

Background: FGF-23, a bone-derived hormone, regulates phosphate and vitamin D in the kidney.
Results: Genetic and pharmacological manipulations of FGF-23 alter erythropoiesis and HSC frequency both in young adult age and embryonically.
Conclusion: Fgf-23 regulates erythropoiesis through Epo and independent of vitamin D.
Significance: These findings provide a new _target for treating blood disorders associated with bone and renal defects.
Keywords: Bone, Bone Marrow, Erythrocyte, Erythropoeisis, Hematopoiesis, Kidney, FGF-23, Anemia
Abstract
Abnormal blood cell production is associated with chronic kidney disease (CKD) and cardiovascular disease (CVD). Bone-derived FGF-23 (fibroblast growth factor-23) regulates phosphate homeostasis and bone mineralization. Genetic deletion of Fgf-23 in mice (Fgf-23−/−) results in hypervitaminosis D, abnormal mineral metabolism, and reduced lymphatic organ size. Elevated FGF-23 levels are linked to CKD and greater risk of CVD, left ventricular hypertrophy, and mortality in dialysis patients. However, whether FGF-23 is involved in the regulation of erythropoiesis is unknown. Here we report that loss of FGF-23 results in increased hematopoietic stem cell frequency associated with increased erythropoiesis in peripheral blood and bone marrow in young adult mice. In particular, these hematopoietic changes are also detected in fetal livers, suggesting that they are not the result of altered bone marrow niche alone. Most importantly, administration of FGF-23 in wild-type mice results in a rapid decrease in erythropoiesis. Finally, we show that the effect of FGF-23 on erythropoiesis is independent of the high vitamin D levels in these mice. Our studies suggest a novel role for FGF-23 in erythrocyte production and differentiation and suggest that elevated FGF-23 levels contribute to the pathogenesis of anemia in patients with CKD and CVD.
Introduction
In vertebrates, hematopoiesis within the bone marrow (BM)2 is established by multiple sequential events involving several anatomical locations. During fetal development, the cells that initiate hematopoiesis, namely hematopoietic stem cells (HSCs), colonize the fetal liver from 9.5 days postcoitum onward where they expand and differentiate (1–3). After birth, these cells migrate from the fetal liver to the BM, which becomes the major site of hematopoiesis during adult life.
Changes in blood cell production are apparent in several chronic diseases including chronic kidney disease (CKD) and cardiovascular disease. Patients with CKD are often diagnosed with severe anemia because of the inability of their kidneys to produce erythropoietin, the hormone responsible for red blood cell (RBC) production in the BM in response to low oxygen levels in the blood (4, 5). Additionally, CKD patients also suffer from osteopenia, osteoporosis, or osteomalacia, giving rise to the term “chronic kidney disease-mineral bone disorder,” affirming a link between mineral metabolism and kidney function. Recent studies indicate that kidney function in CKD mineral bone disorder patients is influenced by a circulating factor produced by the skeleton namely, fibroblast growth factor-23 (FGF-23) (6–8). In CKD, circulating levels of FGF-23 gradually increase as renal function declines reaching 1000-fold above the normal range in advanced renal failure (9, 10). High levels of FGF-23 have also been associated with an increase in cardiovascular disease events in hemodialysis patients and development of left ventricular hypertrophy (11, 12).
FGF-23 is an osteocyte-produced 30-kDa secreted protein that is crucial for phosphate homeostasis and vitamin D metabolism. Serum phosphate levels are regulated by FGF-23 via two key pathways: 1) directly by inhibiting sodium-dependent phosphate reabsorption in the kidneys and 2) through indirect suppression of renal 1,25(OH)2D3 production (13, 14). Circulating FGF-23 levels are increased in CKD patients, as well as several bone disorders including autosomal dominant hypophosphatemic rickets and X-linked hypophosphatemia (15–17). In contrast, decreased levels of biologically active FGF-23 result in tumoral calcinosis, which is characterized by elevated serum phosphate and 1,25(OH)2D3 levels and soft tissue calcifications (18, 19). Similar to humans, mice deficient in Fgf-23 (Fgf-23−/−) exhibit hyperphosphatemia and hypervitaminosis D and also present with tissue and vascular calcifications (14, 20). Additionally, Fgf-23−/− mice display aberrant bone mineralization accompanied by decreased bone mineral density, trabeculae, and osteoblast numbers (14, 20). Elimination of vitamin D in Fgf-23−/− mice reversed the hyperphosphatemia and hypercalcemia and abolished the soft tissue and vascular calcifications (21). These data indicate that vitamin D partly mediates the function of Fgf-23 to regulate phosphate homeostasis and bone mineralization.
Bone components such as osteoblasts, extracellular matrix, and minerals are involved in the regulation of hematopoietic stem cell function in the adult mammal. Postnatal depletion of osteoblasts results not only in progressive bone loss but also in widespread hematopoietic failure manifested as severe reduction in erythrocytes, HSCs, and B-lymphocytes (22–24), and impaired bone mineralization results in a defect in HSC localization to the endosteal niche (25). Because normal osteogenesis is required for hematopoiesis and Fgf-23−/− mice display severe bone abnormalities as well as significant reduction in lymphatic organ size such as spleen and thymus, in the present study we hypothesized that FGF-23 plays a key role in regulating erythropoiesis. We characterized the hematopoietic cellular composition of several hematopoietic tissues from Fgf-23−/− mice and determined that loss of Fgf-23 in mice results in specific changes in early hematopoietic progenitors and erythroid populations. More importantly, these changes are also detected prenatally, suggesting that FGF-23 affects erythropoiesis independent of the mineral composition in the bone marrow environment or secondary diseases that arise as part of the Fgf-23−/− mouse phenotype (i.e., emphysema and renal insufficiency). Furthermore, our data demonstrate that exogenous administration of FGF-23 in WT mice results in erythropoietic changes opposite to those observed in Fgf-23−/− mice. Finally, we show that elimination of vitamin D from Fgf-23−/− mice does not influence the HSC or erythroid populations in these mice. Taken together, our study establishes a novel role for FGF-23 in hematopoiesis that links it to erythrocyte production and differentiation.
MATERIALS AND METHODS
Animals
Heterozygous Fgf-23 and 1α(OH)ase mice (both in C57BL/6 background) were interbred at 6–12 weeks to attain wild-type, Fgf-23−/−, Fgf-23−/−/1α(OH)ase−/−, and 1α(OH)ase−/− animals. Heterozygous 1α(OH)ase−/− mice were generously provided by Dr. René St-Arnaud (Genetics Unit of Shriners Hospital, Montreal, Canada). All mice were housed in New York University College of Dentistry Animal Facility, kept on a light/dark (12h/12h) cycle at 23 °C, and received food (standard lab chow) and water ad libitum. Genomic DNA was obtained from tail snips, and routine PCR was used to identify the genotypes as previously described (20, 21). All animal studies were approved by the institutional animal care and use committee at New York University.
RNA Isolation
Total RNA was extracted from crushed whole tibiae, bone marrow, and kidney from 6-week-old mice using TRIzol reagent (Ambion; Invitrogen) according to the manufacturer's protocol (Molecular Research Center Inc., Cincinnati, OH). Synthesis of cDNA was performed using a high capacity cDNA reverse transcription kit as described by the manufacturer (Applied Biosystems, Foster City, CA). cDNA was amplified by quantitative real time PCR using the PerfecCTa® SYBR® Green SuperMix (Quanta Biosciences, Gaithersburg, MD). The following primers were used: Fgf-23, 5′-ACT TGG CCT TTA TTA GCC GGG TCT-3′ and 5′-AGA TGG CCT CCT CCC TGT GTT CAA-3′; FGFR1, 5′-TGA TGG GAG AGT CCG ATA GAG-3′ and 5′-CCT GAA GAC TGC TGG AGT TAA T-3′; FGFR2, 5′-CCA GCA CTG GAG CCT TAT TAT-3′ and 5′-GTG GTT GAT GGA CCC GTA TT-3′; FGFR3, 5′-CTA AAT GCC TCC CAC GAA GAT-5′ and 5′-CTG AGG ATG GAG CAT CTG TTA C-3′; FGFR4, 5′-CCT GAG GCC AGA TAC ACA GAT A-3′ and 5′-GGA TGA CTT GCC GAT GAT ACA C-3′; klotho, 5′-AAA TGG CTG GTT TGT CTG GGG-3′ and 5′-TAT GCC ACT CGA AAC CGT CCA-3′; erythropoietin (Epo), 5′-TCT ACG TAG CCT CAC TTC ACT-3′ and 5′-ACC CGG AAG AGC TTG CAG AAA-3′; hypoxia-induced factor 1α (HIF-1α), 5′-TCT CGG CGA AGC AAA GAG TCT-3′ and 5′-TAG ACC ACC GGC ATC CAG AAG-3′; hypoxia-induced factor 2-α (HIF-2α), 5′-GGG AAC ACT ACA CCC AGT GC-3′ and 5′-TCT TCA AGG GAT TCT CCA AGG-3′; transferrin, 5′-CCC TCT GTG ACC TGT GTA TTG-3′ and 5-CTT TCT CAA CGA GAC ACC TGA A-3′; transferrin receptor, 5′-TCC TGT CGC CCT ATG TAT CT-3′ and 5′-CGA AGC TTC AAG TTC TCC ACT A-3′; glucose transporter-1 (glut-1), 5′-CCC AGG TGT TTG GCT TAG A-3′ and 5′-CAG AAG GGC AAC AGG ATA CA-3′; phosphoglycerate kinase-1 (Pgk-1), 5′-CAC AGA AGG CTG GTG GAT AT-3′ and 5′-CTT TAG CGC CTC CCA AGA TAG-3′; and hypoxanthine-guanine phosphoribosyl transferase (HPRT-housekeeping gene), 5′-AAG CCT AAG ATG AGC GCA AG-3′ and 5′-TTA CTA GGC AGA TGG CCA CA-3′. Forty cycles (95 °C for 15 s; 60 °C for 30 s; and 72 °C for 30 s) were run on a Mastercycle Realplex2 (Eppendorf, Hamburg, Germany), and reactions were analyzed. Melting curve and gel analyses (sizing and sequencing) verified single products of the appropriate base pair size.
