

Abstract
Bacterial type II CRISPR-Cas9 systems have been widely adapted for RNA- guided genome editing and transcription regulation in eukaryotic cells, yet their in vivo _target specificity is poorly understood. Here we mapped genome-wide binding sites of a catalytically inactive Cas9 (dCas9) from Streptococcus pyogenes loaded with single guide RNAs (sgRNAs) in mouse embryonic stem cells (mESCs). Each of the four sgRNAs tested _targets dCas9 to tens to thousands of genomic sites, characterized by a 5-nucleotide seed region in the sgRNA, in addition to an NGG protospacer adjacent motif (PAM). Chromatin inaccessibility prevents dCas9 binding to other sites with matching seed sequences, and consequently 70% of off-_target sites are associated with genes. _targeted sequencing of 295 dCas9 binding sites in mESCs transfected with catalytically active Cas9 identified only one site mutated above background. We propose a two-state model for Cas9 binding and cleavage, in which a seed match triggers binding but extensive pairing with _target DNA is required for cleavage.
Many bacterial and archaeal genomes encode clustered regularly interspaced short palindromic repeats (CRISPR), which are transcribed and processed into short RNAs that guide CRISPR-associated (Cas) proteins to cleave foreign nucleic acids1–5. To _target particular genomic loci in eukaryotic cells, the type II CRISPR-Cas system from Streptococcus pyogenes has been adapted so that it requires the nuclease Cas9 and one sgRNA6–9. The first ∼20 nucleotides of the sgRNA (the guide region) are complementary to the _target DNA site, which also needs to contain a sequence called the protospacer adjacent motif (PAM), typically NGG10.
The simplicity of _targeting any locus with a single protein and a programmable sgRNA has quickly led to widespread use of Cas911,12 in applications such as genome editing7,8,13–16, disease gene repair17,18 and knock-in of specific tags8,19. The catalytically inactive dCas9 (D10A and H840A mutations) alone or when fused to activators or repressors has been used to modulate transcription20–25 and dCas9 has also been fused to GFP to allow imaging of genomic loci in living cells26.
However, the mechanism of _target recognition and _target specificity of the Cas9 protein remains poorly understood8,9,24,27–32. Most previous studies have analyzed a set of candidate off-_target sites with up to five mismatches to the designed on-_target. These studies have examined in vitro cleavage, cleavage induced indels or reporter gene expression change as the readEout rather than direct binding9,24,27,32. Base pairing in the first 10-12 nucleotides adjacent to PAM (defined as the “seed”) was found to be generally more important than pairing in the rest of the guide region6,8,16,33. However, large variations were observed across _target sites, cell types and species regarding the importance of base pairing at each position28. Some studies have shown that Cas9 is highly specific21,30,31, whereas other studies have demonstrated substantial Cas9 off-_target activity9,24,27,29,32. Epigenetic features such as CpG methylation and chromatin accessibility have been reported to have little effect on _targeting9,23.
To our knowledge, there has been no previous report of genome-wide binding maps of dCas9. Our data reveal a well-defined seed region for _target binding and a very large number of off-_target binding sites, most of which do not seem to undergo substantial cleavage by Cas9. Our observations explain some of the previously observed heterogeneity, provide insights into _target recognition and the cleavage process and could guide future _target design.
Results
Genome-wide binding of dCas9-sgRNA
To map dCas9 in vivo binding sites, we generated mESCs with a stably integrated vector encoding HA-tagged dCas9 (Fig. 1a), and performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) with cells transfected with either no sgRNA or one of each of 4 sgRNAs (Phc1-sg1, Phc1-sg2, Nanog-sg2 and Nanog-sg3) _targeting the promoters of Phc1 or Nanog, respectively. For each sgRNA, we observed ∼100 fold enrichment for dCas9 at the on-_target site compared to flanking regions, and the spatial resolution is sufficient to distinguish between two binding sites separated by 22 base pairs (bps) (Nanog-sg2 and Nanog-sg3) (Fig. 1b).
Figure 1.

Genome-wide in vivo binding of dCas9-sgRNA. (a) Schematic of dCas9 ChIP. EF1a promoter-driven HA-tagged dCas9 with nuclear localization signal (NLS) is integrated into the genome of mESCs via the piggyBac system. Plasmids containing U6 promoter-driven sgRNAs were transfected and ChIP was carried out two days later with HA antibody. (b) ChIP signals (normalized read counts) around on-_target sites. Vertical dashed lines indicate designed _target sites (the region complementary to the sgRNA). (c) Peak calling. Reads were sampled from each library, and peaks were called using each other library as a control (Online Methods). Only peaks called over all other five controls were retained. The numbers at the bottom indicate the numbers of peaks called for each library using these criteria.
Using the standard ChIP-seq peak-calling procedure MACS34 – comparing immunoprecipitated material and input (whole cell extract) DNA – we identified between 2,000 and 20,000 peaks in each sequencing library (Supplementary Fig. 1a). Cells expressing dCas9 but not transfected with sgRNAs (dCas9-only ChIP) exhibited 2,115 peaks. Most (77%) of the peaks detected in the dCas9-only ChIP were also detected in libraries prepared from dCas9-sgRNA immunoprecipitations (Supplementary Fig. 1b). The peaks in dCas9-only ChIP were enriched in open chromatin regions (Supplementary Fig. 2a) and 41% contained GG/CC-rich motifs that closely resemble CTCF binding motifs (Supplementary Fig. 2b-d). Such peaks could either represent ‘sampling’ by dCas9 of accessible sites containing NGG33, or transcription-dependent artifacts as previously reported for GFP ChIP in yeast35.
To identify sgRNA-dependent dCas9 binding sites, we matched sequencing depth by randomly sampling an equal number of reads from all six libraries and then performed pair-wise peak calling with MACS using each of the other five libraries as the control; we only retained peaks that were enriched over all five controls (Fig. 1c). Only 3 background peaks were called using this approach for dCas9-only ChIP. The number of sgRNA-specific peaks varied substantially; for example there were nearly 6,000 peaks for Nanog-sg3 but only 26 peaks for Nanog-sg2 (Fig. 1b). Many of the off-_target peaks showed high binding levels, as defined by the peak height relative to on-_target peaks after subtracting dCas9-only reads. For example, there were 91 off-_target peaks with more than 50% of the binding level of the on-_target site for Nanog-sg3 (Supplementary Table 1). These results suggest that there are substantial numbers of off-_target binding sites and the majority of the dCas9-sgRNA complex binds outside the designed _target site.
