

Abstract
Objective:
We investigated the association between circulating biomarkers of inflammation and MRI markers of small vessel disease.
Methods:
We performed a cross-sectional study relating a panel of 15 biomarkers, representing systemic inflammation (high-sensitivity C-reactive protein, interleukin-6, monocyte chemotactic protein-1, tumor necrosis factor α, tumor necrosis factor receptor 2, osteoprotegerin, and fibrinogen), vascular inflammation (intercellular adhesion molecule 1, CD40 ligand, P-selectin, lipoprotein-associated phospholipase A2 mass and activity, total homocysteine, and vascular endothelial growth factor), and oxidative stress (myeloperoxidase) to ischemic (white matter hyperintensities/silent cerebral infarcts) and hemorrhagic (cerebral microbleeds) markers of cerebral small vessel disease (CSVD) on MRI in 1,763 stroke-free Framingham offspring (mean age 60.2 ± 9.1 years, 53.7% women).
Results:
We observed higher levels of circulating tumor necrosis factor receptor 2 and myeloperoxidase in the presence of cerebral microbleed (odds ratio [OR] 2.2, 95% confidence interval [CI] 1.1–4.1 and OR 1.5, 95% CI 1.1–2.0, respectively), higher levels of osteoprotegerin (OR 1.1, 95% CI 1.0–1.2), intercellular adhesion molecule 1 (OR 1.7, 95% CI 1.1–2.5), and lipoprotein-associated phospholipase A2 mass (OR 1.5, 95% CI 1.1–2.1), and lower myeloperoxidase (OR 0.8, 95% CI 0.7–1.0) in participants with greater white matter hyperintensity volumes and silent cerebral infarcts.
Conclusions:
Our study supports a possible role for inflammation in the pathogenesis of CSVD, but suggests that differing inflammatory pathways may underlie ischemic and hemorrhagic subtypes. If validated in other samples, these biomarkers may improve stroke risk prognostication and point to novel therapeutic _targets to combat CSVD.
Cerebral small vessel disease (CSVD) assessed using brain MRI is characterized by 3 main findings that include cerebral microbleeds (CMBs), lacunes of presumed vascular origin (silent cerebral infarcts [SCIs]), and white matter hyperintensity (WMH). The vascular changes underlying ischemic and hemorrhagic CSVD likely include activation of the inflammatory cascade with endothelial failure and resulting neurovascular unit dysfunction.1–3 However, the specific mediators implicated in ischemic and hemorrhagic CSVD may differ.
The Framingham Offspring Cohort provides a large, middle-age, community-based sample in which to investigate the association between systemic biomarkers of inflammation and MRI markers of CSVD. Given the complexity of inflammatory pathways, it is likely that several molecules within the inflammatory cascade are involved. Furthermore, the specific biomarkers that are associated with ischemic and hemorrhagic CSVD is unclear, and such knowledge could lead to more precise risk prediction and identification of novel preventive and therapeutic _targets. Accordingly, we aimed to assess the association of a comprehensive panel of systemic inflammatory markers with MRI measures of CSVD.
METHODS
Sample.
The Framingham Offspring Cohort was enrolled in 1971, and participants have been examined every 4 to 8 years since.4 In 1999, participants underwent brain MRI, and sequences allowing for CMB detection were added in the year 2000. Among offspring participants attending examination cycle 7 (1998–2001; n = 3,539 participants), a comprehensive list of inflammatory biomarkers was measured as part of the broader aim of investigating biomarkers of cardiovascular disease. In the present analysis, we excluded participants who did not have biomarker data (n = 209), did not undergo MRI (n = 1,021), did not have MRI data for the imaging markers of interest (n = 468, all missing CMB data because they had MRI without gradient echo sequences), or had prevalent TIA/stroke or other neurologic disease (severe head injury, multiple sclerosis, brain tumors, hemicraniectomy, etc.) that could affect the estimation of the MRI measures (n = 78).
Standard protocol approvals, registrations, and patient consents.
The institutional review board of Boston University Medical Center approved the study protocol and we obtained informed consent from all subjects.
Clinical characteristics.
