

Abstract
In most colorectal cancer (CRC) patients, outcome cannot be predicted because tumors with similar clinicopathological features can have differences in disease progression and treatment response. Therefore, a better understanding of the CRC biology is required to identify those patients who will benefit from chemotherapy and to find a more tailored therapy plan for other patients. Based on unsupervised classification of whole genome data from 188 stages I–IV CRC patients, a molecular classification was developed that consist of at least three major intrinsic subtypes (A-, B- and C-type). The subtypes were validated in 543 stages II and III patients and were associated with prognosis and benefit from chemotherapy. The heterogeneity of the intrinsic subtypes is largely based on three biological hallmarks of the tumor: epithelial-to-mesenchymal transition, deficiency in mismatch repair genes that result in high mutation frequency associated with microsatellite instability and cellular proliferation. A-type tumors, observed in 22% of the patients, have the best prognosis, have frequent BRAF mutations and a deficient DNA mismatch repair system. C-type patients (16%) have the worst outcome, a mesenchymal gene expression phenotype and show no benefit from adjuvant chemotherapy treatment. Both A-type and B-type tumors have a more proliferative and epithelial phenotype and B-types benefit from adjuvant chemotherapy. B-type tumors (62%) show a low overall mutation frequency consistent with the absence of DNA mismatch repair deficiency. Classification based on molecular subtypes made it possible to expand and improve CRC classification beyond standard molecular and immunohistochemical assessment and might help in the future to guide treatment in CRC patients.
Keywords: colorectal cancer, molecular subtypes, EMT, mismatch repair, chemotherapy benefit
The development of chemotherapy regimens that include fluoropyrimidines, irinotecan and oxaliplatin has improved the overall survival (OS) of patients with colorectal cancer (CRC),1 but many patients experience relapses after adjuvant treatment. In most patients, outcome cannot be predicted upfront because tumors with similar clinicopathological features can have significant differences in disease progression and treatment response.
The traditional model for development of CRC involves a progressive stepwise accumulation of genetic alterations.2,3 However, detailed molecular analyses have revealed that CRC is heterogeneous with regard to genetic and molecular alterations.
Molecular characteristics of the tumor play an important role in their response to treatment, and patients with microsatellite instable (MSI) cancers seem to have different prognosis and benefit from chemotherapy compared to microsatellite stable (MSS) patients.4–8 MSI has been attributed predominantly to silencing of DNA mismatch repair (MMR) genes9 and the terms MSI and dMMR (deficient MMR) have often been used interchangeably. Although a role of MMR proteins in influencing sensitivity to 5-fluorouracil (5FU) has been demonstrated,8 MSI accounts for only 7–20% of all patients and cannot alone explain why some patients have significant benefit from chemotherapy while others do not. Further understanding and consideration of the molecular heterogeneity of CRC is essential to develop better classification methods that are associated with disease progression and response to therapies.
What’s new? —
Even when tumors look the same, they may behave differently. But patients can be treated more effectively if clinicians know how aggressively a cancer will progress or how well it will respond to treatment. That’s why this study investigated molecular and genetic differences in colorectal cancer to find out how to distinguish different subtypes. They classified tumors into three categories based on three biological hallmarks: epithelial to mesenchymal transition, high mutation frequency, and proliferation. Each of the tumor types has a different prognosis and response to chemotherapy, and a patient’s tumor type can help determine what treatment should be attempted.
Unbiased molecular classification of breast cancer into clinically relevant subtypes10 has stimulated the development of treatment plans that are tailored to key oncogenic hallmarks in the tumor.11 We only begin to appreciate similar hallmarks in CRC. Recently, several groups have reported molecular classifications beyond MSI, either by identifying patients with distinct CpG island methylation phenotypes (CIMP),12 distinct mutation profiles13 or distinct expression patterns.14,15 However, it is not yet clear how they relate to one another.
In a previous study, we observed three intrinsic subtypes based on the tumor’s full genome expression patterns.16 In this study, we aim to characterize these intrinsic subtypes to understand how their biology differs and how this influences the patient’s outcome. The CRC intrinsic subtypes showed distinct features regarding the presence of oncogenic mutations, epithelial-to-mesenchymal transition (EMT), MMR phenotypes and cellular proliferation, and most importantly, showed a marked difference in prognosis and benefit from chemotherapy.
Methods
Patients
The CRC molecular subtypes were identified on a previously analyzed cohort of 188 CRC patients16 and were validated in 543 stages II and III CRC patients (Table 1, Supporting Information Table S1). Seventy-three percentage of the stage III patients in the validation cohort, for which treatment and outcome data was available, had been treated with 5FU-based chemotherapy, none with oxaliplatin. All tissue samples were collected from patients with appropriate informed consent. The study was performed in accordance with the ethical standards of the Helsinki Declaration and was approved by the Medical Ethical Board of the participating hospitals.
Table 1.
