

Abstract
In recent years, it has become clear that the current standard therapeutic options for pancreatic cancer are not adequate and still do not meet the criteria to cure patients suffering from this lethal disease. Although research over the past decade has shown very interesting and promising new therapeutic options for these patients, only minor clinical success was achieved. Therefore, there is still an urgent need for new approaches that deal with early detection and new therapeutic options in pancreatic cancer. To provide optimal care for patients with pancreatic cancer, we need to understand better its complex molecular biology and thus to identify new _target molecules that promote the proliferation and resistance to chemotherapy of pancreatic cancer cells. In spite of significant progress in curing cancers with chemotherapy, pancreatic cancer remains one of the most resistant solid tumour cancers and many studies suggest that drug-resistant cancer cells are the most aggressive with the highest relapse and metastatic rates. In this context, activated Notch signalling is strongly linked with chemoresistance and therefore reflects a rational new _target to circumvent resistance to chemotherapy in pancreatic cancer. Here, we have focused our discussion on the latest research, current therapy options and recently identified _target molecules such as Notch-2 and the heparin-binding growth factor midkine, which exhibit a wide range of cancer-relevant functions and therefore provide attractive new therapeutic _target molecules, in terms of pancreatic cancer and other cancers also.
Linked Articles
This article is part of a themed section on Midkine. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-4
Keywords: pancreatic cancer, PDAC, chemotherapy resistance, midkine, tumour, therapy, Notch, gemcitabine
Introduction
Pancreatic cancer remains one of the most lethal cancers worldwide, and nearly all newly diagnosed patients are faced with the question not of whether they will die from the cancer, but rather when they will die. In fact, no real progress in the establishment of new therapeutic options for pancreatic cancer has been made in the last two decades. Therefore, pancreatic cancer seems to be one of the biggest challenges in tackling the fight against cancer in the 21st century.
These unabated high mortality rates are the result of a very aggressively progressing cancer that is clinically accompanied by inadequate tools for early diagnosis and few therapeutic options. In recent years, major advances in molecular technologies, including whole-exome sequencing or sophisticated in vivo mouse models, have significantly enhanced our knowledge of pancreatic (pre-) malignancy and have identified highly specific gene mutations that accumulate during cancer progression as well as relevant signalling pathway molecules, which may help to establish new _targeted treatments.
The primary aim of this review is to highlight recent advances in the therapeutic management of pancreatic ductal adenocarcinoma (PDAC) and to discuss the latest findings that provide a better understanding of its complex molecular biology, which in turn will promote the development of more effective and fruitful therapeutic strategies. In particular, we limit our attention to recently identified signalling pathways and relevant molecules that promote the progression of pancreatic cancer and may therefore serve as ideal _target structures in our fight against this deadly disease.
Pancreatic cancer
An unusual aggressiveness and early metastatic locoregional, as well as distant, spread of pancreatic cancer cells is the basis of the urgent need for new therapeutic options for this cancer, as its incidence is still nearly equal to its mortality, in Western countries (Siegel et al., 2012). The failure of clinical treatment in patients with PDAC is often attributed to the early metastatic growth, a high level of drug resistance to standard therapy options and high rates of local recurrence (Strobel et al., 2013). However, inadequate diagnostic tools as well as the limited therapeutic options for PDAC, also known as pancreatic cancer, also account for its ranking as the fourth leading cause of cancer-related death worldwide (Siegel et al., 2012). Moreover, this complex genetic disease has one of the highest mortality rates of the known solid malignancies, and the overall 5 year survival rate among patients being less than 5%. The latter is, at least, partially due to an almost symptomless progression and evaluation of a patient in whom pancreatic cancer is suspected, frequently results in the diagnosis of tumours at locally advanced stages and consequently renders the majority of cases (∼75%) inoperable (Philip, 2008). For the remaining ∼25% of patients, the only chance of cure is surgical resection, which, in turn, improves the 5 year survival rate from ∼5%, for patients left untreated to ∼20–25% after resection (Bilimoria et al., 2007). Even those patients who are qualified for surgical resection accompanied by tumour-free margins frequently develop recurrent disease and eventually require palliative treatment, which largely depends on the overall health status of the patient (Saif, 2007). For the management of PDAC with adjuvant therapy, chemotherapy with the nucleotide analogue gemcitabine (Hertel et al., 1990) slightly improved overall survival compared with treatment with 5-fluorouracil (5-FU; Burris et al., 1997). In fact, the majority of patients, so far, receive little or no benefit from adjuvant therapies mainly because most of the cancer cells are either intrinsically resistant to chemotherapy and /or radiotherapy or they become resistant during therapy (O'Reilly and Abou-Alfa, 2007).
In most cases, the cause of PDAC is unknown and, although several risk factors and environmental factors such as diabetes, chronic inflammation (pancreatitis), alcohol and cigarette consumption have been discussed, evidence for a definitive causative role exists only for tobacco smokers, with the risk for development of PDAC in smokers being three times higher than in non-smokers (Herreros-Villanueva et al., 2013). More recently, infection with hepatitis B or C virus has emerged as an additional risk factor for PDAC, but the small number of available data involving mainly patients of Asian ethnicity, limit the full meta-analysis at this time (Fiorino et al., 2013). In conclusion, we do not have any truly preventative therapies for PDAC at this time.
Interestingly, up to 10% of patients with PDAC may have a hereditary causation. This possibility is based on three criteria: (i) individuals with two or more immediate relatives (first degree: parent or child) with PDAC, (ii) individuals with three or more close relatives (aunts/uncles, cousins, grandparents) with PDAC and (iii) individuals with even one relative with PDAC who were diagnosed before the age of 50, are all at higher risk of developing PDAC (Klein, 2013). A recently published study has identified lipocalin-2 and tissue inhibitor of metalloproteinase 1 as novel potential serum markers for the early detection of familial pancreatic cancer (Slater et al., 2013). Other studies have shown that individuals with the rare hereditary pancreatitis are also at higher risk of developing cancer (Lerch and Mayerle, 2013), although the relationship between chronic pancreatitis and PDAC is still a matter of debate.
Molecular genetics and signalling in pancreatic cancer
In the past few years, there have been important advances in understanding the complex biology of this form of cancer. Several investigations revealed that PDAC is a complex genetic disease and originates from the successive accumulation of several gene mutations that involve KRAS2, TP53, CDKN2A and SMAD4. Although it has been suggested for many years that PDAC evolved from ductal epithelial cells, recent research revealed the neoplastic involvement of acinar cells in this process as well (Ohike and Morohoshi, 2011). Acinar cell carcinoma is a rare neoplasm of the pancreas and accounts only for approximately 1% of the exocrine pancreas in adults (Klimstra, 2007), and compared with PDAC, acinar cell carcinoma shows lower rates of vascular invasion and lymph node involvement (Kitagami et al., 2007), and therefore extends the 5 year survival rates of these patients to 40–70% (Wisnoski et al., 2008). Moreover, the molecular features differ significantly from those of PDAC as acinar cell carcinoma typically does not carry mutations of the KRAS2, TP53, CDKN2A and SMAD4 genes (de Wilde et al., 2011).
