lngstrm B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-b. Annals of Neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Koole M., Lewis D.M., Buckley C., Nelissen N., Vandenbulcke M., Brooks D.J., Vandenberghe R., Van Laere K. Whole-body biodistribution and radiation dosimetry of 18F-GE067: a radioligand for in vivo brain amyloid imaging. Journal of Nuclear Medicine. 2009;50:818–822. doi: 10.2967/jnumed.108.060756. [DOI] [PubMed] [Google Scholar]
- La Joie R., Perrotin A., Barr L., Hommet C., Mzenge F., Ibazizene M., Camus V., Abbas A., Landeau B., Guilloteau D., de La Sayette V., Eustache F., Desgranges B., Chételat G. Region-specific hierarchy between atrophy, hypometabolism, and beta-amyloid load in Alzheimer's disease dementia. Journal of Neuroscience. 2012;32:16265–16273. doi: 10.1523/JNEUROSCI.2170-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J.-C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B., Letenneur L., Bettens K., Berr C., Pasquier F., Fivet N., Barberger-Gateau P., Engelborghs S., De Deyn P., Mateo I., Franck A., Helisalmi S., Porcellini E., Hanon O., the European Alzheimer's Disease Initiative Investigators, de Pancorbo M.M., Lendon C., Dufouil C., Jaillard C., Leveillard T., Alvarez V., Bosco P., Mancuso M., Panza F., Nacmias B., Boss P., Piccardi P., Annoni G., Seripa D., Galimberti D., Hannequin D., Licastro F., Soininen H., Ritchie K., Blanch H., Dartigues J.-F., Tzourio C., Gut I., Van Broeckhoven C., Alprovitch A., Lathrop M., Amouyel P. Genome-wide association study identifies variants at clu and cr1 associated with Alzheimer's disease. Nature Genetics. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Landau S.M., Harvey D., Madison C.M., Koeppe R.A., Reiman E.M., Foster N.L., Weiner M.W., Jagust W.J., and the Alzheimer's Disease Neuroimaging Initiative Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiology of Aging. 2011;32:1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-J., Desplats P., Sigurdson C., Tsigelny I., Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nature Reviews. Neurology. 2010;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Kim S.H., Kim G.H., Seo S.W., Park H.K., Oh S.J., Kim J.S., Cheong H.K., Na D.L. Identification of pure subcortical vascular dementia using 11c-Pittsburgh Compound B. Neurology. 2011;77:18–25. doi: 10.1212/WNL.0b013e318221acee. [DOI] [PubMed] [Google Scholar]
- Leinonen V., Alafuzoff I., Aalto S., Suotunen T., Savolainen S., Nagren K., Tapiola T., Pirttilä T., Rinne J., Jääskeläinen J.E., Soininen H., Rinne J.O. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Archives of Neurology. 2008;65:1304–1309. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- Leinonen V., Koivisto A.M., Savolainen S., Rummukainen J., Tamminen J.N., Tillgren T., Vainikka S., Pyykkö O.T., Mölsä J., Fraunberg M., Pirttilä T., Jääskeläinen J.E., Soininen H., Rinne J., Alafuzoff I. Amyloid and tau proteins in cortical brain biopsy and Alzheimer's disease. Annals of Neurology. 2010;68:446–453. doi: 10.1002/ana.22100. [DOI] [PubMed] [Google Scholar]
- Leinonen V., Rinne J.O., Virtanen K.A., Eskola O., Rummukainen J., Huttunen J., von Und Zu Fraunberg M., Nerg O., Koivisto A.M., Rinne J., Jääskeläinen J.E., Buckley C., Smith A., Jones P.A., Sherwin P., Farrar G., McLain R., Kailajärvi M., Heurling K., Grachev I.D. Positron emission tomography with [(18) F]flutemetamol and [(11) C]PIB for in vivo detection of cerebral cortical amyloid in normal pressure hydrocephalus patients. European Journal of Neurology. 2013 doi: 10.1111/ene.12102. [DOI] [PubMed] [Google Scholar]