A 5-nucleotide seed for dCas9 binding
Sequence motifs enriched within 50 bps of peak summits were identified using MEME-ChIP36. The top motif found for each ChIP library matched the PAM-proximal region of the transfected sgRNA plus the PAM NGG (Fig. 2a and Supplementary Fig. 3). For 3 of the 4 sgRNAs, only PAM-proximal positions 1 to 5 in the _target DNA showed preference of base match to the guide (Fig. 2a). We therefore define position 1-5 as the ‘seed’ region of the sgRNA. For Nanog-sg2, the guide match extends to about 10-12 bases to the 5′ end, possibly due to the presence of multiple Us in the seed that lowers the thermodynamic stability of the sgRNA-DNA interaction. For Nanog-sg3 and Phc1-sg2, exact match to the 5-nucleotide seed followed by NGG (seed+NGG) within 50 bps of peak summits explained 96% and 97% of the peaks, respectively. When the 50 nucleotides flanking peak summits were shuffled preserving dinucleotide frequency, less than 5.7% of the shuffled sequences contained seed+NGG (Fig. 2a) for all four sgRNAs. Moreover, the seed+NGG sites were highly enriched at the center of the peak (Fig. 2a, right), suggesting the _target sites identified are directly bound by sgRNA-guided dCas9.
Figure 2.
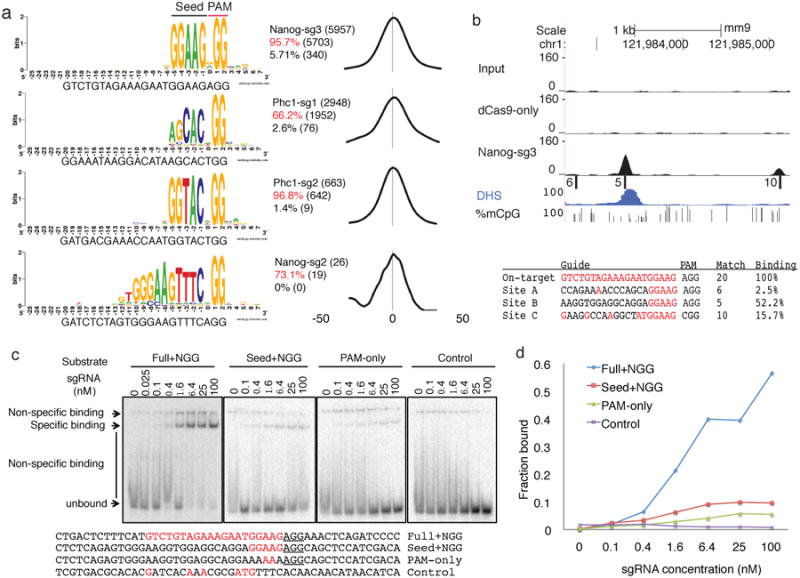
A 5-nucleotide seed for dCas9 binding. (a) Most peaks are associated with seed+NGG matches. The best match to the sgRNA followed by NGG within 50bp flanking peak summits were aligned to generate the sequence logo using WebLogo46. The text to the right of the logos indicates the total number of peaks (top line), percentage and number of peaks with exact 5-nucleotide seed+NGG match within 50 bps of peak summits (middle line, in red) or when the 100 nucleotides sequence were shuffled while maintaining dinucleotide frequency (bottom line). The distribution of the exact seed+NGG match relative to the peak summit was shown on the right (the numbers indicate nucleotide positions) (b) Example of binding at seed+NGG only sites. On the top are six tracks: Input, dCas9- only IP, and Nanog-sg3 IP read density, seed+NGG sites (position indicated by bars, named as A/B/C, and the numbers to the left indicates the number of matches to the guide), DHS read density and fraction of methylated alleles at CpG sites. Below are the _target sequences, PAM, number of matches to the sgRNA and relative binding at each site. Guide-matched bases are in red. (c) Gel shift assay for 50 bp double-stranded DNA substrates with sequences matching the Nanog-sg3 on-_target site (“Full+NGG”) and a seed+NGG only off-_target site (“Seed+NGG”, site B in Fig 2b). “PAM only” is the “Seed+NGG” substrate with a mutated seed. The negative control substrate (“Control”) was designed to contain no NGG or NAG. Complete substrate sequences are shown at the bottom, with PAM underlined and guide-matched bases in red. (d) The quantification of the gels in (c). Shown is the percentage of the specific binding band relative to the entire lane at each sgRNA concentration.
We found that seed+NGG alone is sufficient for Cas9 binding in vivo and in vitro. For example, there were 92 peaks in the Nanog-sg3 sample containing only seed+NGG matches, i.e. mismatches at all the other 15 positions. The strongest peak containing only seed+NGG showed 52% binding activity relative to the on-_target (Fig. 2b). In vitro gel shift assays confirmed specific binding to seed+NGG only substrates but with lower affinity than the on-_target site (Fig. 2c).
The peak motif analysis (Supplementary Fig. 3) revealed no enrichment of binding at seed sites followed by NAG, an alternative PAM previously reported to function in Cas9-mediated cleavage9,16,27. For example, of all 996 (33%) Phc1-sg1 ChIP peaks without seed+NGG sites, only 18 had seed+NAG within 50-bp of the peak summit, even less than expected by chance (Supplementary Fig. 4). ChIP-seq in human HEK293FT cells transfected with dCas9 and the same sgRNAs used in a previous study9, where NAG cleavage was reported, also failed to detect binding at those NAG off-_target sites (Supplementary Fig. 5). In vitro we observed >10 fold decrease in affinity when NGG was mutated to NAG in the on-_target substrate (Supplementary Fig. 6). Our in vivo and in vitro binding data are consistent with previous in vitro cleavage data showing that NAG or other variants rarely function as PAMs under enzyme-limiting conditions27.