As already demonstrated for other tumours such as the breast and colon, non-invasive stages were identified also for pancreatic cancer. These pre-malignant lesions occur in pancreatic ductal cells and are further characterized as three different histological subtypes for which there are clear classifying guidelines. These lesions are so-called pancreatic intraepithelial neoplasia (PanIN), intraductal pancreatic mucinous neoplasia (IPMN) and mucinous cystic neoplasia (MCN; Cooper et al., 2013). Because IPMN and MCN are considered as specific entities that are less well characterized, they fall beyond the scope of this review. Nevertheless, of these lesions, PanIN is the best characterized and most common histological precursor of PDAC. The starting point of these lesions occurs in smaller pancreatic ductal cells and can be further classified, depending on the degree of dysplasia [cytonuclear atypia, cell morphology, (in-) frequent mitosis], into the four grades: 1A, 1B, 2 and 3. The increasing grades of dysplasia in the different PanIN lesions reflect the morphological steps of tumour progression that precede the invasive cancer over time and are genetically accompanied by successive accumulation of mutations, involving activation of the KRAS2 oncogene and inactivation of the tumour suppressor genes TP53, SMAD4 and CDKN2A (Makohon-Moore et al., 2013; Table 1). However, PDAC is by far the most common and most lethal histological subtype. Moreover, mouse models of PDAC revealed a typical interplay between mutations in suppressor genes, oncogenes and genome maintenance genes that ultimately results in the development of PDAC, which, in turn, is a close model of the cognate human disease (Hingorani et al., 2005; Ling et al., 2012). Interestingly, gene mutations activating KRAS2 seem to be one of the earliest detectable genetic abnormalities in the progression model of PDAC and are readily detectable in the precursor lesion PanIN-1, whereas inactivation of the tumour suppressor CDKN2A is mainly detected in PanIN-2 and inactivation mutations of TP53 and SMAD4 genes are rarely detectable before PanIN-3. Additionally, KRAS2-activating mutations are present in more than 90% of cases and are therefore probably the single most common genetic abnormality that takes place very early in the progression of PDAC (Almoguera et al., 1988). This key genetic event would enhance the PanIN-1 to PanIN-2 transition, leading to an irreversible development into PDAC. Interestingly, oncogenic Kras is also required for the maintenance of PDAC in mice, suggesting that KRAS is not only required for the initiation of cancer (Collins et al., 2012). However, inactivating mutations in the tumour suppressor gene CDKN2A are present in virtually all PDAC specimens, whereas TP53- and SMAD4-inactivating mutations are detectable in 50–75% and 50% respectively (Hahn et al., 1996; Schutte et al., 1997; Morton et al., 2010).
Table 1.
Commonly mutated genes in pancreatic cancer
| Gene type | Gene | Intracellular function | Frequency in PDAC (%) |
|---|---|---|---|
| Oncogenes | KRAS2 | ERK–MAPK signalling | >90 |
| CyclinD | Cell cycle progression | 65 | |
| BRAF | ERK–MAPK signalling | ∼5 | |
| Tumour suppressor genes | CDKN2A | G1/S phase Cell cycle inhibition | >95 |
| SMAD4 | TGF-β-signalling | 50 | |
| TP53 | Cell cycle arrest | ∼75 | |
| Genome maintenance genes | MLH1 | DNA damage repair | 5 |
| BRCA2 | DNA damage repair | ∼10 |
The KRAS2 gene encodes a member of the RAS family of GTP binding proteins that display a wide range of cellular activities including survival, cytoskeletal remodelling, motility and proliferation. A variety of different stimuli, such as growth factor receptor/ligand interactions, result in receptor activation and are accompanied by signal transduction through intermediary proteins that promote the activation of KRAS. The known KRAS2-activating gene mutation is principally limited to a point mutation in codon 12 (G → D; Caldas and Kern, 1995), and transcription of this gene leads to the formation of an abnormal KRAS protein that is ‘trapped’ in its activated form. This constitutively active KRAS further induces the ‘uncontrolled’ activation of several downstream effector pathways, including RAF-MAPK, MEK1/2, Akt and PI3K. The PI3K-Akt pathway is an essential cell survival pathway that has various roles in several solid malignancies. This pathway is constitutively active in the vast majority of PDACs, and _targeting this pathway with small molecule inhibitors or knock-down strategies results in growth inhibition in vitro and in vivo (Eser et al., 2013). Interestingly, dual _targeting of PI3K-AKT2 oncogenes with RNA interference strategies, compared with inhibition of each oncogene alone, resulted in a significantly higher percentage of apoptosis and inhibited proliferation and colony formation in vitro and in vivo, suggesting that simultaneous _targeting of key players in the progression of PDAC will be a useful strategy to circumvent higher-order cancer cell signalling (Pawaskar et al., 2013). Thus, new effective treatment modalities will probably need to attack several _targets simultaneously or sequentially, and may therefore require personalized therapy regimens.
However, identification of additional mutations and an estimate of the prevalence of specific mutations are essential for development of new therapies for pancreatic cancer. Moreover, global expression platforms such as microarray technology or next-generation sequencing have undoubtedly helped the understanding of the biology of PDAC and the precursor lesions (Güngör et al., 2011; Chang et al., 2013). These studies provide multiple insights into differential expression patterns from a global view and may affect, in combination with in vivo mouse models, significantly the establishment of translational therapy applications in future studies. Thus, an analysis of pancreatic cancers by global genomic sequencing through the simultaneous screening of 24 pancreatic cancers by sequencing of ∼21 000 protein-coding exons per tumour revealed on average 63 genetic alterations, reflecting an extreme complexity of this disease (Jones et al., 2008). Therefore, ‘digging’ deeper into pancreatic cancer genomes by global sequencing is very important, because it will help to translate this knowledge into improved treatment modalities. However, there is still a lack of understanding of how such genetic alterations act in concert to induce development of PDAC. Interestingly, the genetic abnormalities affected in most of the analysed tumours comprise 12 partially overlapping core signalling pathways, although the pathway components that are altered in any one of the analysed tumours vary widely. Within these abnormal pathways, five of them were previously identified in other tumour entities as well and are linked to DNA damage repair, apoptosis, G1/S phase cell cycle progression, cell–cell adhesion and migration/invasion. The remaining pathways are considered as signalling cascades and can be further divided into three core groups: (i) embryonically relevant pathways such as sonic hedgehog, Wnt/Catenin and Notch; (ii) MAPK; and (iii) TGF-β signalling (Table 2).