Blood Collection and Hematologic Analysis
Peripheral blood was collected posteuthanasia from 6-week-old mice by cardiac puncture into EDTA-coated tubes to prevent clotting. Blood samples were then shipped overnight to Cornell University Veterinary Clinic for automated complete blood count.
Fetal Liver Collection
Time breeding of Fgf-23+/− mice was carried out. Pregnant females were sacrificed at E15.5, and embryos were obtained by C-section. Genomic DNA was obtained from tail snips, and routine PCR was used to identify the genotypes of the embryos as previously described (20, 21). Fetal livers were isolated and fetal liver cell suspensions were prepared from WT and Fgf-23−/− mice.
Isolation and Assessment of Blood, Bone Marrow, Spleen, and Liver Cells by Flow Cytometry
Bone marrow was isolated from dissected tibiae and femora from 6-week-old mice by flushing in Iscove's modified Dulbecco's medium (IMDM) (Sigma-Aldrich) supplemented with 20% fetal bovine serum (20% IMDM) through a 26-gauge Becton Dickinson needle. Marrow cells were dispersed by manual agitation and then filtered to remove foreign particles. Spleens from 6-week-old mice and fetal livers from E15.5 embryos were surgically removed and homogenized into a cell suspension in 20% IMDM. Flow cytometry analysis for peripheral blood, bone marrow, spleen, and fetal liver cells were carried out in a BD FACSortTM flow cytometer equipped with 488 argon lasers (BD Biosciences, San Jose, CA). For immunostaining, cells were washed and resuspended in 1× PBS containing 0.1% BSA. Mouse Fc receptor was blocked prior to staining using CD16/32 antibody to reduce nonspecific binding. After the addition of antibodies, cells were incubated for 40 min on ice; for peripheral blood, red blood cells were further lysed using BD FACS lysing solution (BD Biosciences). Labeled cells were then washed with 1× PBS and analyzed by flow cytometry. Appropriate isotype controls were kept for each set. Forward and side scatter patterns were gated, excluding the debris. A total of 20,000 events were collected and analyzed using FlowJo software version 7.6.5. Erythroid lineage was assessed using Ter119 APC/CD71 PE markers combined with the forward scatter properties (26). Hematopoietic stem/progenitor cells were differentiated using SLAM markers (CD150 PE/CD48 APC), Sca-1 FITC (Ly6A-E), c-Kit Percp Cy5.5 (CD117), CD90 PE (Thy-1), and APC-tagged lineage mixture comprising of antibodies against CD3, B220 (CD45R), Ly6G and Ly6C (Gr-1), CD11b (Mac-1), and TER119. c-Kit+Sca1+ cells were gated on lineage negative fraction to analyze LSK (Lin−c-Kit+Sca1+). The LSK cells were then analyzed on a Thy-1low gate to obtain KTLS population (LSK Thylow). CD45.1 PE and CD45.2 FITC antibodies were used to differentiate donor and recipient populations after transplantation. All antibodies except SLAM markers were purchased from BD Pharmingen. SLAM markers CD150 and CD48 were purchased from e-Biosciences (San Diego, CA). Cell apoptosis was assessed using the TACS annexin V kit (Trevigen Inc., Gaithersburg, MD).
Methylcellulose Cultures
Cell suspensions were prepared from 6-week-old bone marrow, spleen, and fetal liver (E15.5) in 20% IMDM. Aliquots were then plated in a methylcellulose medium supplemented with recombinant cytokines for colony assays of murine cells (Methocult M3434; StemCell Technologies, Vancouver, Canada). Cytokines included: IL-3 (4 ng/10 μl), stem cell factor (20 ng/10 μl), granulocyte macrophage-colony stimulating factor (2 ng/10 μl), and erythropoietin (2 units/10 μl). Fetal liver and bone marrow cells were plated at a concentration of 1 × 104, whereas spleen cells were plated at a concentration of 1 × 105 cells/35-mm tissue culture dish. Cells were incubated in a humidified chamber at 37 °C with 5% CO2. Three different types of colonies were scored 8 and 12 days postculture including: burst-forming unit of erythroid cells (BFU-E) and the most primitive colony-forming units of granulocyte-erythrocyte-macrophage-megakaryocyte cells (CFU-GEMM).
Serum FGF-23 and Epo Measurements
Serum FGF-23 and erythropoietin (Epo) concentrations were measured in samples from 6-week-old mice using the mouse FGF-23 C-terminal ELISA kit (Immutopics International, San Clemente, CA) and the Quantikine rat/mouse Epo immunoassay kit (R&D Systems, Minneapolis, MN), according to the manufacturer's protocols.
FGF-23 Protein Injections
Wild-type C57BL/6 mice (6–8 weeks old) or 1α(OH)ase−/− and WT littermates were given a single injection intraperitoneally of recombinant human FGF-23 protein (5 μg) (R&D Systems) or vehicle (PBS) and analyzed 24 h later for hematopoietic cellularity in peripheral blood, bone marrow, and spleens as well as for hematology, as described above.
Oxygen Treatment
Fgf-23−/− mice and WT littermates were placed in a sealed Plexiglass chamber where oxygen was administered through a tube at a rate of 4 liters/min for 1 h. The room and chamber temperatures were maintained at ∼20 °C throughout the experiment. Mice were not anesthetized or restrained, and food and water were available ad libitum during the entire treatment.
Adhesion Assay
Bone marrow cell adherence was determined using 96-well plates coated with 5 μg of fibronectin (Sigma) overnight. Whole bone marrow cells from 6-week-old WT and Fgf-23−/− mice were plated at a density of 105 cells in 100 μl of 2% IMDM, seeded in triplicate, and incubated for 40 min at 37 °C. Cells were then fixed in 4% paraformaldehyde (Sigma), stained using 0.25% crystal violet (Sigma), and lysed with 0.1% Triton X-100 (Sigma) in 1× PBS. The plates were read at an absorbance of 550 nm, where readings represented that higher optical density values corresponded to a higher adhesion.
In Vitro Transmigration Assay
Chemotaxis toward stromal-derived factor 1 (SDF-1α) was assessed using a dual chamber Transwell with a pore size insert of 8 μm. 105 whole bone marrow cells from 6-week-old WT and Fgf-23−/− mice in 100 μl of 2% IMDM were seeded in triplicate to the upper chamber of 24-well plates containing inserts (BD Falcon). 100 ng of SDF-1α (Peprotech, Rocky Hill, NJ) was added to the lower chamber containing 600 μl of medium. Bone marrow cells were incubated for 3 h at 37 °C, 5% C02 in the presence or absence (to detect spontaneous migration) of SDF-1α. Afterward, the nonmigrated cells remaining in the Transwells and the migrated fraction were both collected and counted. Migrated cells were expressed as a percentage of migrated cells per total cells seeded.
In Vivo Homing
Homing experiments were performed using tail vein injection of 2.5 × 106 whole bone marrow cells from 6-week-old wild-type or Fgf-23−/− (CD45.2; Ly5.2) mice into 8-week-old myeloablated B6.SJL (CD45.1; Ly5.1) recipient mice. Myeloablation was achieved by a lethal dose of irradiation (900 rads). Recipient mice were sacrificed 24 h post-transplantation, and peripheral blood, spleen, and bone marrow cells were collected. Flow cytometry was performed on cell suspensions from each tissue collected and analyzed for the presence of CD45.2+ donor cells to determine the numbers of homed cells to the BM in comparison with spleen and peripheral blood. A minimum of 50,000 events were acquired in a BD FACSort TM flow cytometer equipped with 488 argon lasers (BD Biosciences).
In Vitro Ter119+ Cell Isolation
Bone marrow was isolated from dissected tibiae and femora from 6-week-old mice by flushing in IMDM (Sigma-Aldrich) supplemented with 20% fetal bovine serum (20% IMDM) through a 26-gauge Becton Dickinson needle. Marrow cells were dispersed by manual agitation and then filtered to remove foreign particles. These cells were sorted using Ter119-conjugated magnetic beads (Miltenyi Biotec) and stained with CD71PE and Ter119FITC antibodies (BD Biosciences) to obtain early to late stage erythroblast populations using flow cytometry. Purified Ter119+ cells were seeded in triplicate in 24-well plates for 24 h at a cell density of 105 cells/well and then treated for 4 h with or without 5 ng/ml of recombinant FGF-23 (R&D Systems).