Molecular subtype characteristics
| Development cohort | Validation cohort | |||||||
|---|---|---|---|---|---|---|---|---|
| A-type | B-type | C-type | A-type | B-type | C-type | All | Total | |
| n | 65 | 98 | 25 | 117 | 336 | 90 | P (X2) | 731 |
| Stage | ||||||||
| I | 14% (9) | 13% (13) | 8% (2) | 0.21 | 24 | |||
| II | 58% (38) | 51% (50) | 48% (12) | 61% (71) | 61% (204) | 50% (45) | 420 | |
| III | 26% (17) | 30% (29) | 40% (10) | 39% (46) | 39% (132) | 50% (45) | 279 | |
| IV | 2% (1) | 6% (6) | 4% (1) | 8 | ||||
| Age | ||||||||
| <70 | 62% (40) | 59% (58) | 60% (15) | 54% (63) | 56% (188) | 67% (60) | 0.23 | 424 |
| ≥70 | 38% (25) | 41% (40) | 40% (10) | 46% (54) | 44% (148) | 33% (30) | 307 | |
| Gender | ||||||||
| male | 40% (26) | 47% (46) | 48% (12) | 48% (56) | 64% (216) | 50% (45) | 1.07E-03 | 401 |
| female | 60% (39) | 53% (52) | 52% (13) | 52% (61) | 36% (120) | 50% (45) | 330 | |
| Location | ||||||||
| left colon | 31% (20) | 61% (59) | 52% (13) | 31% (36) | 64% (210) | 56% (50) | 5.42E-16 | 388 |
| right colon | 63% (40) | 29% (28) | 36% (9) | 67% (77) | 27% (88) | 41% (37) | 279 | |
| rectum | 6% (4) | 10% (10) | 12% (3) | 2% (2) | 9% (29) | 3% (3) | 51 | |
| not available | (1) | (1) | (0) | (2) | (9) | (0) | 13 | |
| Grade | ||||||||
| low | 5% (3) | 7% (7) | 4% (1) | 16% (19) | 22% (75) | 7% (6) | 5.23E-08 | 111 |
| intermediate | 72% (47) | 80% (78) | 64% (16) | 44% (51) | 62% (208) | 66% (59) | 459 | |
| high | 22% (14) | 9% (9) | 28% (7) | 40% (47) | 16% (53) | 28% (25) | 155 | |
| not available | (1) | (4) | (1) | (0) | (0) | (0) | 6 | |
| BRAF | ||||||||
| activating mutation | 47% (30) | 0% (0) | 21% (5) | 21% (18) | 2% (5) | 13% (6) | <2.2E-16 | 64 |
| wildtype / other | 53% (34) | 100% (91) | 79% (19) | 79% (69) | 98% (223) | 87% (40) | 476 | |
| not available | (1) | (7) | (1) | (30) | (108) | (44) | 191 | |
| KRAS | ||||||||
| activating mutation | 25% (16) | 26% (24) | 35% (8) | 46% (40) | 28% (63) | 31% (14) | 0.08 | 165 |
| wildtype / other | 75% (47) | 74% (67) | 65% (15) | 54% (47) | 72% (165) | 69% (31) | 372 | |
| not available | (2) | (7) | (2) | (30) | (108) | (45) | 194 | |
| PIK3CA | ||||||||
| activating mutation | 16% (10) | 7% (6) | 19% (4) | 31% (17) | 10% (18) | 13% (3) | 9.23E-04 | 58 |
| wildtype / other | 84% (54) | 93% (85) | 81% (17) | 69% (37) | 90% (160) | 87% (20) | 373 | |
| not available | (1) | (7) | (4) | (63) | (158) | (67) | 300 | |
| Microsatellite | ||||||||
| stable (MSS) | 63% (24) | 100% (42) | 90% (9) | 44% (29) | 98% (176) | 90% (38) | <2.2E-16 | 318 |
| instable (MSI) | 37% (14) | 0% (0) | 10% (1) | 56% (37) | 2% (3) | 12% (5) | 60 | |
| not available | (27) | (56) | (15) | (51) | (157) | (48) | 354 | |
Note: Percentages might not add up due to rounding
Molecular subtype classification
Unsupervised clustering of whole genome data revealed three mayor groups with distinct gene expression pattern.16 In this study, three distinct gene expression signatures representative for the molecular subtypes were used for sample classification (Table 2). Signature development and sample classification is described in the Supporting Information Methods. Subtype-related gene expression measurements were performed on custom made Agilent microarrays.16,17 Microarray data are available in the Gene Expression Omnibus (GSE42284).
Table 2.