Table 2.
Commonly affected signalling pathways in pancreatic cancer
| Signalling pathway | Altered genes in PDAC |
|---|---|
| DNA damage repair | TP53 |
| Apoptosis | TP53 |
| G1/S transition | CDKN2A |
| Cell adhesion | CDH1/Cadherin-1 |
| Regulation of invasion | ADAM11 / 12 |
| Embryonic signalling | |
| Hedgehog signalling | GLI1, SOX3, CREBBP |
| Notch signalling | TCF4 |
| Wnt signalling | WNT9A, MYC |
| MAPK signalling | |
| JNK | MAP4K3, ATF2 |
| TGF-β signalling | TGFBR2, SMAD4 |
| ERK | KRAS2 |
To discuss all the relevant and abnormal PDAC signalling pathways is beyond the scope of this review, but we will discuss in more detail some factors, especially the heparin-binding growth factor midkine (MK), which has been previously identified as being aberrantly expressed in three, patient-derived, highly chemotherapy-resistant, pancreatic cancer specimens (Güngör et al., 2011). Moreover, MK interferes with at least some of the abnormal pathways in PDAC, and research over the last two decades has revealed that this growth factor plays several critical roles in different aspects of cancer biology and therefore displays an attractive profile as a therapeutic _target for other cancers as well.
The growth factor MK
Growth factors are involved in the regulation of a number of cellular processes, especially proliferation and differentiation (Sounni and Noel, 2013). They are often overexpressed and have become effective _targets for cancer treatment (Yoong et al., 2011).
MK is a heparin-binding growth factor and cytokine, rich in basic amino acids and cysteines, with a molecular mass of 13 kDa (Tomomura et al., 1990). It was first described as a retinoic acid-inducible gene product during embryogenesis, more than twenty years ago (Kadomatsu et al., 1990). Since then, a range of MK functions have been described in normal and transformed tissue. MK shares 50% homology in amino acid sequence with pleiotrophin, the only other member of a unique two-member growth factor family (Zhang and Deuel, 1999). MK expression is restricted in healthy tissues and, in these, it is relatively high in small intestine, moderate in thyroid and weak in lung, colon, stomach, kidney and spleen (Maeda et al., 2007). MK plays also an important role in reproduction, development and repair, and is involved in the onset and/or progression of inflammatory diseases and malignancy (Muramatsu, 2011). Interestingly, MK is expressed at high levels in many different types of cancer (Kurtz et al., 1995; Muramatsu, 2002) and is involved in a number of biological activities that promote cell growth, survival, migration and angiogenesis (Dai, 2009). Interestingly, when MK is transfected into NIH3T3 cells, it is able to transform the cells, inducing the cells to form agar colonies and tumours in nude mice (Kadomatsu et al., 1997). The knock-down of MK in xenograft mouse models with colorectal and prostate cancer cells showed significant suppression of tumour growth (Takei et al., 2001; 2006,). The human MK gene is located on chromosome 11p11.2 with five exons: one non-coding and four coding exons (Uehara et al., 1992). MK is largely composed of two domains, each of which is compactly held by two or three disulfide bridges (Fabri et al., 1993). A truncated form of MK mRNA lacking exon 3, which encodes the N-terminal portion, has been found in a number of progressed cancers, including breast, gastric, liver, pancreas, oesophagus and colon (Kaname et al., 1996; Miyashiro et al., 1997; Nobata et al., 2005). This truncation does not generate a frameshift mutation and has not been detected in non-cancerous adult tissues (Kurtz et al., 1995; Kaname et al., 1996). MK activates various signalling pathways in different cells by interacting with the receptor-like protein tyrosine phosphatase-ζ, the low-density lipoprotein receptor-related protein 1 (LRP-1), anaplastic lymphoma kinase, and α4β1-and α6β1-integrins (Muramatsu, 2010). In particular, activation of Notch signalling by MK was recently reported in the context of transformed tissue (Güngör et al., 2011; Kishida et al., 2013).
MK is a secreted protein and blood levels can be easily monitored. In 87% of human adult cancers, serum MK levels are elevated and decrease after removal of the tumour (Ikematsu et al., 2003). In oesophageal cancer, urinary excretion of MK is elevated (Ikematsu et al., 2003), and high levels of serum MK are associated with tumour progression (Obata et al., 2005), tumour size and poor survival (Shimada et al., 2003).
MK and pancreatic cancer: receptors and signalling
Many signalling pathway aberrations contribute to the complicated pathogenesis of pancreatic tumorigenesis. Within this review, we would like to focus on pancreatic cancer signalling in terms of MK interaction and its contribution to the development of resistance to chemotherapy.
MK binds a variety of different receptors and signalling molecules, and initiates, thereby, many different higher order signalling pathways in pancreatic cancer cells. These receptors and signalling molecules include Notch-2 (Güngör et al., 2011; Kishida et al., 2013), JAK/STAT (Ratovitski et al., 1998; Huang et al., 2008a), LRP-1 (Sakamoto et al., 2011) and integrins (α4, α6, β1; Kadomatsu et al., 2013).
Notch signalling
The Notch pathway is highly conserved among most multicellular species, plays an important role in cell–cell communication and regulates embryonic development. Notch functions during development by preventing terminal cell differentiation until appropriate conditions are reached, or maintains a population of undifferentiated cells as progenitors in normal tissue. The activation of Notch receptors (Notch-1–4) by canonical ligands (delta-like ligand and Jagged families) results in γ–secretase-mediated cleavage and nuclear localization of its intracellular domain to engage other DNA-binding proteins and thus to regulate expression of its _target genes (Kopan, 2012; Figure 1). Notch is primarily activated during embryogenesis, but is, in common with other embryonic pathways, commonly reactivated in many cancers (McCleary-Wheeler et al., 2012). In the context of PDAC, increased expression of Notch receptors and ligands promoted the constitutive activation of Notch in early PanIN lesions (Miyamoto et al., 2003) and a loss of Notch-2 in a KrasG12D mouse model of pancreatic cancer resulted in inhibition of PanIN progression (Mullendore et al., 2009).
Figure 1.