Statistics
Statistical significance was evaluated by Student's t test for comparison between two groups or by one-way analysis of variance followed by Tukey's test for multiple group comparisons. All analyses were performed using GraphPad Prism 4.0, and all values were expressed at means ± S.E. p values less than 0.05 were considered significant.
RESULTS
Expression of Fgf-23 and Its Signaling Components in Erythroid Cells
High expression of Fgf-23 in bone has been reported by several groups, confirming that bone is the principal source of Fgf-23 production (20, 27, 28). However, expression of Fgf-23 in specific bone marrow cells remains unknown. Here, we determined mRNA expression of Fgf-23 and several fgf-23 signaling components (klotho and FGFR1–4) in isolated BM erythroid cells (Ter119+) of adult WT mice. Real time quantitative RT-PCR revealed that WT Ter119+ erythroid cells highly express Fgf-23, klotho, and FGFR1, 2, and 4 but showed minimal FGFR3 expression (Fig. 1). These data suggest that erythroid cells are capable of undergoing active Fgf-23 signaling.
FIGURE 1.
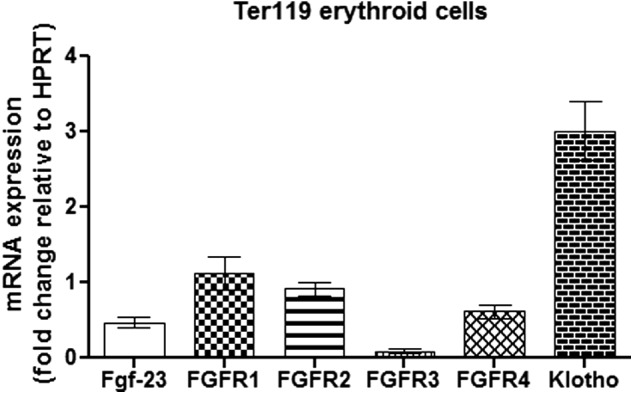
Erythroid cells express Fgf-23 signaling components. Quantitative real time RT-PCR show changes in Fgf-23, klotho, and FGFR1–4 mRNA expression in isolated Ter119+ erythroid cells from WT BM (n = 8–9). The data are represented as mean fold change ± S.E. relative to housekeeping gene HPRT.
Analysis of the Fgf-23−/− Mice Hematologic Characteristics
To assess the impact of FGF-23 on erythropoiesis, we performed complete blood count analysis in peripheral blood (PB) of 6-week-old WT, heterozygous (Fgf-23+/−), and Fgf-23 null (Fgf-23−/−) mice. Fgf-23−/− mice were found to exhibit significantly elevated erythrocyte (RBC) numbers in comparison with WT and Fgf-23+/− mice (Fig. 2A). In addition, a significant increase in RBC distribution width (Fig. 2B) and a marked reduction in the red blood cell indices, mean cell volume, and mean corpuscular hemoglobin were detected in Fgf-23−/− mice (Fig. 2, C and D), characteristic of hypochromic microcytic anemia. No differences were found in any of the examined hematologic parameters between WT and Fgf-23+/− mice.
FIGURE 2.

Peripheral blood complete blood count analysis of 6-week-old WT (n = 15), heterozygous (Fgf-23+/−, n = 5), and Fgf-23 null (Fgf-23−/−, n = 15) mice. A, RBC counts. B, RBC distribution width (RDW). C, mean cell volume (MCV). D, mean corpuscular (cell) hemoglobin (MCH). **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Fgf-23 Deficiency Results in Aberrant Erythropoiesis
Because of the significant increase in circulating red blood cell numbers, we further investigated by flow cytometry the effect of Fgf-23 deficiency in erythrocyte differentiation in PB and BM as the origin of postnatal hematopoiesis. Based on the known role of osteoblasts in the regulation of hematopoiesis (22, 23, 29) and published data showing severe reduction in bone mineral density and osteoblast numbers in Fgf-23−/− mice (14, 20, 21, 30), we expected to find severely altered erythropoiesis in the BM of Fgf-23−/− mice.
We confirmed the increase in total RBC numbers by analysis of erythroid cells and determined the maturation stage of erythroblasts (Ter119+) by the loss of CD71 expression, as reported by Asari et al. (31). A representative dot plot illustrating different erythroid populations is shown in Fig. 3A. Our results show a significant increase in the differentiation of immature (pro-E; Ter119+med CD71+hi), as well as mature erythroid cells (Ter119hi+) in PB (Fig. 3, B and C) and BM (Fig. 3, D and E) of Fgf-23−/− mice. In addition, the capacity of Fgf-23-deficient HSCs to generate erythroid colonies (BFU-E) in vitro was considerably higher compared with WT BM cells (Fig. 4A), suggesting that loss of Fgf-23 results in increased erythroid progenitor cell activity. Furthermore, we examined whether the increase in erythrocyte numbers is due to increased Epo being released by the kidneys. Our data show that circulating Epo levels were elevated in Fgf-23−/− mice compared with WT littermates (Fig. 4B). In addition, we found that Epo mRNA expression was significantly increased in bone marrow, liver, and kidney of Fgf-23-deficient mice (Fig. 4C) because of induced HIF signaling, as determined by significant up-regulation of the hypoxia-inducible transcription factors HIF-1α and HIF-2α mRNA expression in all three tissues examined (Fig. 4, D and E). However, HIF-1α and HIF-2α mRNA expression was suppressed in Fgf-23−/− bone (Fig. 4, D and E), leading to significantly reduced Epo expression in bone (Fig. 4C, inset) both of which are not surprising because fgf-23-deficient mice have severely decreased osteoblast numbers. Recent reports have indicated that osteoblasts are a source of local Epo production in the bone, which would account for our observed decrease in bone Epo and HIF expression (32). Additionally, mRNA expression of hypoxia-responsive genes including transferrin, transferrin receptor, glucose transporter-1, and phosphoglycerate kinase-1 was significantly elevated both in liver and bone marrow of Fgf-23−/− mice (Fig. 5, A–H). These findings suggest that a lack of Fgf-23 in mice results in increased erythrocyte production in the bone marrow and release into the circulation by altering the BM environment and rendering it hypoxic. Hypoxia in turn induces HIF signaling, which activates local and systemic Epo production that stimulates erythropoiesis in Fgf-23 mutant mice. To further address the role of hypoxia in mediating the observed erythroid cell changes, we treated Fgf-23−/− mice and WT littermates with 100% oxygen for 1 h and measured several erythroid cell parameters. We have found that oxygen treatment significantly reduced serum Epo levels in Fgf-23−/− mice and returned them to control levels (Fig. 4F). Furthermore, after only 1 h of oxygen treatment, mRNA expression of renal and BM HIF-1α was significantly decreased in Fgf-23−/− mice compared with untreated Fgf-23−/− mice (Fig. 4, G and I). Renal Epo mRNA was also found significantly decreased with oxygen treatment in Fgf-23−/− mice (Fig. 4H). Importantly, BM Ter119+ erythroid populations were also rescued in Fgf-23−/− mice treated with oxygen compared with untreated mice (Fig. 4J). These data further support our hypothesis that a lack of Fgf-23 stimulates a hypoxic BM environment, which activates Epo-induced erythropoiesis in our Fgf-23 mutant mice.
FIGURE 3.

Flow cytometry analysis of peripheral blood and bone marrow from 6-week-old WT and Fgf-23−/− mice. A, representative FACS dot plots illustrating different RBC populations. PB and BM cells from WT and Fgf-23−/− mice are stained for Ter119 and CD71 for erythroid cells. B and C, peripheral blood. B, percentage of primitive pro-erythroblasts (pro-E) stained positive for Ter119med and CD71high (WT, n = 6; Fgf-23−/−, n = 7). C, percentage of mature erythroid cells stained positive for Ter119 (WT, n = 9; Fgf-23−/−, n = 11). D and E, bone marrow. D, percentage of primitive pro-erythroblasts (pro-E) stained positive for Ter119med (WT, n = 7; Fgf-23−/−, n = 8). E, percentage of mature erythroid cells stained positive for Ter119 and CD71 (WT, n = 15; Fgf-23−/−, n = 13). The data are represented as means ± S.E. *, p < 0.05; **, p < 0.01.
FIGURE 4.

Increased erythropoiesis in Fgf-23−/− mice. A, colony-forming assay for erythroid (BFU-E) progenitors (WT, n = 5; Fgf-23−/−, n = 12). Cells from each mouse were plated in duplicate, and the number of colonies in each plate was counted. B, Epo concentrations measured by ELISA in serum of WT (n = 12) and Fgf-23−/− mice (n = 8). Samples were measured in duplicate. C, quantitative real time RT-PCR showing changes in Epo mRNA expression in bone marrow (WT, n = 7; Fgf-23−/−, n = 5), bone (inset) (WT, n = 5; Fgf-23−/−, n = 6), liver (WT, n = 7; Fgf-23−/−, n = 7), and kidney (WT, n = 7; Fgf-23−/−, n = 6) in WT and Fgf-23−/− mice. Bone graph is amplified in the inset because of small values compared with other tissues. D and E, quantitative real time RT-PCR showing changes in HIF-1α (D) and HIF-2α (E) mRNA expression in bone marrow, bone, liver, and kidney in WT and Fgf-23−/− mice (WT, n = 7; Fgf-23−/−, n = 7). F–J, oxygen treatment. WT (n = 4) and Fgf-23−/− (n = 5) mice were subjected to 100% oxygen at 4 liters/min for 1 h. F, Epo concentrations measured by ELISA in serum of WT (n = 4) and Fgf-23−/− mice (n = 5). Samples were measured in duplicate. G–I, quantitative real time RT-PCR showing changes in renal HIF-1α (G), renal EPO (H), and BM HIF-1α mRNA expression (I) in WT and Fgf-23−/− mice (WT, n = 4; Fgf-23−/−, n = 5). J, flow cytometry analysis of bone marrow from 6-week-old WT and Fgf-23−/− mice with or without oxygen treatment. A percentage of mature erythroid cells stained positive for Ter119 (WT, n = 4; Fgf-23−/−, n = 5). The data are represented as mean fold change ± S.E. relative to housekeeping gene HPRT. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 5.