Chromatin accessibility is the major determinant of in vivo binding
There are hundreds of thousands of seed+NGG sites in the genome for each sgRNA, for example, 621,651 for Nanog-sg3. To understand why only a small fraction of sites (<1%) were bound, we first looked for a correlation between the number of base match to the 20-nucleotide guide region and binding levels of ChIP peaks. Overall the correlation was very weak (Pearson correlation coefficient r=0.03, 0.12, 0.15 and 0.55 for Nanog-sg3, Phc1-sg2, Phc1-sg1 and Nanog-sg2, respectively (Fig. 3a and Supplementary Fig. 7)).
Figure 3.

Chromatin accessibility is the major determinant of binding in vivo. (a) Scatter (center) and histogram (top and right) plots of the number of matches to the sgRNA guide region (x-axis) and binding relative to the on-_target site (y-axis) for all Nanog- sg3 peaks. Relative binding levels (0 to 1) are divided into 10 equal bins and the number of peaks in each bin is shown on the right of the scatter plot. (b) Ranking of features based on R2, the percent of variation in binding explained by each feature in a linear regression model (using R, one feature a time). DHS: DNase I hypersensitivity read density; Tm: melting temperature; Match: number of bases that match the sgRNA; E(F) min/max/avg: minimum, maximum, and average tetranucleotide energy (flexibility) score within the guide+NGG region; A/C/G/T or their combination indicates mono- and di-nucleotide frequency in the guide+NGG region; %mCpG: average fraction of methylated CpG in the guide+NGG region. (c) Scatter plot and linear regression between the number of dCas9 ChIP peaks and the number of accessible seed+NGG sites (i.e. sites overlapping with DHS peaks). (d) As for (b) but only plotting the top five features after regression was done using sites containing CpG dinucleotides. (e) Off-_target peaks are preferentially associated with genes. Shown is the percentage of Nanog-sg3 seed+NGG sites (top) or ChIP peaks (bottom) that fall in each region category. (f) Example of off-_target binding at the Dusp19 promoter. Tracks are the same as Fig. 2b. On the right is the alignment of the off-_target site with 7 matches (bottom) to the guide sequence (top).
To identify determinants influencing in vivo binding, we applied a linear regression model of a set of sequence (mono- and di-nucleotide frequency), structural (melting temperature, DNA energy and flexibility37) and epigenetic (chromatin accessibility as assayed by DNase I hypersensitivity (DHS)38 and DNA CpG methylation39) features around the seed+NGG sites for each sgRNA (Online Methods). We found that chromatin accessibility (DHS) is the strongest indicator of binding in vivo, explaining up to 19% of the variation in binding when considering all individual seed+NGG sites in the genome (Fig. 3b). The difference in the number of seed+NGG sites in DHS peaks (i.e. accessible seed+NGG sites) explained 92% of the variation in the number of dCas9 peaks among the four sgRNAs (Fig. 3c, n = 4, p<0.05, F-test). Although this is based on a limited set of sgRNAs, it suggests that it might be possible to predict the approximate number of off-_target peaks based on the seed sequence in cell types where chromatin accessibility data are available.
Previous data suggested that Cas9 cleavage activity is not affected by DNA CpG methylation9. However, for the 17% of seed+NGG sites in the genome that contain CpG dinucleotides within the 20mer guide match and NGG, CpG methylation became the strongest predictor of dCas9 binding and negatively correlated with binding (Fig. 3d, Supplementary Fig. 8a-b). In a regression model, adding CpG methylation to DHS for sites containing CpGs almost doubled the amount of variation explained (Supplementary Fig. 8c). Our data suggests that CpG methylation likely reflects an aspect of chromatin accessibility not fully captured by DHS or that when combined with extensive mismatches, CpG methylation may impede binding.
The correlation with chromatin accessibility suggested that dCas9 off-_target binding might preferentially occur at active genes. For Nanog-sg3, 70% of the off-_target sites were associated with genes, including 18% in promoter region (< 2 kb upstream of gene TSS), 6% near enhancer regions and 46% within genes (Fig. 3e). For example, an off-_target peak that co-localized with the Dusp19 gene TSS and a DHS peak showed 74% binding relative to the on-_target with only 7 base matches to NanogE sg3 (Fig. 3f).
Seed sequences influence sgRNA abundance and specificity
The Nanog-sg2 sgRNA had substantially fewer off-_target binding sites than predicted by accessible seed+NGG sites (Fig. 3c). Although the same amount of sgRNA plasmids were transfected, the abundance of Nanog-sg2 was more than seven fold lower than the other three sgRNAs as determined by Northern blot (Fig. 4a). The same pattern of sgRNA abundance was observed when cells were transfected with sgRNA expression plasmids without co-transfecting dCas9, although all four sgRNAs showed substantially decreased levels of abundance, consistent with previous reports that Cas9 stabilizes sgRNA in cells13.
Figure 4.

Seed sequences influence sgRNA abundance and specificity. (a) Northern blot showing the abundance of sgRNAs. Lanes 1-10: from cells transfected with dCas9 (lanes 6-10) or without dCas9 (lanes 1-5), and with either no sgRNA (lanes 1 and 6) or one of the four sgRNAs (P1: Phc1-sg1; P2: Phc1-sg2; N2: Nanog-sg2; N3: Nanog-sg3). Lanes 11-14: Nanog-sg3 abundance from dCas9-mESCs transfected with 20, 2, 0.2 or 0.02 μg Nanog-sg3 plasmid. Lanes 15-17: Nanog-sg2 abundance from dCas9-mESCs transfected with 40, 20 or 10 μg Nanog-sg2 plasmid. (b) The number of ChIP peaks detected from cells transfected with decreasing amount of sgRNA plasmids. (c) U-rich seed limits sgRNA abundance. Northern blot from dCas9 cells transfected with the sgRNAs listed below. Consecutive Us are highlighted in bold black.
To test if sgRNA abundance influences the number of off-_target sites bound, we repeated the ChIP experiments after transfection with various amounts of sgRNA plasmids. Northern blots confirmed the decrease in sgRNA when less plasmid was transfected (Fig. 4a) and we identified decreased numbers of peaks with decreased amounts of plasmid (Fig. 4b). When the level of Nanog-sg3 was reduced to a similar level as Nanog-sg2 (Fig 4a, comparing lane 13 to lanes 16 and 17), the number of peaks for Nanog-sg3 was still much higher than for Nanog-sg2, presumably due to the presence of more accessible Nanog-sg3 seed+NGG sites in the genome (Fig. 3c). When 0.02 μg plasmid was transfected, Nanog-sg3 RNA was barely detected (lane 14); the 122 peaks identified in this library showed low overlap (9%) with our previous Nanog-sg3 ChIP, suggesting these were mostly non-specific signals.