Gene signatures for classification of intrinsic CRC subtypes
| A-type | B-type | C-type | ||||
|---|---|---|---|---|---|---|
| HSPA4L | BG114486 | VAPB | THBS2 | GPSM1 | LOC338328 | ASPM |
| SLC7A11 | THC2669157 | HNRNPA1L2 | SPOCK1 | VWF | ANKRD35 | ORC6L |
| NUDT6 | QPRT | KIF3B | COL5A2 | WISP1 | KIAA1442 | ZNF367 |
| ME1 | PLA2G12B | ARFGEF2 | FBLN1 | SLIT3 | THY1 | NIPSNAP1 |
| DLG7 | VAV3 | PIWIL2 | MGP | MC1R | FES | SPBC25 |
| KNTC2 | PTPRO | FANCF | MXRA8 | LAMB2 | PGF | DIAPH3 |
| PRC1 | RNF43 | THC2644861 | DCN | PCOLCE | MAP3K3 | |
| ECHS1 | DDC | MOCS3 | AEBP1 | GPX7 | GPSM3 | |
| DEPDC1 | AXIN2 | PIGU | BASP1 | COX7A1 | NPC2 | |
| ACADSB | C13orf18 | CEP250 | COL6A1 | FGFR1 | C14orf139 | |
| EIF4A2 | TSPAN6 | IFT52 | COL1A2 | AK021531 | THC2532155 | |
| MREG | GGH | CXorf56 | HTRA1 | CALD1 | C1orf198 | |
| NIPA1 | PLAGL2 | COBLL1 | LOXL1 | JAK3 | FLT4 | |
| TIAL1 | ACSL6 | EPOR | COL5A1 | TRO | SNRP70 | |
| URM1 | RBP2 | MAPRE2 | FSTL1 | TGFB3 | KIAA1602 | |
| ZNF167 | SLC6A4 | SLC41A1 | RARRES2 | C1QTNF6 | ELMO1 | |
| RARA | CTSL2 | KCTD1 | MSN | DTX3 | RNF207 | |
| SNX21 | AMACR | TRIB2 | SPARC | NID2 | POLE | |
| NRXN2 | POFUT1 | PLK2 | PDGFRB | COL18A1 | CPSF6 | |
| ARFGAP1 | CEBPA | RAMP1 | TUBB6 | SLC27A1 | BCL2L14 | |
| PAPLN | PARD6B | LOC388610 | SERPINF1 | JAM2 | TOM1L1 | |
| SMARCC2 | PRDX5 | TPM2 | EFHA2 | SNRPC | ||
| AS3MT | SEPHS2 | CD248 | GGTLA1 | SYNCRIP | ||
| DKFZp547K054 | C20orf142 | LGALS1 | LAMC1 | NDUFAB1 | ||
| RGN | GPSM2 | CRYAB | ROBO4 | RABL3 | ||
| CTSF | SLC5A6 | CXCL12 | IGFBP5 | XRCC2 | ||
| SORBS1 | TP53RK | CLDN5 | FAM20C | NDUFA10 | ||
| FCGRT | NCOA6 | LOC387763 | TSPYL5 | PA2G4 | ||
| LARP6 | C20orf111 | BNC2 | VAMP5 | RFC4 | ||
| FHOD3 | C20orf43 | OBSL1 | FBXO17 | ZNF695 | ||
| NINL | HNF4A | EVL | CLEC11A | PPARA | ||
| SRPX2 | PSMA7 | COL6A3 | PDLIM4 | FBXO5 | ||
Gene signatures specific for each of the three CRC intrinsic subtypes. Genes of each of the subtype profile (A-type 32 genes, B-type 53 genes and C-type 102 genes) are ranked (top to bottom and left to right) according to their relative up-regulation (green) or down-regulation (blue) compared to the other two subtypes.
Mutation analysis
Mutations in BRAF(V600E), KRAS codons 12, 13 and 61 and PIK3CA exons 9 and 20 were assessed in cDNA by means of Sanger sequencing (Supporting Information Methods). In addition, 615 genes including all genes encoding the kinases (kinome) were analyzed by deep-sequencing on 73 patients (selected to include all subtypes, Supporting Information Table S1). Sample preparation, sequencing procedure (HiSeq2000, Illumina) and sequence data analysis variant calling (somatic point mutations and small insertions and deletions) are described in Supporting Information Methods.
MSI and dMMR phenotype assessment
Assessment of MSI-status was performed according to the local standard at each participating hospital and is described in Supporting Information Methods. In addition, all samples were analyzed for their MSI/dMMR associated gene expression pattern by a previously reported 64-gene signature that accurately identifies CRC tumors that are MSI and/or show a dMMR phenotype18 (Supporting Information Methods).
Readout of EMT phenotype
Epithelial and mesenchymal characteristics were analyzed based on relative gene expression levels (median centered per gene) of marker genes known to be upregulated in mesenchymal and upregulated in epithelial cells. For each marker, a Student’s t-test was performed for comparison of A versus B+C, B versus A+C and C versus A+B (development cohort).
In addition, gene expression of the previously reported EMT signature from Loboda et al. was investigated in the development cohort.14 Ninety-six of their top 100 genes that were previously reported to be strongly associated with the EMT program were analyzed for differential expression across the three molecular subtypes, together with a multigene profile readout in which a higher EMT index represented a more mesenchymal phenotype (Supporting Information Methods).