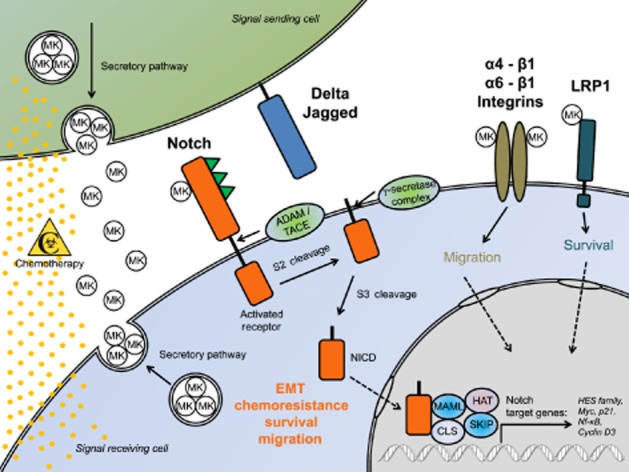
Elevated MK expression promotes activation of Notch signalling and chemoresistance in pancreatic cancer. Expression of the heparin-binding growth factor MK is frequently up-regulated in PDAC. MK expression is inducible by chemotherapy with gemcitabine (yellow dots), accompanied by its elevated secretion in chemoresistant cells. Secreted MK activates extracellular Notch-2 in a paracrine/autocrine fashion, followed by ADAM/TACE cleavage and shedding of the extracellular domain and subsequent γ–secretase-mediated generation of the soluble Notch-2 intracellular domain and its translocation into the nucleus to activate transcription of _target genes. Secreted MK-mediated Notch-2 activation is highly linked to epithelial-mesenchymal transition (EMT), chemoresistance and migration of PDAC cells, whereas MK depletion reverses EMT to mesenchymal–epithelial transition, a reversal strongly linked to increased chemosensitivity and decreased migratory potential of chemoresistant cells. Notch signalling contributes to regulation of cell death through crosstalk with NF-κB signalling. Blockade of the secreted MK–Notch-2 interaction or MK down-regulation is therefore a rational new strategy to circumvent chemoresistance in PDAC.
MK was recently identified, by global gene expression analyses, as being highly up-regulated in three independent, patient-derived, primary PDAC cell lines, which were highly resistant to chemotherapy (Güngör et al., 2011). The aim of the study was to identify the relevant factors that are involved in and/or induce resistance to chemotherapy in pancreatic cancer. Interestingly, chemotherapy of resistant PDAC cell lines with gemcitabine induced expression and secretion of MK in a dose-dependent manner, whereas depletion of MK resulted in strong sensitization to gemcitabine. Additionally, MK expression was not inducible in gemcitabine-treated chemosensitive PDAC cell lines, suggesting that MK is indeed necessary to promote survival during chemotherapy. Moreover, pathway analyses revealed that chemotherapy-induced MK secretion triggered activation of extracellular Notch-2, which in turn, promoted the epithelial–mesenchymal transition (EMT) that accompanies increased resistance to chemotherapy and migration potential (Figure 1). Interestingly, down-regulation of MK in chemoresistant PDAC cells reversed the mesenchymal phenotype to an epithelial phenotype, a transition linked to increased chemosensitivity and decreased migration. Soluble recombinant MK also triggers activation of the Notch-2 pathway that resulted in up-regulation of two Notch-2 downstream _targets, Hes1 and NF-κB. Down-regulation of Notch-2 was also linked to increased chemosensitivity to gemcitabine, suggesting that secretion of MK triggers Notch-2 activation and leads to resistance to chemotherapy in pancreatic cancer. Thus, Notch signalling not only contributes to proliferation and differentiation, but also to regulation of cell death through crosstalk with NF-κB signalling (Güngör et al., 2011). In support of such findings, pathological interaction between MK and Notch-2 was confirmed very recently in a mouse model of neuroblastoma (Kishida et al., 2013). Here, MK deficiency resulted in attenuated Notch-2 activation, whereas in pre-cancerous lesions, the genetic ablation of MK showed defects in Notch-2 activation and decreased expression of the Notch-2 _target gene Hes1. Interestingly, deletion of MK also delayed tumour formation and reduced tumour incidence in vivo (Kishida et al., 2013).
Furthermore, immortalized keratinocytes expressing MK or treated with recombinant MK displayed dramatic changes in cell morphology consistent with EMT and resulted in decreased expression of epithelial markers, such as E-cadherin, β-catenin and γ-catenin and an increase in mesenchymal markers such as fibronectin, vimentin and smooth muscle actin; (Huang et al., 2008b). This process was mediated by an interaction between MK and the ligand-binding domain of Notch-2. MK also mediated the nuclear accumulation of Notch-2 in HaCaT cells and complex formation between the Notch-2 downstream intermediates, Hes1 and STAT3 (Huang et al., 2008b). The effectiveness of Notch inhibition with γ-secretase inhibitors is currently under investigation in clinical trials.
JAK/STAT signalling
IL–6-type cytokines usually trigger tyrosine kinases of the JAK family as mediators of signal transduction (Lütticken et al., 1994) and STAT3 activation plays a significant role in the progression of PDAC (Lesina et al., 2011). Interestingly, there was crosstalk between Notch and JAK/STAT signalling pathways in epithelial cells of Drosophila and mammals (Kamakura et al., 2004; Assa-Kunik et al., 2007) and STAT3 was activated in the presence of active Notch, as well as the Notch effectors Hes1 and Hes5. The Hes proteins associate with JAK2 and STAT3, facilitating complex formation and thus promoting STAT3 phosphorylation and activation. Notch-mediated activation of STAT3 seems to be essential for maintenance of radial glial cells and differentiation of astrocytes in the developing CNS. Moreover, exposure to recombinant MK enhanced formation of a protein complex between Hes1 and STAT3. This interaction led to phosphorylation of STAT3, thereby activating signalling. MK also facilitated recruitment of JAK2 into Hes1-containing complexes in nuclear fractions (Huang et al., 2008b). These results suggest that direct protein–protein interactions are involved in the crosstalk between the Notch–Hes and JAK–STAT pathways (Huang et al., 2008b).
LRP-1
MK binds and signals via the low-density LRP-1 and mediates survival of embryonic neurons (Muramatsu et al., 2000). This is achieved either by LRP–1-mediated endocytosis of MK, which resulted in suppression of apoptosis in the nucleus (Shibata et al., 2002), or by signal transduction through the LRP-1 receptor itself. This direct signalling is mediated by a cytoplasmic domain to which adaptor proteins can bind (Muramatsu et al., 2000). The cytoplasmic tail of LRP-1 is associated with SHC and modulates JNK, a member of the MAPK family (Barnes et al., 2001; Lutz et al., 2002). Recently, LRP-1 was identified in a model of the progression of IPMN of the pancreas, as a potential mediator of cancer progression (Jiang et al., 2013). Furthermore, in an isografted PDAC mouse model, recruitment of monocytes into orthotopic and subcutaneous tumours was significantly increased in animals with LRP–1–knock-out in their myeloid lineage cells, compared with that in WT mice (Staudt et al., 2013).