Lack of Fgf-23 results in up-regulation of HIF _target genes. A–D, quantitative real time RT-PCR showing changes in liver transferrin (A, WT, n = 6; Fgf-23−/−, n = 5), transferrin receptor (B, WT, n = 6; Fgf-23−/−, n = 5), glucose transporter-1 (C, WT, n = 6; Fgf-23−/−, n = 5), and phosphoglycerate kinase-1 (D, WT, n = 6; Fgf-23−/−, n = 5) mRNA expression in WT and Fgf-23−/− mice. E–H, quantitative real time RT-PCR showing changes in bone marrow transferrin (E, WT, n = 6; Fgf-23−/−, n = 6), transferrin receptor (F, WT, n = 6; Fgf-23−/−, n = 6), glucose transporter-1 (G, WT, n = 6; Fgf-23−/−, n = 6), and phosphoglycerate kinase-1 (H, WT, n = 5; Fgf-23−/−, n = 5) mRNA expression in WT and Fgf-23−/− mice. The data are represented as mean fold change ± S.E. relative to housekeeping gene HPRT. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Fgf-23 Deficiency Alters Early Hematopoietic Progenitors
Because our data suggest that Fgf-23 results in aberrant erythropoiesis, we investigated whether these changes also occur in early hematopoietic progenitors. The frequency of CD150+CD48− (SLAM) cells, enriched for HSCs, was 2-fold higher in Fgf-23−/− mice compared with WT littermates, both in peripheral blood (2.3 ± 0.3 and 1.2 ± 0.3, respectively; Fig. 6A) and bone marrow (0.9 ± 0.1 and 0.41 ± 0.1, respectively; Fig. 6B), indicating that a loss of Fgf-23 results in increased HSC production. Similarly, other HSC markers, LSK (Lin−Sca-1+c-Kit+) and KTLS (c-Kit+Thy-1+Lin−Sca-1+), were also markedly elevated in PB and BM of Fgf-23−/− mice compared with WT littermates (data not shown). We have also found that the increase in HSCs coincided with a significant decrease in HSC apoptosis in Fgf-23−/− BM, as assessed by a combination of SLAM and annexin V staining (Fig. 6C). To determine whether the perturbations in the bone marrow and peripheral blood arise at the progenitor level, we characterized the capacity of hematopoietic progenitor cells from WT and Fgf-23−/− mice for myeloid differentiation within their local BM environment. As assessed by colony-forming assay, the number of multilineage colony-forming cells (CFU-GEMM) per femur was significantly increased in Fgf-23−/− bone marrow compared with WT littermates (Fig. 6D). Similarly, the number of total colonies formed in Fgf-23−/− BM was also increased (data not shown). Therefore, our results demonstrate not only that the frequency of HSCs was drastically elevated in peripheral blood and bone marrow of Fgf-23−/− mice, but also that this increase correlated with a significant increase in the in vitro proliferative capacity and absolute numbers of hematopoietic progenitor cells from Fgf-23−/− mice, providing evidence for increased function and activity of Fgf-23-deficient HSCs.
FIGURE 6.

Flow cytometry analysis of HSC populations in peripheral blood and bone marrow from 6-week-old WT and Fgf-23−/− mice. A, percentage of HSC population in peripheral blood stained for SLAM (CD150+CD48−) (WT, n = 7; Fgf-23−/−, n = 10). B, percentage of HSC population in bone marrow stained for SLAM (CD150+CD48−) (WT, n = 6; Fgf-23−/−, n = 11). C, percentage of HSC population undergoing apoptosis in the bone marrow stained for SLAM (CD150+CD48−) and annexin V (WT, n = 6; Fgf-23−/−, n = 11). D, colony-forming assay for granulocyte-erythrocyte-monocyte-megakaryocyte (CFU-GEMM) progenitors (WT, n = 5; Fgf-23−/−, n = 12). Cells from each mouse were plated in duplicate, and the number of colonies in each plate was counted. The data are represented as means ± S.E. *, p < 0.05.
Migration and Homing of Fgf-23-deficient BM Cells
Elevated HSCs in the bone marrow could be due to alterations in HSC homing or retention of the cells in the correct microenvironment and/or defects in their migratory function. To address this, we assessed surface expression of molecules required for HSC homing/migration such as the chemokine SDF-1α/CXCL-12. Serum SDF-1α levels were considerably elevated in Fgf-23−/− mice compared with in WT mice (Fig. 7A). It has been shown that elevation of SDF-1 levels in peripheral blood results in hematopoietic progenitor and stem cell mobilization to the peripheral circulation (33). Our data also showed that BM cells from Fgf-23-deficient mice exhibited no change in chemotaxis toward an SDF-1α gradient compared with WT BM cells (Fig. 7B).
FIGURE 7.

Migration and homing of Fgf-23−/− bone marrow cells. A, SDF-1α concentration measured by ELISA in serum of WT (n = 9) and Fgf-23−/− mice (n = 8). Samples were measured in duplicate. B, in vitro migration experiment revealed no change in migratory capacity of Fgf-23−/− BM cells toward an SDF-1 gradient (WT, n = 18; Fgf-23−/−, n = 14). C, adherence assay showed no changes in Fgf-23−/− BM cells to adhere to fibronectin (WT, n = 10; Fgf-23−/−, n = 9). D, graphic representation of flow cytometry analysis of peripheral blood, bone marrow, and spleen (SPL) after transplantation of WT or Fgf-23−/− BM cells in irradiated B6.SJL recipient mice (WT, n = 10; Fgf-23−/−, n = 11). E–G, flow cytometry profile of peripheral blood, bone marrow, and spleen after transplantation. The data are represented as means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We further carried out in vivo homing experiments and transplanted WT and Fgf-23−/− bone marrow cells (CD45.2+) into lethally irradiated B6.SJL WT (CD45.1+) recipient mice. Studies have shown that lethal irradiation creates a noncompetitive host HSC pool that can be easily replaced by donor HSCs (34, 35). Homing efficiency was assessed 24 h post-transplantation in the bone marrow, peripheral blood, and spleen of the recipient mice. We found a significant increase in the number of Fgf-23−/− cells seeding the host bone marrow (Fig. 7, D and F) compared with transplanted WT BM cells. Furthermore, these experiments showed a marked reduction in Fgf-23−/− BM cell localization in peripheral blood (Fig. 7, D–E). However, localization of Fgf-23−/− and WT transplanted BM cells was similar in the spleens of irradiated recipient mice (Fig. 7, D and G). Finally, we assessed the ability of BM cells to adhere to fibronectin, a substance found in the bone microenvironment. We detected no significant differences between WT and Fgf-23−/− BM cell adherence capabilities, indicating that Fgf-23−/− BM cells are as capable as WT BM cells of migrating from the BM to the peripheral blood (Fig. 7C). Collectively, our results confirm that loss of Fgf-23 results in increased production of HSCs in the bone marrow and no evidence of a defect in their migratory function or in physical association with the regulatory components of the microenvironment niche.
Assessment of Spleen Cellularity
To assess whether loss of Fgf-23 results in hematopoietic abnormalities in hematopoietic organs other than the bone marrow, we investigated the cellular composition of the spleens of Fgf-23−/− mice. Hematopoietic differentiation of homogenized spleen cells showed that the frequency of both immature and mature erythroid cells was significantly lower in Fgf-23−/− mice (Fig. 8, A and B). These findings were confirmed by CFU assays showing a marked reduction in the number of erythroid colonies (BFU-E; Fig. 8C), as well as the primitive granulocyte-erythrocyte-macrophage-megakaryocyte (CFU-GEMM) progenitors and, subsequently, the number of total colonies formed (data not shown) in the spleens of Fgf-23−/− mice compared with in WT mice, ruling out the possibility that extramedullary erythropoiesis is the cause of the elevated erythroid populations in Fgf-23−/− mice. Furthermore, the frequency of HSC-enriched CD150+CD48− (SLAM) and their colony forming ability were greatly higher in Fgf-23−/− than in WT spleens (Fig. 8, D and E). It is possible that increased HSC frequency in our Fgf-23-deficient mice results in increased differentiation into the lymphoid lineage, specifically T-lymphocytes, because of heighten immune response. This may occur at the expense of the myeloid lineage, explaining the decrease in splenic erythroid populations. Further investigation of the different lymphoid populations is required to properly understand this phenomenon.
FIGURE 8.