Statistical and survival analysis
All analyses and statistical tests were performed in R 2.14.1 (www.r-project.org), and were considered significant with a p-value <0.05 (two-sided). Association of the subtypes with clinical and molecular makers was analyzed using a Pearson’s Chi-squared test. Survival analysis was performed on the validation cohort using Cox proportional hazard models with two end-points: 10-year distant metastasis-free survival (DMFS) and 10-year cancer-related OS. Average follow-up time was 70 months (range 3–270 months). Investigation of benefit from 5FU-based adjuvant chemotherapy was performed by comparing OS rates of patients with and without chemotherapy. An interaction analysis was performed to analyze the differences in benefit between the subtypes based on 5-year OS. Most (76%) stage II patients were untreated, therefore this analysis was limited to the stage III patients of the validation cohort.
Results
Three intrinsic molecular subtypes
Unsupervised clustering of whole genome expression data revealed three intrinsic CRC molecular subtypes16 (Fig. 1a). To further characterize and understand the biological and clinical differences between these intrinsic subtypes, we have developed a diagnostic single sample classifier based on three “core” gene profiles representative for each of the subtypes (A-, B- and C-type) (Fig. 1b, Table 2). On the development cohort, 35% was classified as A-type, 52% as B-type and 13% as C-type CRC (Fig. 1c, Table 1), with a very high concordance of 97% compared to the unsupervised clustering method.16
Figure 1.
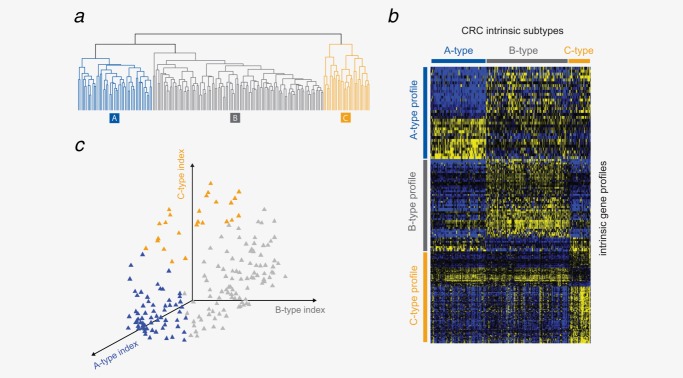
Single sample predictor for molecular subtype classification. (a) Unsupervised hierarchical clustering of the development cohort shows three distinct CRC intrinsic subtypes. (b) Gene signatures specific for A-type (32 genes), B-type (53 genes) and C-type (102 genes) CRC. Yellow indicates relative up-regulation and blue down-regulation of the genes across the 188 development samples. (c) A single sample classifier for identification of the three CRC subtypes.
Subtype classification and characteristics were confirmed in a validation cohort of 543 stages II and III CRC samples (Table 1, Supporting Information Table S1). Twenty-two percent of patients were classified as A-type, 62% as B-type and 17% as C-type. Subtype classification was not associated with tumor stage (p = 0.21) or patient age (p = 0.23), but was significantly associated with tumor grade (p = 5.23e-8), gender (p = 1.07e-3) and microsatellite status (p < 2.2e-16) (both cohorts combined, Table 1). A significant difference in colon tumor location was observed (left vs. right colon, p = 1.04e-15; colon vs. rectum, p = 0.027).
Association with molecular markers
Molecular characterization of the intrinsic subtype indicated that activating BRAF(V600E) mutations were unequally distributed between the subtypes (p = 1.9e-7) (Table 1). Not only A-type (32%, both cohorts) but also C-type patients (16%) were enriched for BRAF mutations, while B-type patients were almost exclusively BRAF wild-type (98%). Subtype classification was also associated with activating PIK3CA mutations (p = 9.23e-4) but not associated with activating KRAS mutations (p = 0.08) (Table 1). Combining the mutually exclusive KRAS and BRAF mutations, a highly significant association was observed with 68% A-type patients harbored an activating BRAF or KRAS mutation (Supporting Information Table S2).
The A-type group showed a significant higher proportion of MSI patients (49%, p < 2.2e-16, both cohorts) while B-type patients were almost exclusively MSS (99%) (Table 1). The association of A-type with MSI-status is in good agreement with other characteristics that have been observed before to be associated with MSI (BRAF mutations, right-sided tumor location, female gender and poor differentiation).19 The lack of statistically significant association of A-type with stage might be explained by the absence of stages I and IV cancers in the validation cohort. However, the development cohort indicates that A-type patients might be more frequent in early stage cancer, in good agreement with better prognosis of A-type and MSI-patients.
Readout of the MSI/dMMR signature that is able to identify MSI and MSI-like tumors representative of a dMMR phenotype18,19 reinforced the strong MSI/dMMR characteristics of A-type patients with 68% showing a MSI/dMMR expression profile and confirmed the exclusive MSS and proficient MMR (pMMR) phenotype (99%) of B-type patients (Fig. 2a). Although not enriched for MSI by the hospital assessment, a significant proportion of C-type patients (36%, p = 5.7e-4) showed a dMMR phenotype based on the MSI/dMMR-signature (Fig. 2a).
Figure 2.