A comparison of the seed regions of the four sgRNAs suggested that UUU in the seed of Nanog-sg2 might be responsible for decreased sgRNA abundance and increased specificity, consistent with recent observation that U in PAM-proximal position 1-4 leads to low gene knockout efficacy14. Indeed, two mutations (U to G and U to A) in the Nanog-sg2 seed region that converted the seed (GUUUC) to the same sequence as the Phc1-sg2 seed (GGUAC), led to higher levels of sgRNA (sgRNA N2b in Fig. 4c). Considering the presence of GUUUUA adjacent to the seed and because sgRNAs are transcribed by RNA polymerase III which is terminated by U-rich sequences40,41, we speculate that together with the downstream U-rich region, multiple Us in the seed might induce termination of sgRNA transcription. Consistent with this, three sgRNAs with seeds UUAUU, ACUUU and UUUUU also showed very low abundance (Fig. 4c, sgRNA P3, N5 and N6). When GUUUC was placed upstream of the seed thus away from GUUUUA in the sgRNA, the sgRNA was well expressed (sgRNA C4 in Fig. 4c).
One of 295 off-_target sites is mutated above background
To test if binding correlates with Cas9 nuclease-induced mutation, we examined the indel frequencies of the four on-_target sites and 295 selected off-_target sites by _targeted PCR and sequencing9. These sites were selected to cover a broad range of binding levels and numbers of mismatches to the sgRNA. We ranked all peaks by binding (background subtracted read counts) and for each binding level selected a peak with the fewest mismatches and another peak with most mismatches to the guide.
We determined the indel frequency of the 299 selected binding sites in wild type mESCs transfected with active Cas9 and each of the four sgRNAs, for three independent biological replicates (Supplementary Table 3). The level of Cas9 protein transiently expressed in the cells was 2.6 fold higher than in cells with stably integrated dCas9 used for ChIP (Supplementary Fig. 9a, comparing lane 1 to lane 8). The same ChIP and peak calling procedures in cells transiently transfected with dCas9 identified 2.7 times more Nanog-sg3 peaks (16,119 versus 5,957 in dCas9 stable cell lines), including 96% (85) of the 89 peaks selected for indel analysis. The amount of Cas9/dCas9 plasmids used for transfection is similar to levels used for genome editing applications by the field (Supplementary Fig. 9b).
Using our previously validated model9, the background indel frequencies due to sequencing noise were determined for each individual _target using two biological replicates transfected with only Cas9 but no sgRNA (“control”). Importantly the control samples showed no evidence of _targeted mutations by Cas9 (note that background indels in the absence of Cas9 might also occur). We manually reviewed sequencing alignments of all loci with indel frequencies above 0.03%. We found 12% to 37% sequencing reads from the on-_target sites contained indels, yet only one off-_target, which was from Nanog-sg2, was mutated at a frequency of 0.7% (Fig. 5). There was no detectable correlation between binding and indel frequency (sites in Fig. 5 are ranked by decreasing binding from left to right for each sgRNA). The selected sites include 7 of the top 10 (including all the top 6) and 36 of the top 50 Nanog-sg3 binding sites with the strongest ChIP signals, and 4 of the 8 Nanog-sg3 off-_target binding sites that have fewer than four mismatches to the sgRNA; none of these off-_target sites showed cleavage significantly above the background level.
Figure 5.

Indel frequencies at on-_target sites and 295 off-_target sites. For each sgRNA, selected _target sites (Supplementary Table 3) were ranked by decreasing ChIP binding relative to on-_target. Dots and gray bars indicate the mean and standard deviation of indel frequency from three biological replicates, respectively. The Y-axis was truncated at 0.001% for visualization at log scale. The indel frequencies for the four on-_target sites are labeled with percentages.
Discussion
We have shown that dCas9 binding is more promiscuous than previously thought. The low binding specificity is explained by the limited requirement for an accessible match to a 5-nucleotide seed followed by an NGG PAM. The position of the seed region next to PAM is consistent with previous observations that base pairing near PAM is critical for _targeting6,8,16,33, but the seeds we identified for 3 of the 4 sgRNAs are shorter than those previously reported; seed lengths of 8-13 nucleotides have been described as required for cleavage by Cas96,8,16,33.
The seed sequence influences the specificity of Cas9-sgRNA binding in several ways. Firstly, seed composition determines the frequency of a seed+NGG site in the genome. Secondly, seed composition determines how likely a seed+NGG site will be in open chromatin. Thirdly, seed composition affects sgRNA abundance, probably at the level of transcription, and thus the effective concentration of Cas9-sgRNA complex. Lastly, seed composition may also affect loading into Cas9 and again tune the level of functional Cas914. Through all four mechanisms U-rich seeds are likely to increase _target specificity.
Our results suggest that applications based on dCas9 or dCas9-effector fusions, such as transcription modulation, imaging, and epigenome editing, could be complicated by substantial off-_target binding. Previous studies suggest that several sgRNAs _targeting the same gene are frequently necessary for gene activation22-24; this could potentially reduce off-_target effects due to the requirement of co-_targeting. However, the use of multiple sgRNAs increases the number of potential off-_target binding sites, which might complicate interpretation. Although we only detected indels at a low frequency (0.7%) above background for one off-_target binding site among 295 selected sites, 295 is a small fraction of all possible binding sites and may not be representative of the complete off-_target mutation profile of each sgRNA. This is an important question as low frequencies of indels could complicate certain CRISPRE-Cas9 applications, such as genome-wide screening that involves selective growth14,15. Therefore, to minimize the likelihood of false positive screening hits resulting from off-_targeting, we recommend using multiple guide RNAs to _target each gene and using the concordance among multiple guides to interpret screening results. We further note that although binding sites with NAG PAMs are not enriched in the ChIP data, a previous study has shown that NAG-flanked genomic loci can contribute to off-_target indel-mutations. Therefore, unbiased and more sensitive detection of genome-wide mutations will be needed to determine Cas9 cutting specificity.