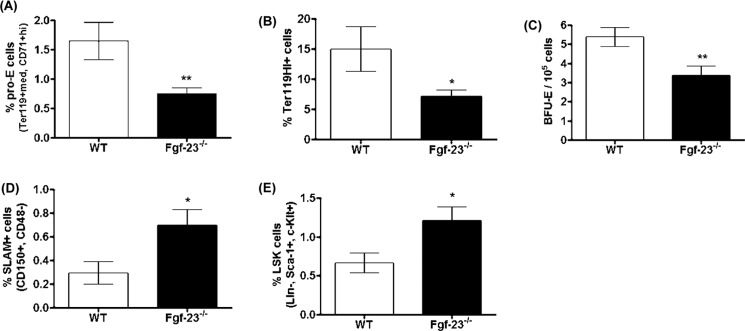
Flow cytometry analysis and colony-forming assays of spleens from 6 week old WT and Fgf-23−/− mice. A and B, flow cytometry. A, percentage of early erythroid cells (pro-E) stained positive for Ter119med and CD71high (WT, n = 6; Fgf-23−/−, n = 9). B, percentage of mature erythroid cells stained positive for Ter119 (WT, n = 6; Fgf-23−/−, n = 7). C, colony-forming assay for erythroid (BFU-E) progenitors (WT, n = 7; Fgf-23−/−, n = 8). Cells from each mouse were plated in duplicate, and the number of colonies in each plate was counted. D and E, flow cytometry. D, percentage of HSC population stained for SLAM (CD150+CD48−) (WT, n = 8; Fgf-23−/−, n = 5). E, percentage of HSC population stained for LSK (Sca-1 FITC (Ly6A-E), c-Kit Percp Cy5.5 (CD117), and APC-tagged lineage mixture comprising of antibodies against CD3, B220 (CD45R), Ly6G and Ly6C (Gr-1), CD11b (Mac-1), and TER119. c-Kit+Sca1+ cells were gated on lineage negative fraction to analyze LSK (Lin−c-Kit+Sca1+). The data are represented as means ± S.E. *, p < 0.05; **, p < 0.01.
FGF-23 Affects Hematopoiesis Independent of Changes in the Bone Marrow Environment
Previous studies have shown that Fgf-23 is expressed in fetal liver, heart, and somites of mice at E11.5 days postcoitum (20). Examining prenatal erythropoiesis allows us to investigate the effects of Fgf-23 prior to inflammatory disease onset commonly observed in these mice (i.e., emphysema and renal insufficiency). To determine whether the hematopoietic changes in Fgf-23−/− mice are specific to the bone marrow environment or due to aberrant production or function of HSCs, we examined whether hematopoietic stem cell populations in Fgf-23−/− mice are affected before their translocation from fetal liver to the bone marrow.
We evaluated fetal liver cell populations at embryonic day E15.5, and we found that Fgf-23 deletion results in a significant increase in mature erythrocyte populations (Fig. 9A). Colony-forming assays also showed that the high frequency of erythroid cells in Fgf-23−/− fetal livers correlated with an actual increase in functional erythroid progenitors and more BFU-E colonies from Fgf-23−/− fetal liver cells (Fig. 9B). Interestingly, similar to the BM data, the frequency of HSC-enriched CD150+CD48− (SLAM) cells was considerably higher in Fgf-23−/− than in WT fetal livers (Fig. 9, C and D). LSK (Lin−Sca-1+c-Kit+) and KTLS (c-Kit+Thy-1+Lin−Sca-1+) cells were also found to be greatly increased in Fgf-23−/− fetal livers (Fig. 9, E–G). Taken together, our data suggest that Fgf-23 regulates hematopoiesis independently of changes in the bone environment.
FIGURE 9.

Flow cytometry analysis and colony-forming assays of fetal liver cells from E15.5 WT and Fgf-23−/− mice. A and B, flow cytometry. A, percentage of mature erythroid cells stained positive for Ter119 (WT, n = 8; Fgf-23−/−, n = 8). B, colony-forming assay for erythroid (BFU-E) progenitors (WT, n = 10; Fgf-23−/−, n = 10). Cells from each mouse were plated in duplicate, and the number of colonies in each plate was counted. C, percentage of HSC population stained for SLAM (CD150+CD48−) (WT, n = 7; Fgf-23−/−, n = 8). D, representative flow cytometry dot plot showing CD150+CD48− populations in WT and Fgf-23−/− mice. E, percentage of HSC population stained for LSK (Lin−Sca-1+c-Kit+) (WT, n = 9; Fgf-23−/−, n = 10). F, representative flow cytometry dot plot showing LSK populations in WT and Fgf-23−/− mice. G, percentage of HSC population stained for KTLS (c-Kit+Thy1+Lin−Sca-1+) (WT, n = 9; Fgf-23−/−, n = 10). The data are represented as means ± S.E. *, p < 0.05; **, p < 0.01.
Effect of FGF-23 Administration on Erythropoiesis
To further investigate the mechanism by which Fgf-23 affects erythropoiesis, we injected intraperitoneally recombinant human FGF-23 protein (or vehicle (PBS)) into wild-type C57BL/6 mice and evaluated in vivo changes in hematopoiesis. Serum FGF-23 levels were markedly elevated after injection of FGF-23, confirming successful delivery of the protein (Fig. 10A). Our data show that exogenous administration of one single dose of FGF-23 protein in WT mice leading to high levels of FGF-23 results in erythropoietic changes opposite to the ones observed in mice lacking Fgf-23. Specifically, we detected a substantial reduction in erythropoiesis in all tissues examined (PB, BM, and spleen) in mice injected with FGF-23 (Fig. 10, C, D, F–H, and K–L). This correlated with an actual decrease in functional erythroid cells, which generated considerably fewer erythroid colonies in the BM (Fig. 10I). We also examined whether the decrease in erythrocyte numbers were due to a decrease in erythropoietin. Our data show that circulating Epo levels were in fact significantly reduced in mice injected with FGF-23 compared with WT littermates (Fig. 10B). These data further emphasize the role of Fgf-23 in regulating Epo-mediated changes in erythropoiesis. Furthermore, the frequency of HSC-enriched cells in peripheral blood (Fig. 10E), bone marrow (Fig. 10J), and spleen (Fig. 10M) was significantly reduced in mice injected with FGF-23. The changes induced by high FGF-23 levels were observed within 24 h of FGF-23 administration, indicating its potency.
FIGURE 10.

FGF-23 injections. A, serum FGF-23 concentrations measured by ELISA pre- and postintraperitoneal injections of recombinant human FGF-23 in WT mice. Vehicle-treated mice were injected with PBS (n = 6 per group). B, Epo concentrations measured by ELISA in serum of WT mice injected with FGF-23 (5 μg) or vehicle (PBS). Samples were measured in duplicate. C–M, flow cytometry analysis and colony-forming assays of hematopoietic populations in FGF-23- and vehicle-treated WT mice. C–E, peripheral blood. C, percentage of early erythroid cells (pro-E) stained positive for Ter119med and CD71high. D, percentage of mature erythroid cells stained positive for Ter119. E, percentage of HSC population stained for LSK (Lin−Sca-1+c-Kit+). F–H, bone marrow. F, percentage of early erythroid cells (pro-E) stained positive for Ter119 and CD71high. G, percentage of mature erythroid cells stained positive for Ter119. H, representative dot plot of flow cytometry analysis of WT and Fgf-23−/− pro-E and Ter119+ cell populations in BM. I, colony forming assay for erythroid (BFU-E) progenitors. Cells from each mouse were plated in triplicate, and the number of colonies in each plate was counted. (n = 6 per group). J, percentage of HSC population stained for SLAM (CD150+CD48−). K–M, spleen. K, percentage of early erythroid cells (pro-E) stained positive for Ter119med and CD71high. L, percentage of mature erythroid cells stained positive for Ter119. M, percentage of HSC population stained for KTLS (Lin−Sca-1+c-Kit+Thy-1+). The data are represented as means ± S.E. *, p < 0.05.
To further determine the significant role of Fgf-23 in regulating erythropoiesis, we isolated and treated BM-derived Ter119+ cells from WT and Fgf-23−/− littermate mice and treated the cells with FGF-23 protein or vehicle (PBS) for 4 h. We found that untreated Fgf-23−/− Ter119+ cells showed significantly elevated mature erythroid cells (Ter119hi+) compared with untreated WT controls, whereas WT cells treated with FGF-23 protein resulted in reduced Ter119hi+ cell frequency (Fig. 11A), similar to our in vivo data. Importantly, FGF-23 treatment of Fgf-23-deficient erythroid cells rescued erythropoiesis and brought Ter119hi+ cell frequency to control levels (Fig. 11A). Furthermore, FGF-23 treatment resulted in a significant decrease in Epo and HIF mRNA expression in Fgf-23−/− cells and similar to untreated WT levels (Fig. 11, C and D). Our data also show that Fgf-23 acts on the Epo-R. Loss of Fgf-23 results in high expression of the Epo-R in isolated Ter119+ cells and FGF-23 treatment restores the frequency of Epo-R positive cells to normal control levels (Fig. 11B). Taken together, our data strongly suggest that Fgf-23 plays an important role in the regulation of erythropoiesis through the HIF/Epo pathway.
FIGURE 11.

In vitro FGF-23 protein treatment of isolated BM Ter119+ cells normalizes erythropoiesis in Fgf-23−/− mice (n = 3 per group). Cells from each mouse were cultured in triplicate. A, percentage of mature erythroid cells stained positive for Ter119. B, percentage of mature erythroid cells stained positive for Ter119 and Epo-R. C and D, quantitative real time RT-PCR showing mRNA levels of Epo (C) and HIF-2α (D) in WT and Fgf-23−/− Ter119+ cells with or without FGF-23 protein treatment. The data are represented as mean fold change ± S.E. relative to housekeeping gene HPRT. *, p < 0.05; **, p < 0.01.