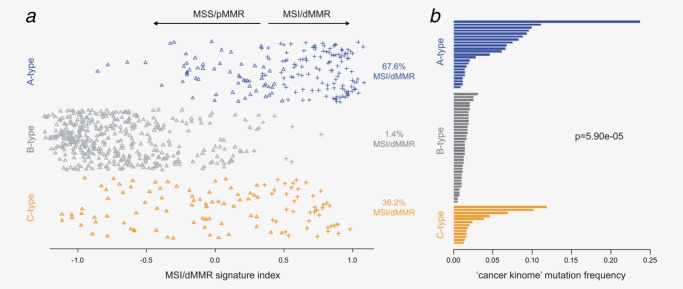
CRC subtypes are associated with MSI and dMMR phenotypes. (a) Readout of the MSI/dMMR signature18 for the three CRC molecular subtypes. The symbols represent the binary MSI/dMMR (crosses) or MSS/pMMR (triangles) calls based on the signature indexes that are plotted on the x-axis. (b) Mutation frequency in the cancer kinome (615 genes) across the three subtypes: 23 A-type, 37 B-type and 13 C-type samples.
To validate the dMMR phenotype, 73 samples were deep-sequenced for their complete kinome. A- and C-type patients showed a significantly higher mutation frequency compared to B-type (p = 5.90e-5). On average, B-type patients showed a low mutation frequency of ten mutated genes of the 615 analyzed (1.6%), while the A-type and C-type groups harbored 38 (6.2%) and 26 (4.2%) mutated genes, respectively (Fig. 2b).
These results are in agreement with a recent publication from the Cancer Genome Atlas Network (TCGA) that shows that some “hypermutated” cancers have high levels of microsatellite instability (MSI) while others are not and are classified as MSS/MSI-L by traditional MSI-methods.13 This might explain why the C-type group has a high level of dMMR characteristics and high mutation rate but many patients are not classified as MSI by hospital methods.
Epithelial-to-mesenchymal characteristics
Gene expression levels of known epithelial and mesenchymal markers were assessed in the subtypes (Fig. 3a). In C-type cancers, almost all the mesenchymal markers, except FLT1, were significantly up-regulated, and three epithelial markers (CDH1, EGFR and MET) were down-regulated. In B-type, four of the five epithelial markers were up-regulated and three mesenchymal markers (CDH2, FGFR1 and TGFB1) were down-regulated. A-type patients showed a significant reduced expression of two mesenchymal markers (TWIST1 and AXL) but also a reduction in one of the epithelial markers (CDH1). These results indicated that C-type tumors have a more mesenchymal phenotype while A- and B-type tumors can be considered as epithelial.
Figure 3.

Molecular subtypes association with EMT characteristics. (a) Relative gene expression levels of eight mesenchymal and five epithelial marker in the three molecular subtypes. Significant levels are indicated for differential expression between the subtypes (pairwise Student’s t-test). (b) Read-out of the EMT signature by Loboda et al.14 across the molecular subtypes. Positive signature indexes are representative for a mesenchymal phenotype. (c) Receiver operating curve of C-type classification using the Loboda signature indexes. (d) Molecular subtype association of the epithelial and mesenchymal genes as presented by Loboda et al. Genes are shown according to the original clustering in Ref.14. Genes without a subtype indication showed no statistical significant differential expression.
The difference in epithelial and mesenchymal characteristics was confirmed by the recently published EMT signature by Loboda et al.14 C-type patients showed a significantly higher (p < 2.2e-16) EMT index representative of a more mesenchymal phenotype (Fig. 3b). Classification of C-type patients by the Loboda signature showed an AUC of 0.92 (Fig. 3c) with mesenchymal genes significant up-regulated in C-type samples and epithelial genes significant up-regulated in A-type or B-type samples (Fig. 3d).
Prognosis
Survival analysis on the validation cohort (n = 543, stages II and III) showed a significant difference in DMFS between the three subtypes (p = 0.004), with hazard ratios (HR) of 2.19 (p = 0.011) for B-type and 2.90 (p = 0.0024) for C-type patients compared to A-type (Fig. 4a). C-type patients showed a significant reduced OS compared to A and B-types with a HR of 2.3 (p = 0.016) (Fig. 4b).
Figure 4.