- Lockhart A., Lamb J.R., Osredkar T., Sue L.I., Joyce J.N., Ye L., Libri V., Leppert D., Beach T.G. PIB is a non-specific imaging marker of amyloid-beta (abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- Masliah E., Terry R.D., Mallory M., Alford M., Hansen L.A. Diffuse plaques do not accentuate synapse loss in Alzheimer's disease. American Journal of Pathology. 1990;137:1293–1297. [PMC free article] [PubMed] [Google Scholar]
- Mathis C.A., Mason N.S., Lopresti B.J., Klunk W.E. Development of positron emission tomography β-amyloid plaque imaging agents. Seminars in Nuclear Medicine. 2012;42:423–432. doi: 10.1053/j.semnuclmed.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N., Andreasson U., Carrillo M.C., Persson S., Shaw L.M., Zegers I., Zetterberg H., Blennow K. Proficiency testing programs for Alzheimer's disease cerebrospinal fluid biomarkers. Biomarkers in Medicine. 2012;6:401–407. doi: 10.2217/bmm.12.41. [DOI] [PubMed] [Google Scholar]
- McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Jr., Kawas C.H., Klunk W.E., Koroshetz W.J., Manly J.J., Mayeux R., Mohs R.C., Morris J.C., Rossor M.N., Scheltens P., Carrillo M.C., Thies B., Weintraub S., Phelps C.H. The diagnosis of dementia due to Alzheimer's disease: recommendations from the national institute on aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers & Dementia. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M.M. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Mesulam M.-M. Oxford University Press; 2000. Principles of Behavioral and Cognitive Neurology, Chapter Aging, Alzheimer's Disease, and Dementia: Clinical and Neurobiological Perspectives; pp. 439–522. [Google Scholar]
- Mesulam M., Shaw P., Mash D., Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Annals of Neurology. 2004;55:815–828. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- Mormino E.C., Kluth J.T., Madison C.M., Rabinovici G.D., Baker S.L., Miller B.L., Koeppe R.A., Mathis C.A., Weiner M.W., Jagust W.J., and the Alzheimer's Disease Neuroimaging Initiative Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J.C., Storandt M., McKeel D., Jr., Rubin E.H., Price J.L., Grant E.A., Berg L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer's disease. Neurology. 1996;46:707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- Mosconi L., Tsui W.H., Herholz K., Pupi A., Drzezga A., Lucignani G., Reiman E.M., Holthoff V., Kalbe E., Sorbi S., Diehl-Schmid J., Perneczky R., Clerici F., Caselli R., Beuthien-Baumann B., Kurz A., Minoshima S., de Leon M.J. Multicenter standardized 18F-fdg PET diagnosis of mild cognitive impairment, Alzheimer's disease, and other dementias. Journal of Nuclear Medicine. 2008;49:390–398. doi: 10.2967/jnumed.107.045385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen N., Vandenbulcke M., Fannes K., Verbruggen A., Peeters R., Dupont P., Van Laere K., Bormans G., Vandenberghe R. Abeta amyloid deposition in the language system and how the brain responds. Brain. 2007;130:2055–2069. doi: 10.1093/brain/awm133. [DOI] [PubMed] [Google Scholar]
- Nelissen N., Van Laere K., Thurfjell L., Owenius R., Vandenbulcke M., Koole M., Bormans G., Brooks D.J., Vandenberghe R. Phase 1 study of the Pittsburgh Compound B derivative 18F-flutemetamol in healthy volunteers and patients with probable Alzheimer disease. Journal of Nuclear Medicine. 2009;50:1251–1259. doi: 10.2967/jnumed.109.063305. [DOI] [PubMed] [Google Scholar]