Genetic Ablation of Vitamin D in Fgf-23−/− Mice
Fgf-23 is a known regulator of 1,25(OH)2D3 and phosphate homeostasis. Fgf-23−/− mice exhibit highly elevated serum vitamin D and phosphate levels along with severe skeletal abnormalities and extraosseous calcifications (14, 20, 21, 30). Elimination of vitamin D in Fgf-23−/− mice reversed the described abnormalities in these mice (21), indicating that vitamin D partly mediates the function of Fgf-23 to regulate phosphate homeostasis and bone mineralization. Therefore, we investigated whether the elevated levels of vitamin D could account for the elevated levels of erythropoiesis in Fgf-23−/− mice. We generated mice deficient in both Fgf-23 and 1α-hydroxylase genes, the enzyme required to convert vitamin D to its active form, 1,25(OH)2D3, (Fgf-23−/−/1α(OH)ase−/−). Here, we compared and analyzed data obtained from wild-type, Fgf-23−/−, Fgf-23−/−/1α(OH)ase−/−, and 1α(OH)ase−/− animals.
Complete blood count analysis revealed that ablation of vitamin D from Fgf-23−/− mice partially rescued the erythroid changes observed in our Fgf-23−/− mice. Specifically, RBC numbers were significantly increased in Fgf-23−/− (as described above), Fgf-23−/−/1α(OH)ase−/−, and 1α(OH)ase−/− animals compared with WT littermates (Fig. 12A); however, RBC counts in both Fgf-23−/−/1α(OH)ase−/− and 1α(OH)ase−/− were markedly lower than in Fgf-23 single knock-out mice (Fig. 12A).
FIGURE 12.

Deletion of vitamin D rescues the hematopoietic phenotype of Fgf-23−/− mice. A–D, peripheral blood complete blood count analysis of 6-week-old WT, Fgf-23−/−, Fgf-23−/−/1α(OH)ase−/−, and 1α(OH)ase−/− mice. A, RBC counts (WT, n = 21; Fgf-23−/−, n = 6; Fgf-23−/−/1α(OH)ase−/−, n = 8; and 1α(OH)ase−/− n = 13). B, percentage of primitive pro-E-stained positive for Ter119med and CD71high (WT, n = 4; Fgf-23−/−, n = 4; Fgf-23−/−/1α(OH)ase−/− n = 5; and 1α(OH)ase−/− n = 9). C, percentage of mature erythroid cells stained positive for Ter119 and CD71low (Ery-C fraction) (WT, n = 8; Fgf-23−/−, n = 7; Fgf-23−/−/1α(OH)ase−/− n = 6; and 1α(OH)ase−/− n = 7). D, percentage of HSC population stained for SLAM (CD150+CD48−) (WT, n = 7; Fgf-23−/−, n = 9; Fgf-23−/−/1α(OH)ase−/− n = 7; and 1α(OH)ase−/− n = 8). E and F, FGF-23 injections. E, percentage of primitive pro-erythroblasts (pro-E) stained positive for Ter119med and CD71high (WT, n = 5; Fgf-23−/−, n = 6; Fgf-23−/−/1α(OH)ase−/− n = 3; and 1α(OH)ase−/− n = 3). F, percentage of mature erythroid cells stained positive for Ter119 (WT, n = 7; 1α(OH)ase−/− n = 12; WT+FGF-23, n = 6; 1α(OH)ase−/− + FGF-23, n = 3). The data are represented as means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared with WT; #, p < 0.05 compared with fgf-23−/−; ##, p < 0.01 compared with fgf-23−/−; $, p < 0.05 compared with fgf-23−/−/1α(OH)ase−/−; $$, p < 0.01 compared with fgf-23−/−/1α(OH)ase−/−.
Because of the significant increase in circulating red blood cell numbers in all three mutant genotypes (Fgf-23−/−, Fgf-23−/−/1α(OH)ase−/−, and 1α(OH)ase−/−), we investigated further the impact of vitamin D deficiency in erythroid cell differentiation in BM, by flow cytometry. Interestingly, both Fgf-23−/− and Fgf-23−/−/1α(OH)ase−/− mice had significantly elevated immature (pro-E) and mature (Ery-C) erythroid cells (Fig. 12, B and C) compared with WT controls. Erythroid cell populations, however, in 1α(OH)ase−/− mice did not differ from WT mice, but they were considerably decreased compared with Fgf-23−/− and Fgf-23−/−/1α(OH)ase−/− mice (Fig. 12, B and C). Evaluation of early SLAM enriched HSC progenitors showed elevated frequency of HSCs in Fgf-23−/− and Fgf-23−/−/1α(OH)ase−/− mice, but not in 1α(OH)ase−/− mice compared with WT littermates (Fig. 12D). SLAM+ cells were, however, greatly decreased in 1α(OH)ase−/− mice compared with Fgf-23−/− mice.
To further address the interplay between Fgf-23 and vitamin D in regulating hematopoiesis, we treated 1α(OH)ase−/− mice with exogenous FGF-23 protein. As we have previously shown, FGF-23 administration significantly reduced both erythroid cell and HSC frequencies in the BM of WT cells (Fig. 12, E and F). However, exogenous FGF-23 significantly reduced the frequency of both immature and mature erythroid cells in 1α(OH)ase−/− mice (Fig. 12, E and F). In summary, these data suggest that elimination of vitamin D cannot rescue the hematopoietic changes observed in our Fgf-23−/− mice, suggesting that these changes are independent of vitamin D, and thus Fgf-23 plays a vital role in regulating hematopoiesis and, particularly, erythropoiesis.
DISCUSSION
Ineffective hematopoiesis is a determinant of morbidity and death in adults and children and a common feature of many disorders. In addition, anemia is a common complication in CKD and is associated with worse long term outcomes. Correcting anemia could become an important and novel therapeutic strategy to improve long term outcomes in such patients. High FGF-23 levels are associated with CKD, greater cardiovascular risk, higher vascular and aortic calcifications, and left ventricular hypertrophy in dialysis patients. However, the involvement of Fgf-23 in the regulation of erythropoiesis is unknown. In the current study, we provide evidence for a regulatory role of Fgf-23 in the development of abnormal red blood cell production.
Over the last decade, the interplay between osteogenesis and hematopoiesis has become increasingly important. In particular, osteoblasts play an essential role in regulating HSCs, myelopoiesis, and lymphopoiesis in the bone marrow, a role supported by studies using genetically altered animal models that could activate or destroy osteoblastic cells (22, 23, 29). Increased osteoblast numbers lead to increased HSC frequency (22, 36, 37), whereas deletion of osteoblasts results in loss of HSCs as well as committed progenitor cells of the B-lymphocyte and erythroid lineages (23). Deletion of Fgf-23 in mice results in significantly decreased bone mineralization, trabeculae, and osteoblast numbers, along with reduced lymphatic organ size (14, 20, 21, 30). In this study, we examined the erythropoietic state of Fgf-23−/− mice in vivo and in vitro, and we show for the first time that Fgf-23 regulates erythropoiesis, and it does so independently of changes in the bone marrow environment, secondary disease onset, or vitamin D. Specifically, we found that ablation of Fgf-23 leads to significant augmentation of erythropoiesis in all tissues examined (PB, BM, and spleen). In addition, we show that short term in vivo and in vitro FGF-23 treatment results in erythropoietic changes opposite to those caused by Fgf-23 deficiency. More importantly, our study is the first to demonstrate that fetal liver erythropoiesis is severely disturbed in Fgf-23-deficient mice, uncovering a novel role of Fgf-23 in hematopoietic maintenance and HSC function during development.
The production of red blood cells (erythrocytes) in the BM is determined by the concentration of Epo, a hormone released by the kidneys in response to low oxygen levels in the blood, which acts on the erythroid progenitors in the BM to stimulate erythrocyte production. In compromised renal function, such as chronic renal failure and end stage renal disease, patients suffer from severe anemia because RBC production is reduced because of the inability of the kidneys to produce sufficient amounts of Epo to maintain RBC homeostasis (4, 5). Here, we observed that a lack of Fgf-23 results in markedly augmented erythropoiesis in PB and BM that can be accounted for by elevated levels of HIF and Epo in BM, liver, and kidney, which can also lead to increased HSC frequency. Furthermore, treatment of Fgf-23-deficient mice with oxygen significantly reduced renal and BM HIF mRNA levels and restored serum Epo levels to normal, resulting in normal number of erythroid cells in the BM in Fgf-23−/− mice.
It has been recently reported that augmented HIF signaling in osteoprogenitor cells by pharmacologic or genetic manipulations increased RBC production by increasing bone-derived Epo expression despite suppressed renal expression of Epo (32). In contrast, inactivation of HIF in osteoblasts impaired Epo expression in bone and numbers of erythroid progenitors in the bone marrow (32, 38). Interestingly, a recent study has shown that activation of HIF-1α by iron deficiency leads to increased Fgf-23 transcription (39). We are currently investigating the role of Fgf-23 in iron metabolism.