Clinical and demographic characteristics were measured at examination cycle 7. Hypertension was defined using JNC-7 (Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure) criteria as systolic blood pressure ≥140 mm Hg, diastolic blood pressure ≥90 mm Hg, or use of antihypertensive medications. We defined prevalent diabetes mellitus as a fasting blood glucose ≥126 mg/dL or use of oral hypoglycemic agents/insulin and current smoking as self-reported smoking of at least one cigarette per day within the year preceding examination. We ascertained medication use by self-report. Nonstroke cardiovascular disease was defined as coronary heart disease, peripheral arterial disease, and/or heart failure. Some participants lacked data on certain variables of interest (e-Methods on the Neurology® Web site at Neurology.org).
Biomarkers.
We investigated a set of 15 biomarkers representing various components of the inflammatory cascade, including systemic inflammation (high-sensitivity C-reactive protein, interleukin-6, monocyte chemotactic protein-1, tumor necrosis factor α [TNF-α], tumor necrosis factor receptor 2 [TNFR2], osteoprotegerin, and fibrinogen), vascular inflammation/endothelial dysfunction (intercellular adhesion molecule 1 [ICAM-1], CD40 ligand, P-selectin, lipoprotein-associated phospholipase A2 [Lp-PLA2] mass and activity, total homocysteine, and vascular endothelial growth factor), and oxidative stress (myeloperoxidase).
Fasting morning samples were collected and plasma and serum aliquots were stored at −70°C. Several biomarkers were measured with commercially available ELISA kits from R&D Systems (Minneapolis, MN) (ICAM-1, interleukin-6, monocyte chemotactic protein-1, P-selectin, TNFR2, TNFα, vascular endothelial growth factor), Bender MedSystems (Vienna, Austria) (CD40 ligand), and Oxis (Beverly Hills, CA) (myeloperoxidase). C-reactive protein was measured using high sensitivity assay (BN100 nephelometer; Dade Behring, Deerfield, IL), and fibrinogen by the Clauss method (Diagnostica Stago Inc., Parsippany, NJ). Lp-PLA2 activity was measured using a colorimetric activity method (diaDexus Inc., San Francisco, CA). Lp-PLA2 mass was measured using a commercially available sandwich enzyme immunoassay (diaDexus Inc.). Total homocysteine was measured with high-performance liquid chromatography with fluorometric detection.
Intra-assay coefficients of variation (all <10% as previously reported)5,6 are listed in e-Methods.
Outcomes: Brain MRI markers of CSVD.
The MRI markers of CSVD of interest were CMBs, SCIs, and WMH. MRI acquisition, measurement techniques, and interrater reliability have been described previously.3,7 Operators blinded to participants' demographic, clinical, and biomarker data rated the images of interest. We determined the volume of WMH according to previously published methods,7 and defined extensive WMH where the natural log of the ratio of WMH volume to total cranial volume was >1 SD above the age-adjusted mean WMH volume.8 We manually determined SCIs on the basis of their size (>3 mm, <15 mm), location, and imaging characteristics, as previously described.8 MRI markers of CSVD were divided into hemorrhagic (CMBs) and ischemic (SCIs and/or extensive WMH). We dichotomized the presence or absence of ischemic CSVD as MRI evidence of SCIs, extensive WMH, or both. We graded CMBs as per previously described methods.9 In addition, we performed secondary analysis on CMB burden and topography. CMB burden was assessed to minimize the potential of confounding from CMB mimics in cases with only one lesion, and to explore the potential additive role of increasing disease severity. Accordingly, CMB burden within individuals was graded as absent, present (≥1 CMBs), ≥2 CMBs, or ≥3 CMBs. These thresholds were influenced by the distribution of CMB number in our sample. CMB topography was divided into lobar only (confined to cortex and subcortical white matter), deep only (internal capsule/external capsule, thalamus, basal ganglia, brainstem), or any deep (deep only or concurrent lobar and deep location, including cerebellum).
Statistical analyses.