- Ng S., Villemagne V.L., Berlangieri S., Lee S.-T., Cherk M., Gong S.J., Ackermann U., Saunder T., Tochon-Danguy H., Jones G., Smith C., O'Keefe G., Masters C.L., Rowe C.C. Visual assessment versus quantitative assessment of 11c-PIB PET and 18F-fdg PET for detection of Alzheimer's disease. Journal of Nuclear Medicine. 2007;48:547–552. doi: 10.2967/jnumed.106.037762. [DOI] [PubMed] [Google Scholar]
- Nordberg A., Carter S.F., Rinne J., Drzezga A., Brooks D.J., Vandenberghe R., Perani D., Forsberg A., Langström B., Scheinin N., Karrasch M., Nagren K., Grimmer T., Miederer I., Edison P., Okello A., Van Laere K., Nelissen N., Vandenbulcke M., Garibotto V., Almkvist O., Kalbe E., Hinz R., Herholz K. A European multicentre PET study of fibrillar amyloid in Alzheimer's disease. European Journal of Nuclear Medicine and Molecular Imaging. 2013;40:104–114. doi: 10.1007/s00259-012-2237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg S., Jönhagen M.E., Cselényi Z., Halldin C., Julin P., Olsson H., Freund-Levi Y., Andersson J., Varnäs K., Svensson S., Farde L. Detection of amyloid in Alzheimer's disease with positron emission tomography using [11C]AZD2184. European Journal of Nuclear Medicine and Molecular Imaging. 2009;36:1859–1863. doi: 10.1007/s00259-009-1182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N., Suemoto T., Furumoto S., Suzuki M., Shimadzu H., Akatsu H., Yamamoto T., Fujiwara H., Nemoto M., Maruyama M., Arai H., Yanai K., Sawada T., Kudo Y. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer's disease. Journal of Neuroscience. 2005;25:10857–10862. doi: 10.1523/JNEUROSCI.1738-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello A., Koivunen J., Edison P., Archer H.A., Turkheimer F.E., Nagren K., Bullock R., Walker Z., Kennedy A., Fox N.C., Rossor M.N., Rinne J.O., Brooks D.J. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology. 2009;73:754–760. doi: 10.1212/WNL.0b013e3181b23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki S., Deptula D., Thurfjell L., Barkhof F., Bohrmann B., Brooks D.J., Klunk W.E., Ashford E., Yoo K., Xu Z.-X., Loetscher H., Santarelli L. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Archives of Neurology. 2012;69:198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- Pao W.C., Dickson D.W., Crook J.E., Finch N.A., Rademakers R., Graff-Radford N.R. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer's Disease and Associated Disorders. 2011;25:364–368. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike K.E., Savage G., Villemagne V.L., Ng S., Moss S.A., Maruff P., Mathis C.A., Klunk W.E., Masters C.L., Rowe C.C. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Prohovnik I., Perl D.P., Davis K.L., Libow L., Lesser G., Haroutunian V. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology. 2006;66:49–55. doi: 10.1212/01.wnl.0000191298.68045.50. [DOI] [PubMed] [Google Scholar]
- Rabinovici G.D., Furst A.J., O'Neil J.P., Racine C.A., Mormino E.C., Baker S.L., Chetty S., Patel P., Pagliaro T.A., Klunk W.E., Mathis C.A., Rosen H.J., Miller B.L., Jagust W.J. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- Rabinovici G.D., Jagust W.J., Furst A.J., Ogar J.M., Racine C.A., Mormino E.C., O'Neil J.P., Lal R.A., Dronkers N.F., Miller B.L., Gorno-Tempini M.L. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Annals of Neurology. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici G.D., Furst A.J., Alkalay A., Racine C.A., O'Neil J.P., Janabi M., Baker S.L., Agarwal N., Bonasera S.J., Mormino E.C., Weiner M.W., Gorno-Tempini M.L., Rosen H.J., Miller B.L., Jagust W.J. Increased metabolic vulnerability in early-onset Alzheimer's disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici G.D., Rosen H.J., Alkalay A., Kornak J., Furst A.J., Agarwal N., Mormino E.C., O'Neil J.P., Janabi M., Karydas A., Growdon M.E., Jang J.Y., Huang E.J., Dearmond S.J., Trojanowski J.Q., Grinberg L.T., Gorno-Tempini M.L., Seeley W.W., Miller B.L., Jagust W.J. Amyloid vs fdg-PET in the differential diagnosis of ad and ftld. Neurology. 2011;77:2034–2042. doi: 10.1212/WNL.0b013e31823b9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne J.O., Brooks D.J., Rossor M.N., Fox N.C., Bullock R., Klunk W.E., Mathis C.A., Blennow K., Barakos J., Okello A.A., Martinez Rodriguez, de Liano S., Liu E., Koller M., Gregg K.M., Schenk D., Black R., Grundman M. 11C-PIB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurology. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- Rinne J.O., Wong D.F., Wolk D.A., Leinonen V., Arnold S.E., Buckley C., Smith A., McLain R., Sherwin P.F., Farrar G., Kailajärvi M., Grachev I.D. 18F-flutemetamol PET imaging and cortical biopsy histopathology for fibrillar amyloid β detection in living subjects with normal pressure hydrocephalus: pooled analysis of four studies. Acta Neuropathologica. 2012;124:833–845. doi: 10.1007/s00401-012-1051-z. [DOI] [PubMed] [Google Scholar]
- Rombouts S.A., Barkhof F., Witter M.P., Scheltens P. Unbiased whole-brain analysis of gray matter loss in Alzheimer's disease. Neuroscience Letters. 2000;285:231–233. doi: 10.1016/s0304-3940(00)01067-3. [DOI] [PubMed] [Google Scholar]
- Rosen R.F., Ciliax B.J., Wingo T.S., Gearing M., Dooyema J., Lah J.J., Ghiso J.A., LeVine H., 3rd, Walker L.C. Deficient high-affinity binding of Pittsburgh Compound B in a case of Alzheimer's disease. Acta Neuropathologica. 2010;119:221–233. doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe C.C., Ng S., Ackermann U., Gong S.J., Pike K., Savage G., Cowie T.F., Dickinson K.L., Maruff P., Darby D., Smith C., Woodward M., Merory J., Tochon-Danguy H., O'Keefe G., Klunk W.E., Mathis C.A., Price J.C., Masters C.L., Villemagne V.L. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Rowe C.C., Ackerman U., Browne W., Mulligan R., Pike K.L., O'Keefe G., Tochon-Danguy H., Chan G., Berlangieri S.U., Jones G., Dickinson-Rowe K.L., Kung H.P., Zhang W., Kung M.P., Skovronsky D., Dyrks T., Holl G., Krause S., Friebe M., Lehman L., Lindemann S., Dinkelborg L.M., Masters C.L., Villemagne V.L. Imaging of amyloid beta in Alzheimer's disease with 18F-bay94-9172, a novel PET tracer: proof of mechanism. Lancet Neurology. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- Rowe C.C., Ellis K.A., Rimajova M., Bourgeat P., Pike K.E., Jones G., Fripp J., Tochon-Danguy H., Morandeau L., O'Keefe G., Price R., Raniga P., Robins P., Acosta O., Lenzo N., Szoeke C., Salvado O., Head R., Martins R., Masters C.L., Ames D., Villemagne V.L. Amyloid imaging results from the Australian imaging, biomarkers and lifestyle (AIBL) study of aging. Neurobiology of Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Rudelli R.D., Ambler M.W., Wisniewski H.M. Morphology and distribution of Alzheimer neuritic (senile) and amyloid plaques in striatum and diencephalon. Acta Neuropathologica. 1984;64:273–281. doi: 10.1007/BF00690393. [DOI] [PubMed] [Google Scholar]