The observation that most of the sites bound by Cas9 do not seem to have substantial cleavage is reminiscent of the eukaryotic Argonaute-microRNA system, in which most _target mRNAs bearing partial microRNA match are bound without cleavage and only a few _targets with extensive pairing are cleaved42. We propose a two-state model (Fig. 6) similar to the Argonaute-microRNA system, in which pairing of a short seed region triggers binding after PAM recognition and subsequent DNA unwinding. _targets with only seed complementary remain bound by Cas9 without cleavage; only those with extensive pairing undergo efficient cleavage. This suggests a conformation change between binding and cleavage as observed for Argonaute-microRNA complexes42,43. While this paper was under review, a pair of Cas9 structural studies were published44,45, including a crystal structure of dCas9 in complex with sgRNA and _target DNA, which not only supports our observation of a PAM-proximal 5-nucleotide seed but also suggests a large conformation change during the inactive-active state transition45.
Figure 6.

A model for Cas9 _target binding and cleavage. (a) In the unbound state, Cas9 is loaded with sgRNA but not bound to DNA. The PAM region in the DNA is colored in red. (b) Recognition of the PAM by Cas9. (c) Cas9 melts the DNA _target near the PAM to allow seed pairing. (d) If base pairing can be propagated to PAM-distal regions, the two Cas9 nuclease domains may be able to ‘clamp’ the _target DNA and cleave it. (e) If only partial pairing occurs, there is no cleavage and Cas9 remains bound to the _target.
Online Methods
Oligonucleotides
All oligonucleotides used in this study were purchased from Integrated DNA Technologies. Sequences are listed in Supplementary Table 2.
Cloning
A two-step fusion PCR was used to amplify Cas9 nickase ORF from pX335 vector (Addgene: 42335) and incorporate H840A mutation to create a nuclease deficient Cas9 (dCas9). This PCR product was inserted into the Gateway donor backbone pCR8/GW/TOPO to create pAC84 (Addgene: 48218). The dCas9 ORF in pAC84 was then transferred to a piggyBac-based destination vector pAC150 (PB-Lox-HygroR- Lox-4×HSInsulators-EF1a-DEST) by LR Clonase reaction (Invitrogen) to create pAC159 (PB-LHL-4×HS-EF1a-dCas9). The sgRNA expression cassette was amplified by PCR from pX335 vector and cloned into a piggyBac vector pAC158 (PB-neo-4×HSInsulators) to create pAC103 (Pbneo-sgExpression). sgRNA was then cloned into BbsI-digested pAC103 by oligo cloning method as described previously8. Cas9 transient transfection constructs consisted CBh-driven WT-Cas9 or Cas9-D10AH840A (dCas9) containing a C-terminal HA-tag.
Cell culture
V6.5 mouse embryonic stem cells (mESCs) were cultured in DMEM supplemented with 10% FBS, pen/strep, L-glutamine, nonessential amino acids and LIF. For generation of cells stably integrating dCas9, cells were transfected in a 6-well and selected using Hygromycin B at 100 ug/mL 24 hours post transfection, then raised to 150 ug/mL 48 hours post transfection. Cells were split onto 10 cm plates, and single clones were isolated, expanded, and used for all experiments described. HEK293FT cells were cultured as previously described9. All transfection were done with Lipfectamine 2000 (Invitrogen).
ChIP
Three million cells were seeded on to 10cm plates on day 1, transfected with sgRNAs plasmids (or together with HA-dCas9 plasmids) on day 2, transferred to 15cm plates on day 3, and crosslinked on day 4 with roughly 50 million cells. Crosslinking is done by adding 2 mL (0.1 volume) 37% formaldehyde to the plate, incubating at room temperature for 15min, and quenched by adding 1 mL 2.5M glycine. Cells were rinsed twice with cold PBS and scraped to collect in cold PBS. Cells were centrifuged at 1,350g for 5min at 4°C and washed again in cold PBS. Cells were flash frozen in a dry ice/ethanol mix and stored at -80°C. Cell pellet was resuspended in 5mL cold Lysis Buffer 1 (50mM HEPES-KOH pH 7.5, 140mM NaCl, 1mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Trition X-100, 1× Roche complete protease inhibitors), rotated at 4°C for 10min followed by centrifugation at 1,350g for 5min at 4°C. Pellet was resuspended in 5mL Lysis Buffer 2 (10mM Tris-Cl pH 8, 200mM NaCl, 1mM EDTA, 0.5mM EGTA, 1× Roche complete protease inhibitors), rotated at 4°C for 10min followed by centrifugation at 1,350g for 5min at 4°C. Nuclear pellet was resuspended in 2mL Sonication Buffer (20mM Tris-Cl pH 8, 150mM NaCl, 2mM EDTA, 0.1% SDS, 1% Triton X-100, 1× Roche complete protease inhibitors) and sonicated (60min total time, 30sec on, 30sec off, in 6 rounds of 10min) in a Bioruptor (Diagenode). Lysate was centrifuged in eppendorf tubes in a microfuge at 4°C a max speed for 20min. Supernatant was collected and 50μL of this was saved as input. Protein G Dynabeads were conjugated to 5 ug rabbit anti-rat antibody (Thermo) in 0.1M Na-Phosphate pH 8 buffer at 4°C with rotation followed by conjugation to 5 ug HA antibody (Roche 3F10, #11867431001). Beads were resuspended in 50 ul Sonication Buffer and added to samples to immunoprecipitate overnight. The next day, beads were washed twice in sonication buffer, once in sonication supplemented with 500mM NaCl, once in LiCl Buffer (10mM Tris-Cl pH 8, 250mM LiCl, 1mM EDTA, 1% NP-40) and once in TE + 50mM NaCl. Each wash was accomplished with rotation at 4°C for 5min. Chromatin was eluted at 65°C for 15min in Elution Buffer (50mM Tris-Cl pH 8, 10mM EDTA, 1% SDS). Input was combined with elution buffer and both input and IP crosslinks were reversed at 65°C overnight. RNA was digested with RNAse A at 0.2mg/ml final concentration (Sigma) at 37 °C for 2hrs and protein was digested with proteinase K at 0.2mg/mL final concentration (Life Technologies) at 55 °C for 45min. DNA was phenol:chloroform:isoamyl alcohol (Life Technologies) extracted and ethanol precipitated. Barcoded libraries were prepared and sequenced on Illumina HiSeq2000.