We obtained descriptive statistics for the clinical, demographic, and biomarker variables. We performed natural logarithmic (ln) transformation on inflammatory biomarkers that had a skewed distribution, as well as on WMH volume to total cranial volume ratios to avoid undue influence of extreme observations. We used logistic regression to obtain odds ratios and 95% confidence intervals for the association of each biomarker and each outcome (CMBs, SCIs/extensive WMH). For both primary outcomes, our primary model was adjusted for age, sex, and time interval between examination 7 and MRI acquisition. Sensitivity analysis confined to participants without any missing biomarker data was also performed. In additional secondary models, we chose clinical covariates previously associated with the neuroimaging markers of interest within the Framingham Offspring Cohort.10,11 For the CMB outcome, model 2a was adjusted for our primary model covariates plus hypertension, statin use, and antithrombotic use, and model 3a was adjusted for our primary model covariates plus MRI markers of CSVD (SCIs and ln WMH volume). For the SCIs and extensive WMH outcome, model 2b was adjusted for the primary model covariates plus hypertension, diabetes mellitus, current smoking, and history of cardiovascular disease, and model 3b was adjusted for primary model covariates plus the presence of any CMB (yes vs no). Receiver operating characteristic (ROC) curves were constructed for the notable associations within our primary analysis to determine whether the grouping of biomarkers was more predictive than individual biomarkers alone. All analyses were performed using SAS version 9.2 (Cary, NC). A p value <0.05 (uncorrected) was considered statistically significant. The threshold for statistical significance after Bonferroni correction for multiple comparisons (primary analysis: 15 biomarkers × 2 outcomes [1 = all CMBs, 2 = composite of SCI and/or extensive WMH volume]) was set at a p value <0.0017.
RESULTS
Of the 3,539 Framingham offspring participants attending examination cycle 7, 1,763 participants met our eligibility criteria for analysis within the current study. Baseline characteristics of the sample are shown in table 1. Baseline characteristics of participants excluded are presented in table e-1. Results of our primary analyses are presented in tables 2 and 3, e-2, and e-3.
Table 1.
Baseline characteristics of the study sample (n = 1,763)
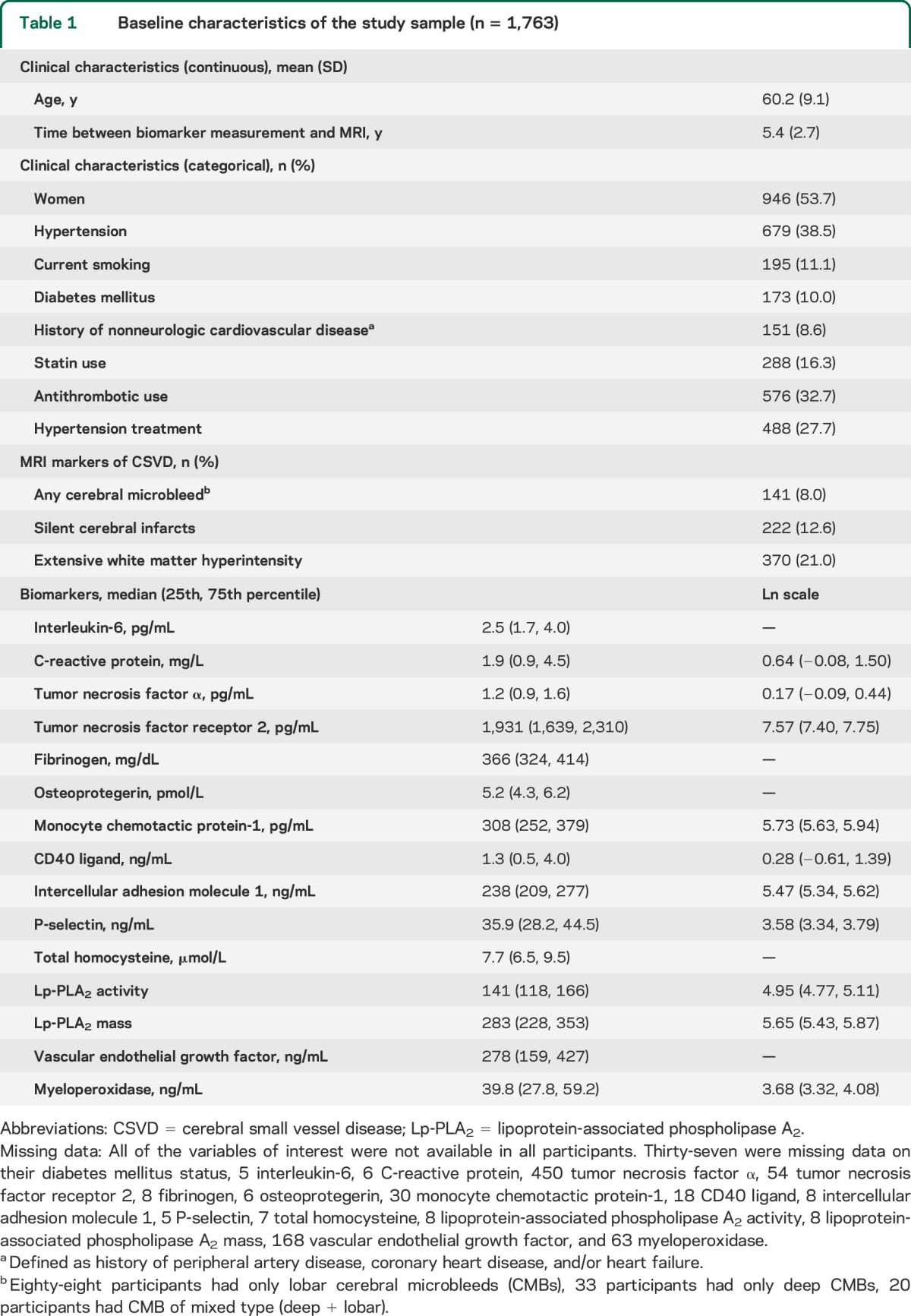
Table 2.