- Savva G.M., Wharton S.B., Ince P.G., Forster G., Matthews F.E., Brayne C., for the MRC Cognitive Function and Ageing Study, Study, A. Age, neuropathology, and dementia. The New England Journal of Medicine. 2009;360:2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- Scheff S.W., Price D.A., Schmitt F.A., Mufson E.J. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiology of Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Scheinin N.M., Aalto S., Koikkalainen J., Lötjönen J., Karrasch M., Kemppainen N., Viitanen M., Nagren K., Helin S., Scheinin M., Rinne J.O. Follow-up of [11C]PIB uptake and brain volume in patients with Alzheimer disease and controls. Neurology. 2009;73:1186–1192. doi: 10.1212/WNL.0b013e3181bacf1b. [DOI] [PubMed] [Google Scholar]
- Schöll M., Wall A., Thordardottir S., Ferreira D., Bogdanovic N., Langström B., Almkvist O., Graff C., Nordberg A. Low PIB PET retention in presence of pathologic csf biomarkers in arctic APP mutation carriers. Neurology. 2012;79:229–236. doi: 10.1212/WNL.0b013e31825fdf18. [DOI] [PubMed] [Google Scholar]
- Schneider J.A., Arvanitakis Z., Yu L., Boyle P.A., Leurgans S.E., Bennett D.A. Cognitive impairment, decline and fluctuations in older community-dwelling subjects with lewy bodies. Brain. 2012;135:3005–3014. doi: 10.1093/brain/aws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Lee S.-Y., Kim S.J., Kim S.-H., Cho S.-J., Kim Y.-B. Voxel-based analysis of Alzheimer's disease PET imaging using a triplet of radiotracers: PIB, FDDNP, and FDG. NeuroImage. 2010;52:488–496. doi: 10.1016/j.neuroimage.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Sleegers K., Lambert J.-C., Bertram L., Cruts M., Amouyel P., Van Broeckhoven C. The pursuit of susceptibility genes for Alzheimer's disease: progress and prospects. Trends in Genetics. 2010;26:84–93. doi: 10.1016/j.tig.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Small G.W., Kepe V., Ercoli L.M., Siddarth P., Bookheimer S.Y., Miller K.J., Lavretsky H., Burggren A.C., Cole G.M., Vinters H.V., Thompson P.M., Huang S.-C., Satyamurthy N., Phelps M.E., Barrio J.R. PET of brain amyloid and tau in mild cognitive impairment. The New England Journal of Medicine. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- Snowdon D.A., Greiner L.H., Mortimer J.A., Riley K.P., Greiner P.A., Markesbery W.R. Brain infarction and the clinical expression of Alzheimer disease. the nun study. Journal of the American Medical Association. 1997;277:813–817. [PubMed] [Google Scholar]
- Sojkova J., Driscoll I., Iacono D., Zhou Y., Codispoti K.-E., Kraut M.A., Ferrucci L., Pletnikova O., Mathis C.A., Klunk W.E., O'Brien R.J., Wong D.F., Troncoso J.C., Resnick S.M. In vivo fibrillar beta-amyloid detected using [11c]PIB positron emission tomography and neuropathologic assessment in older adults. Archives of Neurology. 2011;68:232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R.A., Laviolette P.S., O'Keefe K., O'Brien J., Rentz D.M., Pihlajamaki M., Marshall G., Hyman B.T., Selkoe D.J., Hedden T., Buckner R.L., Becker J.A., Johnson K.A. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R.A., Jack C.R., Jr., Black S.E., Frosch M.P., Greenberg S.M., Hyman B.T., Scheltens P., Carrillo M.C., Thies W., Bednar M.M., Black R.S., Brashear H.R., Grundman M., Siemers E.R., Feldman H.H., Schindler R.J. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's association research roundtable workgroup. Alzheimers & Dementia. 2011;7:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankoff B., Freeman L., Aigrot M.-S., Chardain A., Doll F., Williams A., Galanaud D., Armand L., Lehericy S., Lubetzki C., Zalc B., Bottlaender M. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-11C]-2-(4′-methylaminophenyl)- 6-hydroxybenzothiazole. Annals of Neurology. 2011;69:673–680. doi: 10.1002/ana.22320. [DOI] [PubMed] [Google Scholar]