ChIP-seq data analysis
Reads were de-multiplexed and mapped to mouse genome mm9 using bowtie47, requiring unique mapping with at most two mismatches (-n 2 -m 1 --best --strata). Mapped reads were collapsed and the same number of reads (about 9 million) was randomly sampled from each library to match sequencing depth. Peaks were called using MACS50 with default settings. For each sample, the other samples are each used as a control and only peaks called over all five controls are defined as _target sites. To quantify relative binding strength, reads were first extended at the 3′ end to the average fragment length (d) estimated by MACS, and then the number of fragments (extended reads) overlapping with the seed+NGG region is counted and normalized by subtracting counts from dCas9-only control. If multiple seed+NGG match sites were found, the one with the highest relative binding was assigned to the peak.
Analysis on determinants of binding
Mouse ES cell DNase Hypersensitivity data (bigwig file and narrow peak file) were downloaded from UCSC genome browser hosting the mouse ENCODE project38. DNA CpG methylation data was downloaded from GEO dataset GSE30202. Melting temperature (Tm) was calculated using the oligotm program in primer3 version 2.3.6. DNA stability and flexibility were calculated using a table of tetranucleotide scores derived from X-ray crystal structures in a previous study37. The linear regression is performed by using the lm function in R, one feature a time to calculate the R2 value for each feature.
Northern blot
Total RNA was isolated using TRIzol (Life Technologies) and 5 ug of total RNA was loaded on 8% denaturing PAGE. Northern blot was done as previously described9, using a probe _targeting the scaffold shared by all sgRNAs.
Protein Purification
Human codon-optimized Cas9 (Addgene plasmid 42230) was subcloned into a custom pET-based expression vector with an N-terminal hexahistidine (6×His) tag followed by a SUMO protease cleavage site. The fusion construct was transformed into E. coli Rosetta 2(DE3) competent cells (Millipore), grown in LB media to OD600 0.6, and induced with 0.2 mM IPTG for 16h at room temperature. Cells were pelleted, resuspended and washed with Milli-Q H2O supplemented with 0.2 mM PMSF, and lysed with lysis buffer (20 mM Trizma base, 500 mM NaCl, 0.1% NP-40, 2 mM DTT, 10 mM imidazole). The lysis buffer was supplemented with protease inhibitor cocktail (Roche) immediately prior to use. Whole lysate was sonicated at 40% amplitude (Biologics Inc., 2s on, 4s off) prior to ultracentrifugation (30,000 rpm for 45m). The clarified lysate was applied to cOmplete His-tag purification columns (Roche), washed with wash buffer 1 (20 mM Trizma base, 500 mM NaCl, 0.1% NP- 40, 2 mM DTT, 10% glycerol, 10 mM imidazole) and wash buffer 2 (20 mM Trizma base, 250 mM NaCl, 0.1% NP-40, 2 mM DTT, 10% glycerol, 50 mM imidazole). The 6xHis affinity tag was released via SUMO protease cleavage and bound protein was eluted with a linear gradient of 150 mM – 500 mM imidazole. Eluted protein was concentrated with Amicon centrifugal filter units with Ultracel membrane (Millipore) and stored at -80°C.
In vitro transcription
A T7 promoter forward oligo was annealed to an sgRNA template oligo by heating to 95°C for 3 min in 1× T4 DNA ligase buffer and then cooled at room temperature for 30 min. The annealed product was used as template and transcribed with MEGAshortscript T7 Kit (Life Technologies). RNAs were purified by MEGAclear Kit (Life Technologies) and frozen at -80 °C.
Gel shift assay
Single stranded DNA oligos of 50 nucleotides were purchased from IDT and PAGE purified. Double stranded substrate were generated by mixing 100 pmol each strand in water (10 ul total), heating to 95°C for 3 min and cool to room temperature. The substrates were then 5 end labeled with [γ-32P]-ATP using T4 PNK (New England Biolabs) for 30 min at 37°C, and free ATP removed by G-25 column (GE Healthcare). For each reaction, 100 nM Cas9 was mixed with a 1:4 dilution series of sgRNA (from 0 to 100 nM) in 1× NEBuffer 3 at 37°C for 10 min, and then about 0.5 nM labeled substrate oligos were added and incubated for 5 min at 37°C in a 10 ul reaction. Reactions were stopped on ice and added 1/2 volume of 50% glycerol. Samples were loaded on to 12% native PAGE and run at 300V for 2 hours at room temperature. Gels were visualized by phosphorimaging. Gel quantification is done with ImageJ. The fraction bound shown in Fig. 2c was calculated as the ratio of intensity from the specific binding band to the total intensity of the entire lane.
_targeted sequencing and indel detection
For replicate 1, cells were seeded in 6-well plates (300,000 cells per well), transfected with 2 ug sgRNA plasmid, 2 ug Cas9 plasmid, using 10 ul Lipofectmine 2000 reagent per sample for 3 hours. For replicate 2 and 3, 50% more plasmids were used. DNA was extracted and selected _target sites were PCR amplified, normalized, and pooled in equimolar proportions. Pooled libraries were denatured, diluted to a 14pM concentration and sequenced using the Illumina MiSeq Personal Sequencer (Illumina). Sequencing data was demultiplexed using paired barcodes, mapped to reference amplicons, and analyzed for indels as described previously9.
Supplementary Material
Supplementary Figure 1 | Conventional peak calling comparing IP to input. (a) The number of peaks called by MACS using default settings. (b) The fraction of dCas9-only peaks that are also detected in one, two, three, or all other four IP samples.
Supplementary Figure 2 | Characteristics of dCas9-only peaks. (a) Peaks are enriched for open chromatin regions. Shown is the average density of Dnase Hypersensitivity reads per 50bp bin in a 2kb window centered on peak summits. Blue area indicates standard error. (b) De novo motif finding within 50bp of peak summits by MEME-ChIP uncovered two related GG/CC-rich motifs. (c) Relative position of the motif within the peak, 0 indicates peak summits. (d) The longer motif (bottom) closely resembles CTCF binding motif (top, p < 1e-23)
Supplementary Figure 3 | De novo motif discovery in ChIP peaks. Motifs detected by MEME-ChIP using default settings and sequences within 50 bps of peak summits. The guide RNA sequences were shown below the motif.