Of note, complete genetic inactivation of Fgf-23 results in a profound increase in HSC frequency and early erythroid cells in BM and peripheral blood, coupled with highly elevated serum Epo levels and severe increase in Epo and HIF mRNA synthesis in kidney, BM, and liver. However, the effect of Fgf-23 deletion in late erythroid cells in peripheral blood and BM and in hematological parameters is modest (∼20% increase). These observations suggest that Fgf-23 affects mainly early rather than late erythroid progenitors; the mechanism of this finding requires further investigation. Similarly, although mice lacking functional Epo (40), Epo-R (41), HIF-1α (42), or HIF-2α (43) genes are embryonically lethal, studies have shown that conditional inactivation of either the Epo (44) or Hif-2α (45) genes postnatally results in extremely low serum Epo levels that are associated with moderate anemia (20–30% reduction in RBC numbers, hemoglobin, and hematocrit) in adult mice, demonstrating the significance of Epo/HIF for adult erythropoiesis.
Fgf-23 inhibits vitamin D synthesis and phosphate reabsorption by down-regulating the expression of renal 25(OH)d-1α-hydroxylase (1α(OH)ase) and sodium phosphate transporters NPT2a and NPT2c, respectively (13, 14). Fgf-23 deficiency leads to hypervitaminosis D, hyperphosphatemia, and hypercalcemia along with tissue and vascular calcifications (14, 20). Genetic elimination of vitamin D from Fgf-23−/− mice reversed the hyperphosphatemia and hypercalcemia and abolished the soft tissue and vascular calcifications (21). In the present study, we show that loss of Fgf-23 also results in markedly increased erythrocyte and HSC populations in PB, BM, and fetal liver. However, abolishing vitamin D signaling in our Fgf-23−/− did not rescue the erythroid and HSC populations, suggesting that Fgf-23 is the main cause of induced erythropoiesis. Our data support a role for Fgf-23 on erythropoiesis by showing 1) a substantial reduction in erythrocytes in both PB and BM after treatment with FGF-23 protein, 2) exogenous FGF-23 protein to Fgf-23-deficient erythroid cells rescued the erythroid phenotype, 3) increased fetal erythropoiesis independent of the bone marrow environment, and 4) deletion of vitamin D from Fgf-23 null mice did not rescue the erythropoietic abnormalities.
Although there are no data available directly linking vitamin D to RBC mass, we have found that abolishing vitamin D activity in Fgf-23−/− mice normalized the mean cell volume and mean cell hemoglobin content (data not shown). Studies have shown that increased intracellular calcium levels in circulating RBCs result in a decrease in RBC volume (46). Therefore, it is possible that the effect on mean cell volume in Fgf-23−/− mice may be mediated by the hypercalcemia these mice exhibit rather than vitamin D. Further studies are needed to properly understand the mechanism by which vitamin D or calcium may influence red cell size and whether this effect is associated with iron metabolism.
In summary, the present study demonstrates for the first time a novel function of Fgf-23 in erythropoiesis and provides new insights into the molecular regulation of hematopoietic maintenance during development. Our data strongly suggest that Fgf-23 mediates HSC differentiation into the erythroid lineage. A broad developmental aberration of HSCs, hematopoietic progenitors, and differentiated hematopoietic cells is evident in Fgf-23-deficient fetal liver, demonstrating an essential role for Fgf-23 in HSC generation and differentiation during development. Importantly, our observation that Fgf-23 deficiency causes a defect in erythropoiesis indicates that elevated FGF-23 levels in chronic kidney disease may contribute to the anemia in these patients. Additional studies are needed to determine which specific FGFR mediates the hematopoietic effects of Fgf-23. It is critical to understand the mechanisms governing control of hematopoiesis and the exact role Fgf-23 plays in the regulation of HSC differentiation. Clarifying the relevant signaling pathways may provide additional therapeutic benefits in several diseases and novel approaches for treatment of hematological disorders associated with bone changes, renal, and cardiovascular function defects.
Our proposed model is shown in Fig. 13. Our data show that loss of Fgf-23 results in induction of erythropoiesis by regulating Epo directly or through hypoxia. We are currently investigating further whether Fgf-23 also acts directly on erythroid cells as well as the mechanism by which it affects fetal liver erythropoiesis. Blocking Fgf-23 activity can be beneficial in treating diseases associated with hematopoietic abnormalities.
FIGURE 13.
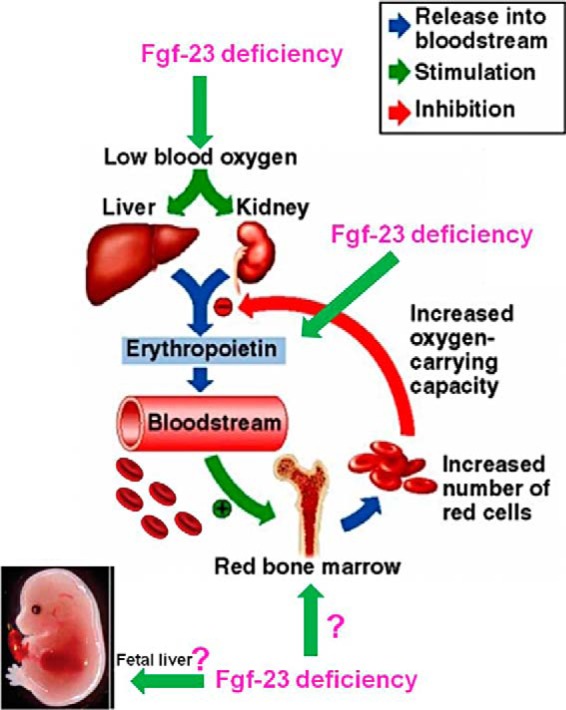
Proposed model showing the mechanism of Fgf-23 actions on erythropoiesis.
Acknowledgments
We are grateful to Dr. René St-Arnaud (Genetics Unit of Shriners Hospital, Montreal, Canada) for kindly providing the 1α(OH)ase+/− breeder mice. We also thank Dr. David Levy and Eric Ohlson (New York University College of Dentistry, New York, NY) for assistance with the use of the flow cytometer and Rick DeFrancisco and Dr. Tracy Stokol (Cornell University College of Veterinary Medicine) for advice on blood sample analysis. We are also very grateful to Prof. Jean-Pierre Saint-Jeannet for critical review of the manuscript and Prof. Nicola C. Partridge for support and valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant P30DE020754. This work was also supported by American Heart Association Scientist Development Grant 12SDG12080152 and American Society for Bone and Mineral Research Career Enhancement Award A13-0052-001 (to D. S.).
- BM
- bone marrow
- CKD
- chronic kidney disease
- HSC
- hematopoietic stem cell
- RBC
- red blood cell
- FGF
- fibroblast growth factor
- EPO
- erythropoietin
- HIF
- hypoxia-induced factor
- IMDM
- Iscove's modified Dulbecco's medium
- BFU-E
- burst-forming unit of erythroid cells
- CFU-GEMM
- colony-forming units of granulocyte-erythrocyte-macrophage-megakaryocyte cells
- SDF
- stromal-derived factor
- PB
- peripheral blood
- pro-E
- pro-erythroblasts.