Logistic regression results for the noteworthy association of biomarkers and prevalent CMB (n = 1,763)a
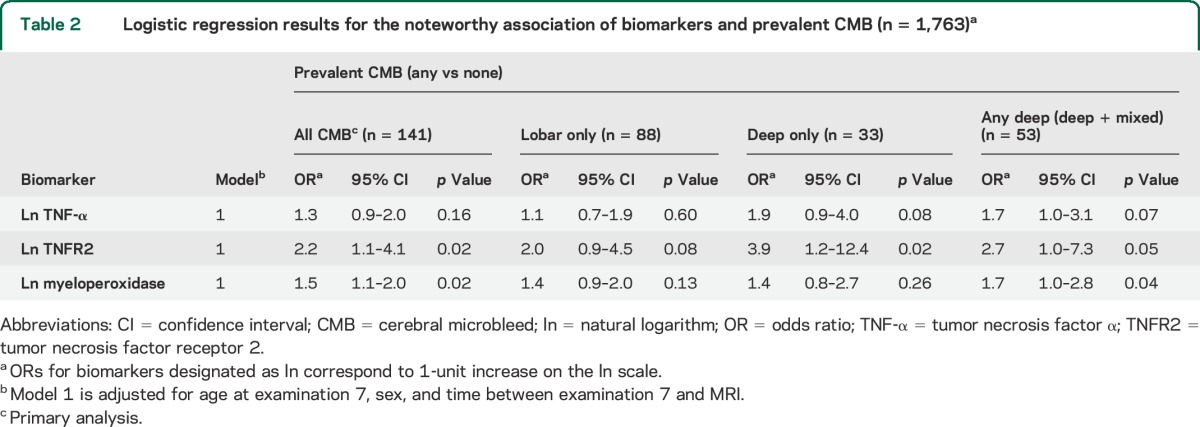
Table 3.
Logistic regression results for the noteworthy association of biomarkers and prevalent silent cerebral infarcts and/or extensive white matter hyperintensities
Cerebral microbleeds.
CMBs were present in 8% (n = 141) of the sample. In our primary analysis, we observed higher levels of circulating TNFR2 and myeloperoxidase in participants with CMBs (table 2). Secondary analysis revealed these associations to be most prominent in persons with only deep CMBs in the case of TNFR2 and any deep CMB in the case of myeloperoxidase. However, similar trends persisted in lobar locations. Grouping of biomarkers was not significantly more predictive of CMB in comparison to the individual biomarkers alone (area under the ROC curve: 0.76 for the composite of TNF-α, TNFR2, and myeloperoxidase vs 0.74 for TNF-α, p = 0.73; 0.74 for TNFR2, p = 0.69; and 0.75 for myeloperoxidase, p = 0.78).