Supplementary Figure 4 | Lack of ChIP enrichment at seed+NAG sites. (a) of the 996 (33%) Phc1-sg1 ChIP peaks without seed+NGG sites, only 18 (1.8%) contain seed+NAG (AGCACNGG) within 50bp of peak summits, which is not higher than random (2.7%). Motif logo of the 18 NAG peaks showing the lack of base pairing outside the seed region. (b) The number of seed+NAG sites in the genome (column 2) and within ChIP peaks (column 3) that contain specific number of mismatches in the guide region (column 1). None of the seed+NAG sites with less than 6 mismatches showed ChIP signals strong enough to be defined as peaks. (c) The best match contains only 1 mismatch and showed no ChIP signals. (d) Only one site with 6 mismatches is within a peak, yet the seed+NAG site is not in the center of the peak. (e-f) Similar to (c-d) but showing the two strongest peaks that are associated with seed+NAG sites. For (c-f), the top track is ChIP signal, and the bottom track is open chromatin. The scale is the same as Fig. 2b.
Supplementary Figure 5 | ChIP signals in HEK293FT cells. ChIP read density at four off-_targets (a-d) and on-_targets (e). Sequences of the guide match and PAM are shown in (f), with mismatches highlighted in red. The four tracks are: input DNA, dCas9 transfected with no sgRNA, dCas9 transfected with EMX1-sg1, and dCas9 transfected with EMX1-sg3.
Supplementary Figure 6 | Gel shift assay for NAG substrates. The assay were done under the same condition as Fig. 2c. Sequences are shown at the bottom, with AG in pink and guide-matched bases in blue. Gels for NGG substrates were taken from Fig. 2c for comparison.
Supplementary Figure 7 | Scatter and histogram plots of guide match and relative binding for sgRNA Phc1-sg1, Phc1-sg2, and Nanog-sg2. The legends were the same as Fig 3a.
Supplementary Figure 8 | CpG methylation is negatively correlated with DHS and ChIP signals. (a) Pearson correlation coefficients between DHS, CpG methylation, and binding. (b) Partial Pearson correlation coefficients between DHS, CpG methylation, and binding. (c) The fraction of variation in binding explained by DHS, CpG methylation, DHS and CpG methylation without interaction, or DHS and CpG methylation with interaction.
Supplementary Figure 9 | Cas9/dCas9 expression. (a) Western blot using lysates from cells with either transiently transfected Cas9 (lanes 1-3), or dCas9 (lanes 4-6), or cells stably integrated with dCas9 (lanes 7-9). All Cas9/dCas9 proteins contain an HA tag. Tubulin was used as loading control. Cells for lanes 2 and 5 were also transfected with Nanog-sg2, and lanes 3 and 6 were from cells transfected with Nanog-sg3. Lanes 7-9 were the same lysate with 1:1, 1:2, and 1:4 dilution. After normalizing to the loading control, the HA band in lane 1 is about 2.6 times of the HA band in lane 8, suggesting that the expression of dCas9 in our stable cells is much lower than the cells with transiently transfected dCas9. (b) Comparison of Cas9 plasmid used in various studies, including the references, type of plates used, and the amount of Cas9/dCas9 plasmids transfected per well, and the equivalent amount on a 10 cm plate based on the area on each plate.
Acknowledgments
We would like to thank Jesse Zamudio and Timothy Kelly for optimizing the ChIP protocol, and the entire Sharp lab for support and discussion. We also thank the Core Facility in the Swanson Biotechnology Center at the David H. Koch Institute for Integrative Cancer Research at MIT for their assistance with high-throughput sequencing. This work was supported by United States Public Health Service grants RO1-GM34277, R01-CA133404 from the National Institutes of Health, and PO1- CA42063 from the National Cancer Institute to P.A.S., and partially by Cancer Center Support (core) grant P30-CA14051 from the National Cancer Institute. F.Z. is supported by an NIH Director's Pioneer Award (1DP1-MH100706), the Keck, McKnight, Poitras, Merkin, Vallee, Damon Runyon, Searle Scholars, Klingenstein, and Simons Foundations, Bob Metcalfe, and Jane Pauley. X.W. is a Howard Hughes Medical Institute International Student Research Fellow. S.C. is a Damon Runyon Fellow (DRG-2117-12). P.D.H. is a James Mills Pierce Fellow. D.A.S. is an NSF pre- doctoral fellow.
Footnotes
Author contributions: X.W., F.Z., and P.A.S. designed experiments; X.W., and A.J.K. performed most experiments; D.A.S. performed _targeted indel sequencing; A.W.C. and D.B.D. cloned the piggyBac dCas9 and sgRNA expressing vectors; A.C.C. generated the dCas9 stable cell line; P.D.H., A.E.T., and S.K. purified Cas9; P.D.H. contributed to in vitro binding assay; S.C. contributed to ChIP experiments with transient transfection. X.W. and PA.S. wrote the manuscript with help from all other authors. R.J., F.Z., and P.A.S. supervised the research.
Competing financial interests: The authors declare no competing financial interests.