Integrins (α4, α6, β1)
MK also interacts with α4β1-and α6β1-integrins. The α4β1-integrin mediates cell migration of lymphocytes (Rose et al., 2001), recruitment of neutrophils to inflammatory sites (Burns et al., 2001) and mediates migration of epicardial progenitors in the heart (Sengbusch et al., 2002). Interestingly, functional blockade of α4-integrin with monoclonal antibodies inhibited MK-induced migration of osteosarcoma cells (Muramatsu et al., 2004). MK binding to α6β1-integrin is also functionally important because it promoted survival and tumorigenesis in head and neck squamous carcinoma cells (Huang et al., 2008a). In PDAC, the expression of α6-integrin is enhanced and redistributed (Halatsch et al., 1997). Inhibition of α6β1-integrin with antibodies inhibited cancer cell adhesion and invasion (Weinel et al., 1995).
Therapy options for pancreatic cancer
Cancer statistics reveal that nearly a quarter of a million people worldwide die annually from pancreatic cancer, and its incidence will increase by ∼2% per year in developed countries. The main cause of these death rates is primarily a combination of a symptomless disease progression and frequent occurrence of unresectable highly therapy-resistant tumours, suggesting that surgery can only be performed in a minority of patients. Notwithstanding, resectable PDAC patients frequently develop recurrent disease and therefore receive adjuvant therapy. For advanced-stage patients, the treatment modality remains palliative. However, the care of advanced-stage patients with currently available treatment options provides a median overall survival of only 8–10 months. These therapy options for PDAC range from systemic chemotherapy alone to combined options with chemoradiation therapy (Guo et al., 2013) and post-operative treatment improves patient outcomes significantly, suggesting that active treatment of pancreatic cancer is principally beneficial. Nearly two decades ago, a randomized clinical trial of gemcitabine versus 5-FU showed chemotherapy with gemcitabine was better than 5-FU for treatment of advanced pancreatic cancer (Burris et al., 1997). Nevertheless, the primary end point of this trial was to determine whether gemcitabine provided any advantage over monotherapy with 5-FU in terms of clinical benefit (pain, performance status, weight), objective response and time to progressive disease or survival. Although gemcitabine only marginally improved median patient survival (5.65 vs. 4.41 months), this drug has entered clinical practice and has replaced 5-FU over time, primarily because the ‘clinical response rate’ was better than that of 5-FU. The use of gemcitabine was accompanied by a favourable toxicity profile and was strongly linked with sustained improvements in pain, analgesic consumption and Karnofsky performance status, observed in ∼24% of gemcitabine-treated patients compared with ∼5% of 5–FU-treated patients (Burris et al., 1997). Thus, gemcitabine alone or in combination with 5–FU-based chemoradiotherapy can now be considered as the standard of care for locally advanced and/or metastatic disease, as first-line therapy (Philip, 2008). Since the late 1990s, several attempts have been made in order to improve the patient outcome by combining gemcitabine with other cytotoxic agents (epirubicin, mitomycin, cisplatin). Unfortunately, most of these combinations failed to achieve statistically significant survival benefit over single-agent gemcitabine treatment, except for the combined treatment with platinum-containing drugs, which showed marginal survival benefit over gemcitabine treatment alone (Heinemann et al., 2007). The majority of patients receive little or no benefit from current chemotherapies mainly because most of the cancer cells are either intrinsically chemoresistant or they become resistant during therapy. Also, extrinsic mechanisms such as the tumour stroma have been discussed as relevant for resistance to chemotherapy in PDAC (Long et al., 2011). Therefore, it is of great interest to identify and characterize novel molecules that promote the development of resistance to chemotherapy in order to establish new therapy options _targeted at these mechanisms of resistance.
One such molecule is MK whose research over the past years reflected an attractive new _target for the treatment of PDAC because its expression and secretion was elevated in more than 50% of pancreatic cancers, and small interfering RNA (siRNA)-mediated down-regulation of MK was strongly linked with the reversal of chemoresistance to gemcitabine. Stress-induced up-regulation of MK was already shown to be highly cytoprotective against various stimuli that tend to result in tissue damage and ischaemic injury (Muramatsu, 2011). However, the external stimuli and intracellular signalling that control MK expression are unknown. It is worth mentioning that MK may serve as a marker during chemotherapy or it may help to stratify those patients who are eligible for _targeted therapy, but this needs further investigations in in vivo models. Many studies have shown that MK promotes various cellular activities during development and cancer. In PDAC, MK promotes not only survival in response to chemotherapy but also proliferation and migration of cells in vitro, suggesting that MK plays many different roles, relevant to cancer progression (Rawnaq et al., unpublished). Because MK was found to be dose-dependently released from PDAC cells by chemotherapy and was accompanied by increased Notch signalling, it is likely that Notch is constitutively activated by a MK-dependent autocrine loop during chemotherapy. Further, it is possible that increased MK secretion may also trigger intercellular chemoresistance by activating Notch signalling in neighbouring cells in order to activate defence mechanisms against drug toxicity through up-regulation of NF-κB (Güngör et al., 2011; Figure 1). Thus, the development of MK inhibitors based on a molecular-driven strategy has been considered as an attractive idea to prevent tumour growth and anti-apoptosis in cancer cells. Potential knock-down strategies of MK in transformed tissue involve antisense oligo-DNA (Dai et al., 2009), siRNA (Takei et al., 2006), RNA aptamers (Kishida et al., 2013), compound inhibition (Matsui et al., 2010) and a conditionally replicating adenovirus containing the MK gene promoter for _targeting MK-positive tumours (Terao et al., 2007). Interestingly, all of these strategies have already shown cancer-specific effects and thus reinforce the concept that MK can be effectively _targeted to reduce tumour growth, metastasis, angiogenesis and survival. Presumably, MK knock-down may enhance the efficacy of current cytotoxic cancer therapies, which, in turn, might be of great benefit for patients suffering from chemoresistant cancers, as is too often the case for PDAC.
Conclusion
The identification of MK as an activator of Notch signalling provided relevant new insights into the functions of MK. The elevated MK expression and secretion frequently found in PDAC promotes survival and proliferation and provides support for cancer cells escaping chemotherapy-induced apoptosis through activation of Notch signalling. Thus, _targeting MK–Notch-2 interactions is a rational, new strategy to circumvent intrinsic as well as acquired resistance to chemotherapy in PDAC.