REFERENCES
- 1. Christensen J. L., Wright D. E., Wagers A. J., Weissman I. L. (2004) Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2, E75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dzierzak E., Speck N. A. (2008) Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat. Immunol. 9, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sugiyama D., Inoue-Yokoo T., Fraser S. T., Kulkeaw K., Mizuochi C., Horio Y. (2011) Embryonic regulation of the mouse hematopoietic niche. ScientificWorldJournal 11, 1770–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lipkin G. W., Kendall R. G., Russon L. J., Turney J. H., Norfolk D. R., Brownjohn A. M. (1990) Erythropoietin deficiency in acute renal failure. Nephrol. Dial. Transplant. 5, 920–922 [DOI] [PubMed] [Google Scholar]
- 5. Zhang F., Laneuville P., Gagnon R. F., Morin B., Brox A. G. (1996) Effect of chronic renal failure on the expression of erythropoietin message in a murine model. Exp. Hematol. 24, 1469–1474 [PubMed] [Google Scholar]
- 6. Damasiewicz M. J., Toussaint N. D., Polkinghorne K. R. (2011) Fibroblast growth factor 23 in chronic kidney disease: New insights and clinical implications. Nephrology 16, 261–268 [DOI] [PubMed] [Google Scholar]
- 7. Fliser D., Kollerits B., Neyer U., Ankerst D. P., Lhotta K., Lingenhel A., Ritz E., Kronenberg F., MMKD Study Group,Kuen E., König P., Kraatz G., Mann J. F., Müller G. A., Köhler H., Riegler P. (2007) Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 18, 2600–2608 [DOI] [PubMed] [Google Scholar]
- 8. Isakova T., Xie H., Yang W., Xie D., Anderson A. H., Scialla J., Wahl P., Gutiérrez O. M., Steigerwalt S., He J., Schwartz S., Lo J., Ojo A., Sondheimer J., Hsu C. Y., Lash J., Leonard M., Kusek J. W., Feldman H. I., Wolf M., and the Chronic Renal Insufficiency Cohort (CRIC) Study Group (2011) Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305, 2432–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Isakova T., Wahl P., Vargas G. S., Gutiérrez O. M., Scialla J., Xie H., Appleby D., Nessel L., Bellovich K., Chen J., Hamm L., Gadegbeku C., Horwitz E., Townsend R. R., Anderson C. A., Lash J. P., Hsu C. Y., Leonard M. B., Wolf M. (2011) Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 79, 1370–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Larsson T., Nisbeth U., Ljunggren O., Jüppner H., Jonsson K. B. (2003) Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 64, 2272–2279 [DOI] [PubMed] [Google Scholar]
- 11. Faul C., Amaral A. P., Oskouei B., Hu M. C., Sloan A., Isakova T., Gutiérrez O. M., Aguillon-Prada R., Lincoln J., Hare J. M., Mundel P., Morales A., Scialla J., Fischer M., Soliman E. Z., Chen J., Go A. S., Rosas S. E., Nessel L., Townsend R. R., Feldman H. I., St John Sutton M., Ojo A., Gadegbeku C., Di Marco G. S., Reuter S., Kentrup D., Tiemann K., Brand M., Hill J. A., Moe O. W., Kuro-O M., Kusek J. W., Keane M. G., Wolf M. (2011) FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 121, 4393–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taylor E. N., Rimm E. B., Stampfer M. J., Curhan G. C. (2011) Plasma fibroblast growth factor 23, parathyroid hormone, phosphorus, and risk of coronary heart disease. Am. Heart J. 161, 956–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perwad F., Zhang M. Y., Tenenhouse H. S., Portale A. A. (2007) Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin d-1α-hydroxylase expression in vitro. Am. J. Physiol. Renal Physiol. 293, F1577–F1583 [DOI] [PubMed] [Google Scholar]
- 14. Shimada T., Kakitani M., Yamazaki Y., Hasegawa H., Takeuchi Y., Fujita T., Fukumoto S., Tomizuka K., Yamashita T. (2004) _targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 113, 561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. ADHRConsortium (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 26, 345–348 [DOI] [PubMed] [Google Scholar]
- 16. Jonsson K. B., Zahradnik R., Larsson T., White K. E., Sugimoto T., Imanishi Y., Yamamoto T., Hampson G., Koshiyama H., Ljunggren O., Oba K., Yang I. M., Miyauchi A., Econs M. J., Lavigne J., Jüppner H. (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N. Engl. J. Med. 348, 1656–1663 [DOI] [PubMed] [Google Scholar]
- 17. Liu S., Guo R., Simpson L. G., Xiao Z. S., Burnham C. E., Quarles L. D. (2003) Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J. Biol. Chem. 278, 37419–37426 [DOI] [PubMed] [Google Scholar]
- 18. Benet-Pagès A., Orlik P., Strom T. M., Lorenz-Depiereux B. (2005) An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet. 14, 385–390 [DOI] [PubMed] [Google Scholar]
- 19. Farrow E. G., Imel E. A., White K. E. (2011) Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and αKlotho). Best Pract. Res. Clin. Rheumatol. 25, 735–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sitara D., Razzaque M. S., Hesse M., Yoganathan S., Taguchi T., Erben R. G., Jüppner H., Lanske B. (2004) Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 23, 421–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sitara D., Razzaque M. S., St-Arnaud R., Huang W., Taguchi T., Erben R. G., Lanske B. (2006) Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am. J. Pathol. 169, 2161–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calvi L. M., Adams G. B., Weibrecht K. W., Weber J. M., Olson D. P., Knight M. C., Martin R. P., Schipani E., Divieti P., Bringhurst F. R., Milner L. A., Kronenberg H. M., Scadden D. T. (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 [DOI] [PubMed] [Google Scholar]
- 23. Visnjic D., Kalajzic Z., Rowe D. W., Katavic V., Lorenzo J., Aguila H. L. (2004) Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 103, 3258–3264 [DOI] [PubMed] [Google Scholar]
- 24. Zhu J., Garrett R., Jung Y., Zhang Y., Kim N., Wang J., Joe G. J., Hexner E., Choi Y., Taichman R. S., Emerson S. G. (2007) Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 109, 3706–3712 [DOI] [PubMed] [Google Scholar]
- 25. Adams G. B., Chabner K. T., Alley I. R., Olson D. P., Szczepiorkowski Z. M., Poznansky M. C., Kos C. H., Pollak M. R., Brown E. M., Scadden D. T. (2006) Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 439, 599–603 [DOI] [PubMed] [Google Scholar]
- 26. Koulnis M., Pop R., Porpiglia E., Shearstone J. R., Hidalgo D., Socolovsky M. (2011) Identification and analysis of mouse erythroid progenitors using the CD71/TER119 flow-cytometric assay. J. Vis. Exp. 54, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mirams M., Robinson B. G., Mason R. S., Nelson A. E. (2004) Bone as a source of FGF23: regulation by phosphate? Bone 35, 1192–1199 [DOI] [PubMed] [Google Scholar]
- 28. Yoshiko Y., Wang H., Minamizaki T., Ijuin C., Yamamoto R., Suemune S., Kozai K., Tanne K., Aubin J. E., Maeda N. (2007) Mineralized tissue cells are a principal source of FGF23. Bone 40, 1565–1573 [DOI] [PubMed] [Google Scholar]
- 29. Zhang J., Niu C., Ye L., Huang H., He X., Tong W. G., Ross J., Haug J., Johnson T., Feng J. Q., Harris S., Wiedemann L. M., Mishina Y., Li L. (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841 [DOI] [PubMed] [Google Scholar]
- 30. Sitara D., Kim S., Razzaque M. S., Bergwitz C., Taguchi T., Schüler C., Erben R. G., Lanske B. (2008) Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 4, e1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asari S., Sakamoto A., Okada S., Ohkubo Y., Arima M., Hatano M., Kuroda Y., Tokuhisa T. (2005) Abnormal erythroid differentiation in neonatal bcl-6-deficient mice. Exp. Hematol. 33, 26–34 [DOI] [PubMed] [Google Scholar]
- 32. Rankin E. B., Wu C., Khatri R., Wilson T. L., Andersen R., Araldi E., Rankin A. L., Yuan J., Kuo C. J., Schipani E., Giaccia A. J. (2012) The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 149, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hattori K., Heissig B., Tashiro K., Honjo T., Tateno M., Shieh J. H., Hackett N. R., Quitoriano M. S., Crystal R. G., Rafii S., Moore M. A. (2001) Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood 97, 3354–3360 [DOI] [PubMed] [Google Scholar]
- 34. Colvin G. A., Lambert J. F., Dooner M. S., Cerny J., Quesenberry P. J. (2007) Murine allogeneic in vivo stem cell homing. J. Cell. Physiol. 211, 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plett P. A., Frankovitz S. M., Orschell-Traycoff C. M. (2002) In vivo trafficking, cell cycle activity, and engraftment potential of phenotypically defined primitive hematopoietic cells after transplantation into irradiated or nonirradiated recipients. Blood 100, 3545–3552 [DOI] [PubMed] [Google Scholar]
- 36. Kuznetsov S. A., Riminucci M., Ziran N., Tsutsui T. W., Corsi A., Calvi L., Kronenberg H. M., Schipani E., Robey P. G., Bianco P. (2004) The interplay of osteogenesis and hematopoiesis: expression of a constitutively active PTH/PTHrP receptor in osteogenic cells perturbs the establishment of hematopoiesis in bone and of skeletal stem cells in the bone marrow. J. Cell Biol. 167, 1113–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stier S., Cheng T., Dombkowski D., Carlesso N., Scadden D. T. (2002) Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 99, 2369–2378 [DOI] [PubMed] [Google Scholar]
- 38. Bernhardt W. M., Wiesener M. S., Scigalla P., Chou J., Schmieder R. E., Günzler V., Eckardt K. U. (2010) Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J. Am. Soc. Nephrol. 21, 2151–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farrow E. G., Yu X., Summers L. J., Davis S. I., Fleet J. C., Allen M. R., Robling A. G., Stayrook K. R., Jideonwo V., Magers M. J., Garringer H. J., Vidal R., Chan R. J., Goodwin C. B., Hui S. L., Peacock M., White K. E. (2011) Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. U.S.A. 108, E1146–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu H., Liu X., Jaenisch R., Lodish H. F. (1995) Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 83, 59–67 [DOI] [PubMed] [Google Scholar]
- 41. Lin C. S., Lim S. K., D'Agati V., Costantini F. (1996) Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes Dev. 10, 154–164 [DOI] [PubMed] [Google Scholar]
- 42. Iyer N. V., Kotch L. E., Agani F., Leung S. W., Laughner E., Wenger R. H., Gassmann M., Gearhart J. D., Lawler A. M., Yu A. Y., Semenza G. L. (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 12, 149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng J., Zhang L., Drysdale L., Fong G. H. (2000) The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. U.S.A. 97, 8386–8391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zeigler B. M., Vajdos J., Qin W., Loverro L., Niss K. (2010) A mouse model for an erythropoietin-deficiency anemia. Dis. Model. Mech. 3, 763–772 [DOI] [PubMed] [Google Scholar]
- 45. Gruber M., Hu C. J., Johnson R. S., Brown E. J., Keith B., Simon M. C. (2007) Acute postnatal ablation of Hif-2α results in anemia. Proc. Natl. Acad. Sci. U.S.A. 104, 2301–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bogdanova A., Makhro A., Wang J., Lipp P., Kaestner L. (2013) Calcium in red blood cells: a perilous balance. Int. J. Mol. Sci. 14, 9848–9872 [DOI] [PMC free article] [PubMed] [Google Scholar]