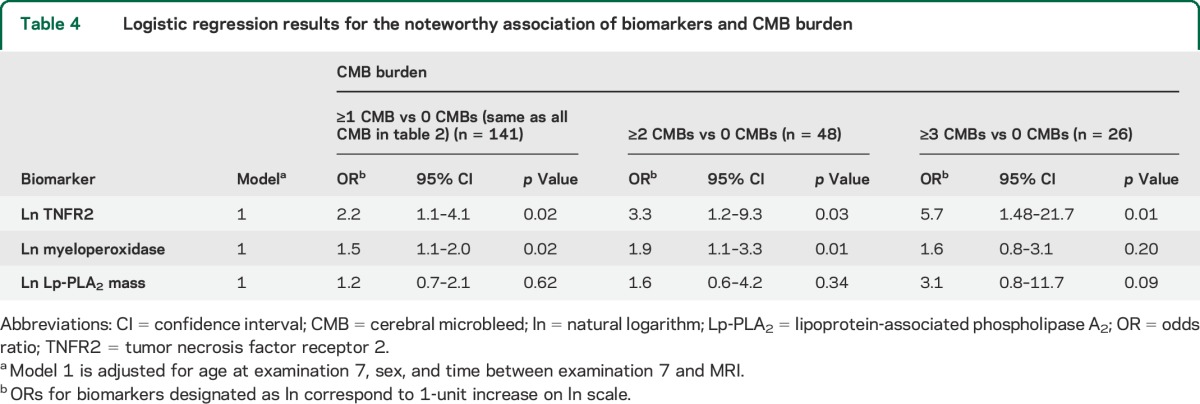
The relation of some of the biomarkers with CMBs increased with greater CMB burden (table 4). Ln TNFR2 related to a 3.3-fold increase in odds of having ≥2 CMBs and 5.7-fold increase in odds of having ≥3 CMBs, each as compared with having no CMBs. In the case of myeloperoxidase, each additional unit increase (ln scale) resulted in a 1.9-fold increase in the odds of ≥2 CMBs and a trend toward a 1.6-fold increase in the odds of having ≥3 CMBs. In addition, a trend existed between higher levels of circulating Lp-PLA2 mass and increasing levels of CMB burden.
Table 4.
Logistic regression results for the noteworthy association of biomarkers and CMB burden
Additional adjustment for clinical covariate and neuroimaging markers of ischemic CSVD did not considerably alter the above findings. There were no noteworthy observations to report with the other biomarkers (table e-2).
SCIs and large WMH volume.
SCIs and/or extensive WMH volume occurred in 30% (n = 522) of the sample. Associations of biomarkers with ischemic CSVD defined as presence of SCIs and/or extensive WMH are listed in table 3. We observed higher levels of circulating osteoprotegerin, ICAM-1, and Lp-PLA2 mass, and lower levels of circulating myeloperoxidase in persons with ischemic CSVD. Grouping of biomarkers was not significantly more predictive of SCIs and/or extensive WMH volume in comparison to the individual biomarkers alone (area under the ROC curve: 0.61 for the composite of osteoprotegerin, ICAM-1, Lp-PLA2 mass, and myeloperoxidase vs 0.58 for osteoprotegerin, p = 0.20; 0.58 for ICAM-1, p = 0.19; 0.58 for Lp-PLA2 mass, p = 0.24; and 0.58 for myeloperoxidase, p = 0.15).
Additional adjustment for clinical covariates and presence of CMBs did not considerably alter the above findings. There were no noteworthy observations to report with the other biomarkers (table e-3).
Only the correlation between higher levels of circulating osteoprotegerin and ischemic markers of CSVD withstood Bonferroni correction for multiple comparisons. Sensitivity analysis confined to participants without any missing biomarker data did not substantially alter our findings.
DISCUSSION
Our results suggest higher levels of various circulating markers of inflammation in persons with MRI markers of CSVD and may support the hypothesis that endothelial failure contributes to the pathogenesis of CSVD.
The strongest association occurred between TNF-α and CMBs. TNF-α is a key regulatory cytokine that is predominantly secreted by macrophages/microglia (the predominant cell type found underlying CMBs in pathologic/autopsy samples)12 and can exert diverse regulatory functions through 2 cell membrane receptors, TNFR1 and TNFR2.13 CMBs are believed to result from a state of increased vascular fragility/permeability and represent hemosiderin-laden macrophages. In addition, they are often associated with a surrounding degree of tissue necrosis.12 Accordingly, their association with TNFR2 has several plausible explanations. TNF-α has been consistently observed to increase vascular permeability and blood-brain barrier dysfunction in animal models.14 Both TNFR1 and TNFR2 seem to be involved in this process.15 Hence, higher levels of TNFR2 may promote the pathogenesis of CMBs. Alternatively, CMB formation could be driving TNF activity through CMB-induced microglial and inflammatory cascade activation. TNFR2 expression may also be increased as a neuroprotective measure in the face of CMB-induced neuronal damage.16
Myeloperoxidase is an inflammatory oxidizing enzyme expressed predominantly by neutrophils in the first 3 days after neuronal injury, followed by macrophages/microglia, which peak within 1 week postinjury.17 Activated leukocytes have been observed to secrete 10 times more myeloperoxidase than their nonactivated counterparts.17 Myeloperoxidase activity results in the generation of hypochlorous acid, a strong oxidant that can cause local tissue damage and amplify the inflammatory cascade. Myeloperoxidase also has been associated with endothelial and blood-brain barrier dysfunction.18 Accordingly, similarly to TNF, myeloperoxidase can either be a cause or effect of CMBs through myeloperoxidase-induced endothelial/blood-brain barrier dysfunction or CMB-induced microglial activation, respectively. Furthermore, myeloperoxidase also may be an active culprit in CMB-induced neuronal injury.