Acknowledgments
CG and BH are funded by the ‘Roggenbuck Foundation’, Hamburg, Germany.
Glossary
- 5-FU
5-fluorouracil
- EMT
epithelial-mesenchymal transition
- IPMN
intraductal pancreatic mucinous neoplasia
- LRP-1
low-density lipoprotein receptor-related protein 1
- MCN
mucinous cystic neoplasia
- MK
midkine
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
Conflict of interest
None.
References
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- Assa-Kunik E, Torres IL, Schejter ED, Johnston DS, Shilo BZ. follicle cells are patterned by multiple levels of Notch signaling and antagonism between the Notch and JAK/STAT pathways. Development. 2007;134:1161–1169. doi: 10.1242/dev.02800. Drosophila. [DOI] [PubMed] [Google Scholar]
- Barnes H, Larsen B, Tyers M, van Der Geer P. Tyrosine-phosphorylated low density lipoprotein receptor-related protein 1 (Lrp1) associates with the adaptor protein SHC in SRC-transformed cells. J Biol Chem. 2001;276:19119–19125. doi: 10.1074/jbc.M011437200. [DOI] [PubMed] [Google Scholar]
- Bilimoria KY, Bentrem DJ, Ko CY, Ritchey J, Stewart AK, Winchester DP, et al. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: report from the National Cancer Database. Cancer. 2007;110:738–744. doi: 10.1002/cncr.22852. [DOI] [PubMed] [Google Scholar]
- Burns JA, Issekutz TB, Yagita H, Issekutz AC. The alpha 4 beta 1 (very late antigen (VLA)-4, CD49d/CD29) and alpha 5 beta 1 (VLA-5, CD49e/CD29) integrins mediate beta 2 (CD11/CD18) integrin-independent neutrophil recruitment to endotoxin-induced lung inflammation. J Immunol. 2001;166:4644–4649. doi: 10.4049/jimmunol.166.7.4644. [DOI] [PubMed] [Google Scholar]
- Burris HA, III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- Chang Z, Li Z, Wang X, Kang Y, Yuan Y, Niu J, et al. Deciphering the mechanisms of tumorigenesis in human pancreatic ductal epithelial cells. Clin Cancer Res. 2013;19:549–559. doi: 10.1158/1078-0432.CCR-12-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CL, O'Toole SA, Kench JG. Classification, morphology and molecular pathology of premalignant lesions of the pancreas. Pathology. 2013;45:286–304. doi: 10.1097/PAT.0b013e32835f2205. [DOI] [PubMed] [Google Scholar]
- Dai LC, Yao X, Wang X, Niu SQ, Zhou LF, Fu FF, et al. In vitro and in vivo suppression of hepatocellular carcinoma growth by midkine-antisense oligonucleotide-loaded nanoparticles. World J Gastroenterol. 2009;15:1966–1972. doi: 10.3748/wjg.15.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L-C. Midkine translocated to nucleoli and involved in carcinogenesis. World J Gastroenterol. 2009;15:412–416. doi: 10.3748/wjg.15.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- Fabri L, Maruta H, Muramatsu H, Muramatsu T, Simpson RJ, Burgess AW, et al. Structural characterisation of native and recombinant forms of the neurotrophic cytokine MK. J Chromatogr. 1993;646:213–225. doi: 10.1016/s0021-9673(99)87023-x. [DOI] [PubMed] [Google Scholar]
- Fiorino S, Chili E, Bacchi-Reggiani L, Masetti M, Deleonardi G, Grondona AG, et al. Association between hepatitis B or hepatitis C virus infection and risk of pancreatic adenocarcinoma development: a systematic review and meta-analysis. Pancreatology. 2013;13:147–160. doi: 10.1016/j.pan.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Güngör C, Zander H, Effenberger KE, Vashist YK, Kalinina T, Izbicki JR, et al. Notch signaling activated by replication stress-induced expression of midkine drives epithelial–mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011;71:5009–5019. doi: 10.1158/0008-5472.CAN-11-0036. [DOI] [PubMed] [Google Scholar]
- Guo XZ, Cui ZM, Liu X. Current developments, problems and solutions in the non-surgical treatment of pancreatic cancer. World J Gastrointest Oncol. 2013;5:20–28. doi: 10.4251/wjgo.v5.i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Halatsch ME, Hirsch-Ernst KI, Kahl GF, Weinel RJ. Increased expression of alpha6-integrin receptors and of mRNA encoding the putative 37 kDa laminin receptor precursor in pancreatic carcinoma. Cancer Lett. 1997;118:7–11. doi: 10.1016/s0304-3835(97)00217-6. [DOI] [PubMed] [Google Scholar]
- Heinemann V, Labianca R, Hinke A, Louvet C. Increased survival using platinum analog combined with gemcitabine as compared to single-agent gemcitabine in advanced pancreatic cancer: pooled analysis of two randomized trials, the GERCOR/GISCAD intergroup study and a German multicenter study. Ann Oncol. 2007;18:1652–1659. doi: 10.1093/annonc/mdm283. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Hijona E, Bañales JM, Cosme A, Bujanda L. Alcohol consumption on pancreatic diseases. World J Gastroenterol. 2013;19:638–647. doi: 10.3748/wjg.v19.i5.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, et al. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) Cancer Res. 1990;50:4417–4422. [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hoque MO, Wu F, Trink B, Sidransky D, Ratovitski EA. Midkine induces epithelial–mesenchymal transition through Notch2/Jak2-Stat3 signaling in human keratinocytes. Cell Cycle. 2008a;7:1613–1622. doi: 10.4161/cc.7.11.5952. [DOI] [PubMed] [Google Scholar]
- Huang Y, Sook-Kim M, Ratovitski E. Midkine promotes tetraspanin–integrin interaction and induces FAK-Stat1alpha pathway contributing to migration/invasiveness of human head and neck squamous cell carcinoma cells. Biochem Biophys Res Commun. 2008b;377:474–478. doi: 10.1016/j.bbrc.2008.09.138. [DOI] [PubMed] [Google Scholar]
- Ikematsu S, Okamoto K, Yoshida Y, Oda M, Sugano-Nagano H, Ashida K, et al. High levels of urinary midkine in various cancer patients. Biochem Biophys Res Commun. 2003;306:329–332. doi: 10.1016/s0006-291x(03)00984-7. [DOI] [PubMed] [Google Scholar]