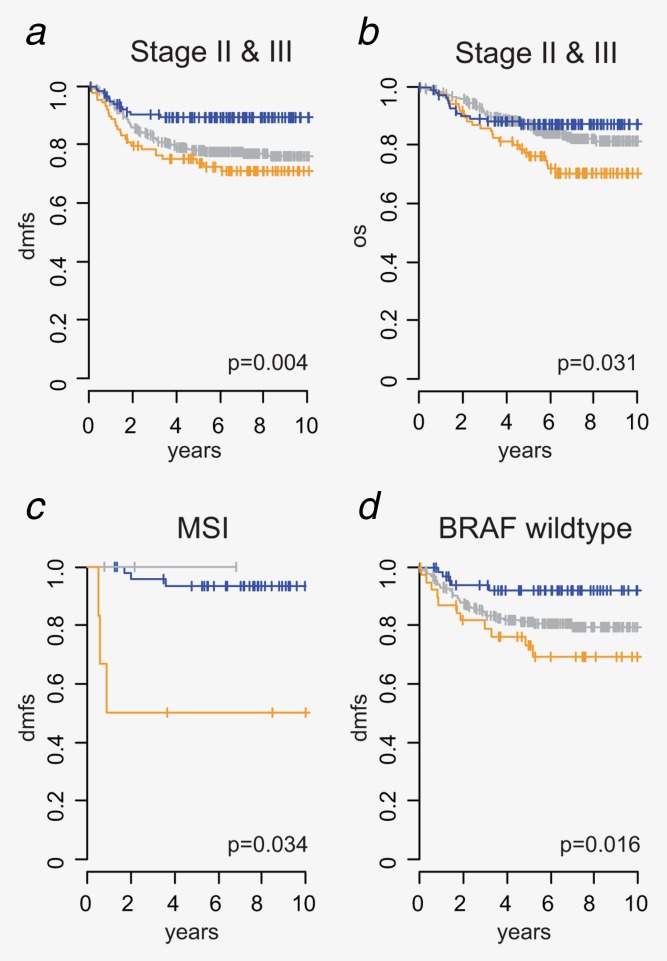
Prognostic value of molecular subtypes in stages II and III. Kaplan–Meier survival analysis of the three molecular subtypes in the validation cohort for (a) DMFS of all stages II and III validation samples, (b) cancer-related OS of all stages II and III, (c) in MSI patients (MSI status is based on the hospital MSI testing or, if not available, on the previously reported MSI gene signature,18 see Methods section for details) and (d) BRAF wildtype patients. Survival curves of A-, B- and C-type samples are indicated in blue, gray and orange, respectively.
Although MSI status is generally associated with a good prognosis, we observed significant differences in DMFS between the subtypes (p = 0.034) with A-type-MSI patients having a very good prognosis but with C-type-MSI-patients having a poor prognosis (93 and 50% 10-year DMFS, respectively) (Fig. 4c). A multivariate analysis, including MSI, BRAF, stage, gender and NCCN classification,20 confirmed that the subtype classification has independent prognostic value (Supporting Information Table S3) that is additive to MSI and BRAF assessment (Fig. 4d).
Adjuvant chemotherapy benefit
To analyze chemotherapy benefit, outcome of 222 stage III patients were compared. Of these patients, 161 received adjuvant 5FU chemotherapy and 61 received no adjuvant chemotherapy. In accordance with data from clinical trials,21 treatment with 5FU-based chemotherapy was beneficial when compared with no additional treatment (HR 0.55, p = 0.033). The benefit from chemotherapy was different in the three molecular subtypes (Figs. 5a–5d). Stage III patients with A- and B-type tumors showed a benefit in OS with a HRs of 0.39 (p = 0.18) and 0.42 (p = 0.014), respectively; the difference not being statistically significant in A-type due to lower number of patients in this type. In contrast to the epithelial-like A- and B-type patients, the mesenchymal-like C-type patients showed no benefit from chemotherapy treatment (HR 1.4, p = 0.542). An interaction analysis between subtypes and chemotherapy with OS as endpoint confirmed that chemotherapy response was significantly different between the molecular subtypes (p = 0.017, Fig. 5d). Since MSI has been associated with nonresponsiveness to 5FU, the analysis for C-type patients was repeated for MSS patients only. C-type-MSS tumors again showed no benefit from chemotherapy (HR 1.08, p = 0.95) (data not shown).
Figure 5.

Molecular subtypes differ for their response to chemotherapy. Kaplan–Meier survival analysis (OS) between patients (validation cohort) treated with and without chemotherapy for (a) A-type, (b) B-type and (c) C-type CRC. (d) Adjuvant chemotherapy benefit for the subtypes as measured by the difference in 5-year OS. (e) Gene expression boxplots of two proliferation markers KI67 and AURKA across the three subtypes. p-Values indicate the significance of differential expression between C-type and A–B-types.
We hypothesized that the difference in chemotherapy benefit might be related to the proliferative activity of the molecular subtypes. Relative gene expression levels of two proliferative markers, Ki-67 (MKI67) and Aurora Kinase A (AURKA), showed a significantly reduced expression of both markers in C-type compared to A- and B-type tumors (MKI67 p = 6.06e-5, AURKA p = 4.53e-6, Student’s t-test) (Fig. 5e). Interestingly, A-type tumors showed the highest expression of MKI67, while AURKA was the strongest proliferative marker for B-type samples.
Discussion
In this study, we have identified, described and validated three CRC intrinsic subtypes for their biological and clinical characteristics in a large set of tumors. As summarized in Figure 6, subtype classification is largely driven by three hallmarks: EMT, higher mutation frequency resulting from dMMR and cellular proliferation.
Figure 6.