Accession code. GSE54745 SRP038774
References
- 1.Van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–7. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–93. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 3.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–70. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 4.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321–7. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11:181–90. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD, et al. DNA _targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the _targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–40. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 11.Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–63. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasiunas G, Siksnys V. RNA-dependent DNA endonuclease Cas9 of the CRISPR system: Holy Grail of genome editing? Trends Microbiol. 2013;21:562–7. doi: 10.1016/j.tim.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Jinek M, et al. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–4. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Y, et al. Correction of a Genetic Disease in Mouse via Use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–62. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Schwank G, et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell. 2013;13:653–8. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–34. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence- specific control of gene expression. Cell. 2013;152:1173–83. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–51. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng AW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–71. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–6. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mali P, et al. CAS9 transcriptional activators for _target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–8. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10:977–9. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen B, et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pattanayak V, et al. High-throughput profiling of off-_target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–43. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carroll D. Staying on _target with CRISPR-Cas. Nat Biotechnol. 2013;31:807–9. doi: 10.1038/nbt.2684. [DOI] [PubMed] [Google Scholar]
- 29.Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems _targeting βEglobin and CCR5 genes have substantial off-_target activity. Nucleic Acids Res. 2013;41:9584–92. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiu H, Schwartz HT, Antoshechkin I, Sternberg PW. Transgene-Free Genome Editing in Caenorhabditis elegans Using CRISPR-Cas. Genetics. 2013;195:1167–71. doi: 10.1534/genetics.113.155879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho SW, et al. Analysis of off-_target effects of CRISPR/Cas-derived RNA- guided endonucleases and nickases. Genome Res. 2014;24:132–41. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y, et al. High-frequency off-_target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–7. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci U S A. 2013;110:18602–7. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–7. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Packer MJ, Dauncey MP, Hunter CA. Sequence-dependent DNA structure: tetranucleotide conformational maps. J Mol Biol. 2000;295:85–103. doi: 10.1006/jmbi.1999.3237. [DOI] [PubMed] [Google Scholar]
- 38.Stamatoyannopoulos JA, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:418. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 40.Orioli A, et al. Widespread occurrence of non-canonical transcription termination by human RNA polymerase III. Nucleic Acids Res. 2011;39:5499–512. doi: 10.1093/nar/gkr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nielsen S, Yuzenkova Y, Zenkin N. Mechanism of eukaryotic RNA polymerase III transcription termination. Science. 2013;340:1577–80. doi: 10.1126/science.1237934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartel DP. MicroRNAs: _target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–12. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 44.Jinek M, et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishimasu H, et al. Crystal Structure of Cas9 in Complex with Guide RNA and _target DNA. Cell. 2014;156:935–49. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Packer MJ, Dauncey MP, Hunter CA. Sequence-dependent DNA structure: tetranucleotide conformational maps. J Mol Biol. 2000;295:85–103. doi: 10.1006/jmbi.1999.3237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 | Conventional peak calling comparing IP to input. (a) The number of peaks called by MACS using default settings. (b) The fraction of dCas9-only peaks that are also detected in one, two, three, or all other four IP samples.
Supplementary Figure 2 | Characteristics of dCas9-only peaks. (a) Peaks are enriched for open chromatin regions. Shown is the average density of Dnase Hypersensitivity reads per 50bp bin in a 2kb window centered on peak summits. Blue area indicates standard error. (b) De novo motif finding within 50bp of peak summits by MEME-ChIP uncovered two related GG/CC-rich motifs. (c) Relative position of the motif within the peak, 0 indicates peak summits. (d) The longer motif (bottom) closely resembles CTCF binding motif (top, p < 1e-23)
Supplementary Figure 3 | De novo motif discovery in ChIP peaks. Motifs detected by MEME-ChIP using default settings and sequences within 50 bps of peak summits. The guide RNA sequences were shown below the motif.
Supplementary Figure 4 | Lack of ChIP enrichment at seed+NAG sites. (a) of the 996 (33%) Phc1-sg1 ChIP peaks without seed+NGG sites, only 18 (1.8%) contain seed+NAG (AGCACNGG) within 50bp of peak summits, which is not higher than random (2.7%). Motif logo of the 18 NAG peaks showing the lack of base pairing outside the seed region. (b) The number of seed+NAG sites in the genome (column 2) and within ChIP peaks (column 3) that contain specific number of mismatches in the guide region (column 1). None of the seed+NAG sites with less than 6 mismatches showed ChIP signals strong enough to be defined as peaks. (c) The best match contains only 1 mismatch and showed no ChIP signals. (d) Only one site with 6 mismatches is within a peak, yet the seed+NAG site is not in the center of the peak. (e-f) Similar to (c-d) but showing the two strongest peaks that are associated with seed+NAG sites. For (c-f), the top track is ChIP signal, and the bottom track is open chromatin. The scale is the same as Fig. 2b.
Supplementary Figure 5 | ChIP signals in HEK293FT cells. ChIP read density at four off-_targets (a-d) and on-_targets (e). Sequences of the guide match and PAM are shown in (f), with mismatches highlighted in red. The four tracks are: input DNA, dCas9 transfected with no sgRNA, dCas9 transfected with EMX1-sg1, and dCas9 transfected with EMX1-sg3.
Supplementary Figure 6 | Gel shift assay for NAG substrates. The assay were done under the same condition as Fig. 2c. Sequences are shown at the bottom, with AG in pink and guide-matched bases in blue. Gels for NGG substrates were taken from Fig. 2c for comparison.
Supplementary Figure 7 | Scatter and histogram plots of guide match and relative binding for sgRNA Phc1-sg1, Phc1-sg2, and Nanog-sg2. The legends were the same as Fig 3a.
Supplementary Figure 8 | CpG methylation is negatively correlated with DHS and ChIP signals. (a) Pearson correlation coefficients between DHS, CpG methylation, and binding. (b) Partial Pearson correlation coefficients between DHS, CpG methylation, and binding. (c) The fraction of variation in binding explained by DHS, CpG methylation, DHS and CpG methylation without interaction, or DHS and CpG methylation with interaction.
Supplementary Figure 9 | Cas9/dCas9 expression. (a) Western blot using lysates from cells with either transiently transfected Cas9 (lanes 1-3), or dCas9 (lanes 4-6), or cells stably integrated with dCas9 (lanes 7-9). All Cas9/dCas9 proteins contain an HA tag. Tubulin was used as loading control. Cells for lanes 2 and 5 were also transfected with Nanog-sg2, and lanes 3 and 6 were from cells transfected with Nanog-sg3. Lanes 7-9 were the same lysate with 1:1, 1:2, and 1:4 dilution. After normalizing to the loading control, the HA band in lane 1 is about 2.6 times of the HA band in lane 8, suggesting that the expression of dCas9 in our stable cells is much lower than the cells with transiently transfected dCas9. (b) Comparison of Cas9 plasmid used in various studies, including the references, type of plates used, and the amount of Cas9/dCas9 plasmids transfected per well, and the equivalent amount on a 10 cm plate based on the area on each plate.