- Suenaga T., Hirano A., Llena J.F., Yen S.H., Dickson D.W. Modified Bielschowsky stain and immunohistochemical studies on striatal plaques in Alzheimer's disease. Acta Neuropathologica. 1990;80:280–286. doi: 10.1007/BF00294646. [DOI] [PubMed] [Google Scholar]
- Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thal D.R., Rb U., Orantes M., Braak H. Phases of a beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- The Ronald and Nancy Reagan Research Institute of the Alzheimer's association and the National Institute on Aging working group Consensus report of the working group on: “Molecular and biochemical markers of Alzheimer's Disease”. Neurobiology of Aging. 1998;19:109–116. [PubMed] [Google Scholar]
- Thurfjell L., Lötjönen J., Lundqvist R., Koikkalainen J., Soininen H., Waldemar G., Brooks D.J., Vandenberghe R. Combination of biomarkers: PET [18F]flutemetamol imaging and structural MRI in dementia and mild cognitive impairment. Neurodegenerative Diseases. 2012;10:246–249. doi: 10.1159/000335381. [DOI] [PubMed] [Google Scholar]
- Tiraboschi P., Hansen L.A., Thal L.J., Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- Tolboom N., van der Flier W.M., Yaqub M., Boellaard R., Verwey N.A., Blankenstein M.A., Windhorst A.D., Scheltens P., Lammertsma A.A., van Berckel B.N.M. Relationship of cerebrospinal fluid markers to 11C-PIB and 18F-FDDNP binding. Journal of Nuclear Medicine. 2009;50:1464–1470. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- Tolboom N., Yaqub M., Boellaard R., Luurtsema G., Windhorst A.D., Scheltens P., Lammertsma A.A., van Berckel B.N.M. Test–retest variability of quantitative [11C]PIB studies in Alzheimer's disease. European Journal of Nuclear Medicine and Molecular Imaging. 2009;36:1629–1638. doi: 10.1007/s00259-009-1129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D., Schuff N., Mathis C.A., Jagust W., Weiner M.W., and the Alzheimer's Disease Neuroimaging Initiative Spatial patterns of brain amyloid-beta burden and atrophy rate associations in mild cognitive impairment. Brain. 2011;134:1077–1088. doi: 10.1093/brain/awr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R., Van Laere K., Ivanoiu A., Salmon E., Bastin C., Triau E., Hasselbalch S., Law I., Andersen A., Korner A., Minthon L., Garraux G., Nelissen N., Bormans G., Buckley C., Owenius R., Thurfjell L., Farrar G., Brooks D.J. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Annals of Neurology. 2010;68:319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- Vandenberghe R., Nelissen N., Salmon E., Ivanoiu A., Hasselbalch S., Andersen A., Korner A., Minthon L., Brooks D.J., Van Laere K., Dupont P. Binary classification of (18)18F-flutemetamol PET using machine learning: comparison with visual reads and structural MRI. NeuroImage. 2013;64:517–525. doi: 10.1016/j.neuroimage.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Vandenbulcke M., Peeters R., Dupont P., Van Hecke P., Vandenberghe R. Word reading and posterior temporal dysfunction in amnestic mild cognitive impairment. Cerebral Cortex. 2007;17:542–551. doi: 10.1093/cercor/bhj179. [DOI] [PubMed] [Google Scholar]