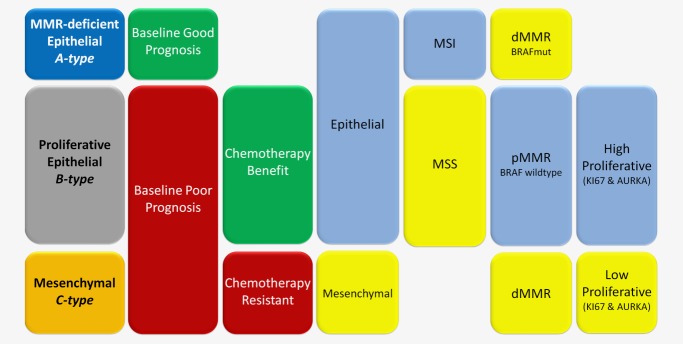
Classification model of CRC by A-, B- and C- subtypes. Classification model that discriminate three distinct subtypes: MMR-deficient epithelial (A-type), proliferative epithelial (B-type) and mesenchymal (C-type). A simplified model is shown for the main clinical and molecular characteristics of each of the three subtypes: baseline prognosis, 5FU-based chemotherapy response, epithelial or mesenchymal-like phenotypes, microsatellite status (MSI or MSS), MMR phenotype (deficient or proficient) and the associating BRAF mutation status and the tumor’s proliferation rate as measured by MKI67 and AURKA expression levels. This representation is a simplified model focused on the core-characteristics of each subtype.
A-type cancers consist of approximately 20–30% of all CRC and were found to be epithelial-like and to display a strong MSI phenotype linked to dMMR,19 resulting in an overall high mutation rate including activating BRAF mutations. A-type patients have a good prognosis with some indication of benefit from 5FU-based adjuvant chemotherapy. Based on these features, A-types can be referred to as MMR-deficient epithelial subtype.
The most prevalent subtype, B-type or the proliferative epithelial subtype, includes 50–60% of tumors that show a strong epithelial phenotype. B-type tumors are almost exclusively MSS, BRAF wild-type and pMMR. Patients with this subtype show a relative poor baseline prognosis but, importantly, they benefit most significantly from adjuvant chemotherapy. This treatment benefit is likely caused by the high proliferative characteristics found in the subtype.
The third class, C-type, is the smallest but most distinct molecular subtype. C-type tumors have undergone an EMT and show dMMR characteristics. Patients with C-type tumors have a poor baseline prognosis and show no benefit from adjuvant 5FU treatment. The poor prognosis and unresponsiveness to chemotherapy is likely linked to their mesenchymal phenotype together with low proliferative activity. It has also been demonstrated by others that the mesenchymal phenotype is linked to low proliferative activity22 and poor response to chemotherapy.23 C-types can thus be referred to as mesenchymal subtype.
One of the hallmarks of the CRC intrinsic subtypes is the MSI or dMMR status. This characteristic is linked to the CIMP phenotype, right-sided location and to hypermutation.13 Clinical studies have demonstrated that MSI rates vary with tumor stage, and in the adjuvant setting, MSI patients have been associated with longer survival than patients with MSS tumors.7 However, the benefit of 5FU in this subgroup is debatable and different clinical studies give conflicting results.5,24,25 The fact that MSI/dMMR patients are classified into two different molecular subtypes, one with a very good prognosis (A-type) and another with a very poor prognosis (C-type), might explain some of the conflicting results about chemotherapy benefit in MSI patients. It is of great interest to further investigate the relationship between MSI by traditional methods,26 dMMR phenotype and molecular subtyping.
The second hallmark, EMT plays a prominent role in development and progression of CRC,27 and might partially explain the difference in benefit from 5FU treatment between B-types (responsive) and C-types (resistant). This hypothesis is supported by an in silico validation of the ABC subtypes in a recently published study by Oh et al. In this study, two subtypes were identified in stages III and IV CRC that were prognostic and predictive for chemotherapy response.28 Their subgroup with main benefit from chemotherapy is strictly associated with our B-type classification, while their nonresponsive subgroup is classified as either A- or C-type (Supporting Information Fig. S2). Other studies have shown that induction of EMT may play a role in acquiring resistance to oxaliplatin,23 so it can be assumed that mesenchymal C-type patients are resistant to most if not all chemotherapies. Together, these results highlight the need for new approaches to treat this poor prognosis patient group.
This study was focused towards stages II and III CRC, therefore further validation of the subtype classification and its clinical relevance on a larger set of stage IV tumors is warranted. The three-way intrinsic colorectal classification demonstrated here is supported by the recently published results by TCGA.13 In this study, 276 CRCs have been characterized by genome-scale analysis, and unsupervised classification based on gene expression levels also identified three distinct subclasses representative of an MSI/CIMP, CIN and an invasive phenotype. These phenotypes match with the respective characteristics of the A-, B- and C-type but here we added prognostic and chemotherapy benefit characteristics to the classification. Further independent studies are necessary to validate these findings and to investigate if the subgroups might divide into further clinical relevant subgroups.
The C-type-specific gene signature contains potential _targets for development of new drugs and therefore might be useful in guiding new clinical studies for the treatment of this CRC subtype that is resistant to 5FU-based chemotherapy. For example, SPARC is upregulated in C-type CRC, and has been shown to be correlative with a response to nab-paclitaxel.29,30 Other interesting potential C-type _targets are JAK3,31 CLDN532 and FLT4 (VEGFR3) for which multiple inhibitors are in development (e.g., cediranib, sunitinib, pazopanib, telatinib and sorafenib), and FGFR1 for which cediranib and the dual inhibitor of FGF and VEGF brivanib have demonstrated antitumor activity.33,34 None of the agents have been used in patients with CRC with molecular selection and appropriate clinical trials are needed to find agents that can benefit C-type patients.