- Jiang P, Zang W, Wang L, Xu Y, Liu Y, Deng SX. Protein–protein interaction and SNP analysis in intraductal papillary mucinous neoplasm. Gene. 2013;513:219–224. doi: 10.1016/j.gene.2012.10.047. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadomatsu K, Huang RP, Suganuma T, Murata F, Muramatsu T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J Cell Biol. 1990;110:607–616. doi: 10.1083/jcb.110.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadomatsu K, Hagihara M, Akhter S, Fan QW, Muramatsu H, Muramatsu T. Midkine induces the transformation of NIH3T3 cells. Br J Cancer. 1997;75:354–359. doi: 10.1038/bjc.1997.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadomatsu K, Kishida S, Tsubota S. The heparin-binding growth factor midkine: the biological activities and candidate receptors. J Biochem. 2013;153:511–521. doi: 10.1093/jb/mvt035. [DOI] [PubMed] [Google Scholar]
- Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6:547–554. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- Kaname T, Kadomatsu K, Aridome K, Yamashita S, Sakamoto K, Ogawa M, et al. The expression of truncated MK in human tumors. Biochem Biophys Res Commun. 1996;219:256–260. doi: 10.1006/bbrc.1996.0214. [DOI] [PubMed] [Google Scholar]
- Kishida S, Mu P, Miyakawa S, Fujiwara M, Abe T, Sakamoto K, et al. Midkine promotes neuroblastoma through Notch2 signaling. Cancer Res. 2013;73:1318–1327. doi: 10.1158/0008-5472.CAN-12-3070. [DOI] [PubMed] [Google Scholar]
- Kitagami H, Kondo S, Hirano S, Kawakami H, Egawa S, Tanaka M. Acinar cell carcinoma of the pancreas: clinical analysis of 115 patients from Pancreatic Cancer Registry of Japan Pancreas Society. Pancreas. 2007;35:42–46. doi: 10.1097/mpa.0b013e31804bfbd3. [DOI] [PubMed] [Google Scholar]
- Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer. 2013;13:66–74. doi: 10.1038/nrc3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimstra DS. Nonductal neoplasms of the pancreas. Mod Pathol. 2007;20:S94–S112. doi: 10.1038/modpathol.3800686. [DOI] [PubMed] [Google Scholar]
- Kopan R. Notch signaling. Cold Spring Harb Perspect Biol. 2012;4(10) doi: 10.1101/cshperspect.a011213. Oct 1; Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Schulte AM, Wellstein A. Pleiotrophin and midkine in normal development and tumor biology. Crit Rev Oncog. 1995;6:151–177. [PubMed] [Google Scholar]
- Lerch MM, Mayerle J. 50 years of progress in pathophysiology, diagnosis and treatment of chronic pancreatitis. Z Gastroenterol. 2013;51:358–362. doi: 10.1055/s-0033-1335278. [DOI] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C, et al. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther _targets. 2011;15:817–828. doi: 10.1517/14728222.2011.566216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lütticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89–92. doi: 10.1126/science.8272872. [DOI] [PubMed] [Google Scholar]
- Lutz C, Nimpf J, Jenny M, Boecklinger K, Enzinger C, Utermann G, et al. Evidence of functional modulation of the MEKK/JNK/cJun signaling cascade by the low density lipoprotein receptor-related protein (LRP) J Biol Chem. 2002;277:43143–43151. doi: 10.1074/jbc.M204426200. [DOI] [PubMed] [Google Scholar]
- Maeda S, Shinchi H, Kurahara H, Mataki Y, Noma H, Maemura K, et al. Clinical significance of midkine expression in pancreatic head carcinoma. Br J Cancer. 2007;97:405–411. doi: 10.1038/sj.bjc.6603879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makohon-Moore A, Brosnan JA, Iacobuzio-Donahue CA. Pancreatic cancer genomics: insights and opportunities for clinical translation. Genome Med. 2013;5:26. doi: 10.1186/gm430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Ichihara-Tanaka K, Lan C, Muramatsu H, Kondou T, Hirose C, et al. Midkine inhibitors: application of a simple assay procedure to screening of inhibitory compounds. Int Arch Med. 2010;21:12. doi: 10.1186/1755-7682-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleary-Wheeler AL, McWilliams R, Fernandez-Zapico ME. Aberrant signalling pathways in pancreatic cancer: a two compartment view. Mol Carcinog. 2012;51:25–39. doi: 10.1002/mc.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- Miyashiro I, Kaname T, Shin E, Wakasugi E, Monden T, Takatsuka Y, et al. Midkine expression in human breast cancers: expression of truncated form. Breast Cancer Res Treat. 1997;43:1–6. doi: 10.1023/a:1005748728351. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res. 2009;15:2291–2301. doi: 10.1158/1078-0432.CCR-08-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu H, Zou K, Sakaguchi N, Ikematsu S, Sakuma S, Muramatsu T. LDL receptor-related protein as a component of the midkine receptor. Biochem Biophys Res Commun. 2000;270:936–941. doi: 10.1006/bbrc.2000.2549. [DOI] [PubMed] [Google Scholar]
- Muramatsu H, Zou P, Suzuki H, Oda Y, Chen GY, Sakaguchi N, et al. alpha4beta1-and alpha6beta1-integrins are functional receptors for midkine, a heparin-binding growth factor. J Cell Sci. 2004;117:5405–5415. doi: 10.1242/jcs.01423. [DOI] [PubMed] [Google Scholar]
- Muramatsu T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J. Biochem. 2002;132:359–371. doi: 10.1093/oxfordjournals.jbchem.a003231. [DOI] [PubMed] [Google Scholar]
- Muramatsu T. Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:410–425. doi: 10.2183/pjab.86.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T. Midkine: a promising molecule for drug development to treat diseases of the central nervous system. Curr Pharm Des. 2011;17:410–423. doi: 10.2174/138161211795164167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobata S, Shinozawa T, Sakanishi A. Truncated midkine induces transformation of cultured cells and short latency of tumorigenesis in nude mice. Cancer Lett. 2005;219:83–89. doi: 10.1016/j.canlet.2004.07.003. [DOI] [PubMed] [Google Scholar]