- Verhoeff N.P.L.G., Wilson A.A., Takeshita S., Trop L., Hussey D., Singh K., Kung H.F., Kung M.-P., Houle S. In-vivo imaging of Alzheimer disease beta-amyloid with [11C]SB-13 PET. The American Journal of Geriatric Psychiatry. 2004;12:584–595. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- Villain N., Chételat G., Grassiot B., Bourgeat P., Jones G., Ellis K.A., Ames D., Martins R.N., Eustache F., Salvado O., Masters C.L., Rowe C.C., Villemagne V.L., and the AIBL Research Group Regional dynamics of Amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer's disease: a voxelwise PIB-PET longitudinal study. Brain. 2012;135:2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- Villemagne V.L., Ong K., Mulligan R.S., Holl G., Pejoska S., Jones G., O'Keefe G., Ackerman U., Tochon-Danguy H., Chan J.G., Reininger C.B., Fels L., Putz B., Rohde B., Masters C.L., Rowe C.C. Amyloid imaging with 18F-florbetaben in Alzheimer disease and other dementias. Journal of Nuclear Medicine. 2011;52:1210–1217. doi: 10.2967/jnumed.111.089730. [DOI] [PubMed] [Google Scholar]
- Villemagne V.L., Mulligan R.S., Pejoska S., Ong K., Jones G., O'Keefe G., Chan J.G., Young K., Tochon-Danguy H., Masters C.L., Rowe C.C. Comparison of 11c-PIB and 18F-florbetaben for Aβ imaging in ageing and Alzheimer's disease. European Journal of Nuclear Medicine and Molecular Imaging. 2012;39:983–989. doi: 10.1007/s00259-012-2088-x. [DOI] [PubMed] [Google Scholar]
- von Gunten A., Bouras C., Kvari E., Giannakopoulos P., Hof P.R. Neural substrates of cognitive and behavioral deficits in atypical Alzheimer's disease. Brain Research Reviews. 2006;51:176–211. doi: 10.1016/j.brainresrev.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Whitwell J.L., Josephs K.A., Murray M.E., Kantarci K., Przybelski S.A., Weigand S.D., Vemuri P., Senjem M.L., Parisi J.E., Knopman D.S., Boeve B.F., Petersen R.C., Dickson D.W., Jack C., Jr. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71:743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk D.A., Price J.C., Saxton J.A., Snitz B.E., James J.A., Lopez O.L., Aizenstein H.J., Cohen A.D., Weissfeld L.A., Mathis C.A., Klunk W.E., DeKosky S.T. Amyloid imaging in mild cognitive impairment subtypes. Annals of Neurology. 2009;65:557–568. doi: 10.1002/ana.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk D.A., Zhang Z., Boudhar S., Clark C.M., Pontecorvo M.J., Arnold S.E. Amyloid imaging in Alzheimer's disease: comparison of florbetapir and Pittsburgh Compound-B positron emission tomography. Journal of Neurology, Neurosurgery, and Psychiatry. 2012;83:923–926. doi: 10.1136/jnnp-2012-302548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack K.B., Diaz-Arrastia R., Aizenstein H.J., Arnold S.E., Barbas N.R., Boeve B.F., Clark C.M., DeCarli C.S., Jagust W.J., Leverenz J.B., Peskind E.R., Turner R.S., Zamrini E.Y., Heidebrink J.L., Burke J.R., DeKosky S.T., Farlow M.R., Gabel M.J., Higdon R., Kawas C.H., Koeppe R.A., Lipton A.M., Foster N.L. Temporoparietal hypometabolism in frontotemporal lobar degeneration and associated imaging diagnostic errors. Archives of Neurology. 2011;68:329–337. doi: 10.1001/archneurol.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.F., Rosenberg P.B., Zhou Y., Kumar A., Raymont V., Ravert H.T., Dannals R.F., Nandi A., Brasic J.R., Ye W., Hilton J., Lyketsos C., Kung H.F., Joshi A.D., Skovronsky D.M., Pontecorvo M.J. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV45. Journal of Nuclear Medicine. 2010;51:913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.F., Moghekar A.R., Rigamonti D., Brasic J.R., Rousset O., Willis W., Buckley C., Smith A., Gok B., Sherwin P., Grachev I.D. An in vivo evaluation of cerebral cortical amyloid with 18F-flutemetamol using positron emission tomography compared with parietal biopsy samples in living normal pressure hydrocephalus patients. Molecular Imaging and Biology. 2013;15:230–237. doi: 10.1007/s11307-012-0583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]