Classification based on these intrinsic subtypes will make it possible to expand and improve the intrinsic classification beyond standard molecular and immunohistochemical assessment and might help in the future to guide treatment in CRC patients.
Acknowledgments
The authors are especially grateful to the Asociación Española contra el Cáncer. PR, ST, MHS, RB and IS are employees of Agendia. CMC and GO are employees of AstraZeneca. PR, ST and IS are inventors on a patent related to the contents of this manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27:3677–83. doi: 10.1200/JCO.2008.20.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–61. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res. 2012;18:1506–12. doi: 10.1158/1078-0432.CCR-11-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel C, Jacob S, Grandjouan S, et al. Frequent alteration of DNA damage signalling and repair pathways in human colorectal cancers with microsatellite instability. Oncogene. 2007;26:5919–26. doi: 10.1038/sj.onc.1210419. [DOI] [PubMed] [Google Scholar]
- Roth AD, Tejpar S, Delorenzi M, et al. Stage-specific prognostic value of molecular markers in colon cancer: results of the translational study on the PETACC 3-EORTC 40993-SAKK 60–00 trial. J Clin Oncol. 2010;28:466–74. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- Warusavitarne J, Schnitzler M. The role of chemotherapy in microsatellite unstable (MSI-H) colorectal cancer. Int J Colorectal Dis. 2007;22:739–48. doi: 10.1007/s00384-006-0228-0. [DOI] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11. [PubMed] [Google Scholar]
- Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins MJ, Baselga J. _targeted therapies for breast cancer. J Clin Invest. 2011;121:3797–803. doi: 10.1172/JCI57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–6. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loboda A, Nebozhyn MV, Watters JW, et al. EMT is the dominant program in human colon cancer. BMC Med Genomics. 2011;4:9. doi: 10.1186/1755-8794-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Villamil B, Romera-Lopez A, Hernandez-Prieto S, et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behavior. BMC Cancer. 2012;12:260. doi: 10.1186/1471-2407-12-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar R, Roepman P, Capella G, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29:17–24. doi: 10.1200/JCO.2010.30.1077. [DOI] [PubMed] [Google Scholar]
- Maak M, Simon I, Nitsche U, et al. Independent validation of a prognostic genomic signature (ColoPrint) for stage II colon cancer patients. Ann Surg. 2013;257:1053–8. doi: 10.1097/SLA.0b013e31827c1180. [DOI] [PubMed] [Google Scholar]
- Tian S, Roepman P, Popovici V, et al. A robust genomic signature for detection of colorectal cancer patients with microsatellite instability phenotype and high mutation frequency. J Pathol. 2012;228:586–95. doi: 10.1002/path.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7:153–62. doi: 10.1038/nrclinonc.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network (NCCN) 2012. Clinical practice guidelines in oncology—colorectal cancer screening. Version 2. Available from: http://www.nccn.org/professionals/physician_gls/pdf/colorectal_screening.pdf.
- Sargent D, Sobrero A, Grothey A, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27:872–7. doi: 10.1200/JCO.2008.19.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AD, Fan F, Camp ER, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12(14 Pt 1):4147–53. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–70. doi: 10.1200/JCO.2010.30.1366. [DOI] [PubMed] [Google Scholar]
- González-García I, Moreno V, Navarro M, et al. Standardized approach for microsatellite instability detection in colorectal carcinomas. J Natl Cancer Inst. 2000;92:544–9. doi: 10.1093/jnci/92.7.544. [DOI] [PubMed] [Google Scholar]
- Bates RC, Mercurio AM. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther. 2005;4:365–70. doi: 10.4161/cbt.4.4.1655. [DOI] [PubMed] [Google Scholar]
- Oh SC, Park YY, Park ES, et al. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut. 2012;61:1291–8. doi: 10.1136/gutjnl-2011-300812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NP, Trieu V, Hwang LY, et al. Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anticancer Drugs. 2008;19:899–909. doi: 10.1097/CAD.0b013e32830f9046. [DOI] [PubMed] [Google Scholar]
- Desai N, Trieu V, Damascelli B, et al. SPARC expression correlates with tumor response to albumin-bound paclitaxel in head and neck cancer patients. Transl Oncol. 2009;2:59–64. doi: 10.1593/tlo.09109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthaler R, Gerspacher M, Holzer P, et al. Inventors; Novartis AG, Assignee. Pyrimidine derivatives. United States patent US 6885550. 2010 Jan 14.
- Kato-Nakano M, Suzuki M, Kawamoto S, et al. Characterization and evaluation of the antitumour activity of a dual-_targeting monoclonal antibody against claudin-3 and claudin-4. Anticancer Res. 2010;30:4555–62. [PubMed] [Google Scholar]
- Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.