- O'Reilly EM, Abou-Alfa GK. Cytotoxic therapy for advanced pancreatic adenocarcinoma. Semin Oncol. 2007;34:347–353. doi: 10.1053/j.seminoncol.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Obata Y, Kikuchi S, Lin Y, Yagyu K, Muramatsu T, Kumai H, et al. Serum midkine concentrations and gastric cancer. Cancer Sci. 2005;96:54–56. doi: 10.1111/j.1349-7006.2005.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohike N, Morohoshi T. Exocrine pancreatic neoplasms of nonductal origin: acinar cell carcinoma, pancreatoblastoma, and solid-pseudopapillary neoplasm. Surg Pathol Clin. 2011;4:579–588. doi: 10.1016/j.path.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Pawaskar DK, Straubinger RM, Fetterly GJ, Hylander BH, Repasky EA, Ma WW, et al. Synergistic interactions between sorafenib and everolimus in pancreatic cancer xenografts in mice. Cancer Chemother Pharmacol. 2013;71:1231–1240. doi: 10.1007/s00280-013-2117-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip PA. _targeted therapies for pancreatic cancer. Gastrointest Cancer Res. 2008;2(4 Suppl. 2):S16–S19. [PMC free article] [PubMed] [Google Scholar]
- Ratovitski EA, Kotzbauer PT, Milbrandt J, Lowenstein CJ, Burrow CR. Midkine induces tumor cell proliferation and binds to a high affinity signalling receptor associated with JAK tyrosine kinases. J Biol Chem. 1998;273:3654–3660. doi: 10.1074/jbc.273.6.3654. [DOI] [PubMed] [Google Scholar]
- Rose DM, Grabovsky V, Alon R, Ginsberg MH. The affinity of integrin alpha(4)beta(1) governs lymphocyte migration. J Immunol. 2001;167:2824–2830. doi: 10.4049/jimmunol.167.5.2824. [DOI] [PubMed] [Google Scholar]
- Saif MW. Controversies in the adjuvant treatment of pancreatic adenocarcinoma. JOP. 2007;8:545–552. [PubMed] [Google Scholar]
- Sakamoto K, Bu G, Chen S, Takei Y, Hibi K, Kodera Y, et al. Premature ligand-receptor interaction during biosynthesis limits the production of growth factor midkine and its receptor LDL receptor-related protein 1. J Biol Chem. 2011;286:8405–8413. doi: 10.1074/jbc.M110.176479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–3130. [PubMed] [Google Scholar]
- Sengbusch JK, He W, Pinco KA, Yang JT. Dual functions of [alpha]4[beta]1 integrin in epicardial development: initial migration and long-term attachment. J Cell Biol. 2002;157:873–882. doi: 10.1083/jcb.200203075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Muramatsu T, Hirai M, Inui T, Kimura T, Saito H, et al. Nuclear _targeting by the growth factor midkine. Mol Cell Biol. 2002;22:6788–6796. doi: 10.1128/MCB.22.19.6788-6796.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H, Nabeya Y, Tagawa M, Okazumi S, Matsubara H, Kadomatsu K, et al. Preoperative serum midkine concentration is a prognostic marker for esophageal squamous cell carcinoma. Cancer Sci. 2003;94:628–632. doi: 10.1111/j.1349-7006.2003.tb01494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics 2012. CA Cancer J Clin. 2012;62:220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- Slater EP, Fendrich V, Strauch K, Rospleszcz S, Ramaswamy A, Mätthai E, et al. LCN2 and TIMP1 as potential serum markers for the early detection of familial pancreatic cancer. Transl Oncol. 2013;6:99–103. doi: 10.1593/tlo.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni NE, Noel A. _targeting the tumor microenvironment for cancer therapy. Clin Chem. 2013;59:85–93. doi: 10.1373/clinchem.2012.185363. [DOI] [PubMed] [Google Scholar]
- Staudt ND, Jo M, Hu J, Bristow JM, Pizzo DP, Gaultier A, et al. Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. 2013;73:3902–3912. doi: 10.1158/0008-5472.CAN-12-4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel O, Hartwig W, Hackert T, Hinz U, Berens V, Grenacher L, et al. Re-resection for isolated local recurrence of pancreatic cancer is feasible, safe, and associated with encouraging survival. Ann Surg Oncol. 2013;20:964–972. doi: 10.1245/s10434-012-2762-z. [DOI] [PubMed] [Google Scholar]
- Takei Y, Kadomatsu K, Matsuo S, Itoh H, Nakazawa K, Kubota S, et al. Antisense oligodeoxynucleotide _targeted to Midkine, a heparin-binding growth factor, suppresses tumorigenicity of mouse rectal carcinoma cells. Cancer Res. 2001;61:8486–8491. [PubMed] [Google Scholar]
- Takei Y, Kadomatsu K, Goto T, Muramatsu T. Combinational antitumor effect of siRNA against midkine and paclitaxel on growth of human prostate cancer xenografts. Cancer. 2006;107:864–873. doi: 10.1002/cncr.22068. [DOI] [PubMed] [Google Scholar]
- Terao S, Shirakawa T, Kubo S, Bishunu A, Lee SJ, Goda K, et al. Midkine promoter-based conditionally replicative adenovirus for _targeting midkine-expressing human bladder cancer model. Urology. 2007;70:1009–1013. doi: 10.1016/j.urology.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Tomomura M, Kadomatsu K, Matsubara S, Muramatsu T. A retinoic acid-responsive gene, MK, found in the teratocarcinoma system. Heterogeneity of the transcript and the nature of the translation product. J Biol Chem. 1990;265:10765–10770. [PubMed] [Google Scholar]
- Uehara K, Matsubara S, Kadomatsu K, Tsutsui J, Muramatsu T. Genomic structure of human midkine (MK), a retinoic acid-responsive growth/differentiation factor. J Biochem. 1992;111:563–567. doi: 10.1093/oxfordjournals.jbchem.a123797. [DOI] [PubMed] [Google Scholar]
- Weinel RJ, Rosendahl A, Pinschmidt E, Kisker O, Simon B, Santoso S. The alpha 6-integrin receptor in pancreatic carcinoma. Gastroenterology. 1995;108:523–532. doi: 10.1016/0016-5085(95)90082-9. [DOI] [PubMed] [Google Scholar]
- de Wilde RF, Ottenhof NA, Jansen M, Morsink FH, de Leng WW, Offerhaus GJ, et al. Analysis of LKB1 mutations and other molecular alterations in pancreatic acinar cell carcinoma. Mod Pathol. 2011;24:1229–1236. doi: 10.1038/modpathol.2011.83. [DOI] [PubMed] [Google Scholar]
- Wisnoski NC, Townsend CM, Jr, Nealon WH, Freeman JL, Riall TS. 672 patients with acinar cell carcinoma of the pancreas: a population based comparison to pancreatic adenocarcinoma. Surgery. 2008;144:141–148. doi: 10.1016/j.surg.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Yoong J, Michael M, Leong T. _targeted therapies for gastric cancer: current status. Drugs. 2011;71:1367–1384. doi: 10.2165/11592530-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Zhang N, Deuel TF. Pleiotrophin and midkine, a family of mitogenic and angiogenic heparin-binding growth and differentiation factors. Curr Opin Hematol. 1999;6:44–50. doi: 10.1097/00062752-199901000-00008. [DOI] [PubMed] [Google Scholar]