The observation that higher levels of TNFR2 and myeloperoxidase were most pronounced in deep or mixed CMB cases, thought to be consequent to hypertensive arteriopathy (arteriolosclerosis), in comparison to lobar CMBs, suggestive of cerebral amyloid angiopathy (CAA), possibly implies the presence of different inflammatory signaling cascades in association with CMBs in these 2 prevalent underlying microangiopathies. Previous radiographic-pathologic concordance studies have not investigated such differences in persons with CMBs,12 and relevant in vivo circulating biomarker data in patients with CAA are lacking. However, pathologic and experimental studies have noted activated macrophages/microglia, reactive astrocytes, and T lymphocytes adjacent to cerebral vascular amyloid deposits, with a broad inflammatory response including complement, reactive oxygen species, and proinflammatory cytokines, such as interleukin-6, CD40L, and TNF-α.19–22 Although we also observed a trend for elevated TNFR2 levels in persons with purely lobar CMBs, we did not observe an association with these other markers present in our panel. Consequently, alternatively, the aforementioned discrepancy between deep/mixed and lobar CMBs may be attributed to the systemic nature of hypertensive arteriopathy, which would result in a greater and more detectable change in circulating inflammatory biomarkers than CAA, which is confined to the cerebrum. To our knowledge, no other studies have previously investigated serum TNF and myeloperoxidase levels in patients with CMBs.
Lp-PLA2, an enzyme secreted by circulating macrophages that hydrolyzes oxidized phospholipids, is involved in inflammation and the metabolism of low-density lipoprotein. It is reported to be atherogenic, and elevated levels previously have been observed in individuals with myocardial infarction and ischemic stroke.23 It has also been associated with MRI markers of CSVD.3,24 Our findings further corroborate these observations. Although in keeping with a previous report,3 we did not note an association between Lp-PLA2 and any CMBs in our primary analysis, secondary exploratory analysis assessing CMB burden demonstrated higher levels of circulating Lp-PLA2 in individuals with greater numbers of CMBs. Furthermore, we noted higher levels of circulating Lp-PLA2 in persons with ischemic markers of CSVD. The particular association of Lp-PLA2 with both ischemic and hemorrhagic markers of CSVD may be explained by its potential role in various forms of vascular pathogenesis, including atherogenesis and endothelial dysfunction.24
Osteoprotegerin is a cytokine secreted by vascular endothelial and smooth muscle cells and regulates processes involved in vascular injury and inflammation. There exists a strong association between osteoprotegerin and large vessel atherosclerosis, but there are conflicting reports as to whether the association is causal or protective.25 Although higher osteoprotegerin levels have been previously reported in persons with lower total brain volume and stroke secondary to atherosclerotic large artery disease,5,26 our findings are the first, to our knowledge, to suggest a relationship between osteoprotegerin and direct neuroimaging markers of CSVD. Alternatively, because we did not account for cervical or intracranial large artery disease within our study, our observations may be secondary to small vessel orifice atheroma or possibly chronic hypoperfusion from proximal large artery stenosis.
ICAM-1 is an adhesion molecule that facilitates the adhesion of leukocytes to the endothelium and microvessel occlusion, as well as their transendothelial migration. Elevated levels, indicating endothelial cell activation, have been reported as risk factors for the presence and progression of SCIs, WMH, and lacunar stroke.27 However, a recent neuropathologic study did not find any relation between CSVD and ICAM-1, or any suggestion of endothelial activation.28 The authors concluded that previously observed elevated levels of ICAM-1 in the setting of CSVD originated from the peripheral vasculature. It is uncertain whether their postmortem tissue analysis is reflective of the in vivo antemortem state. Nevertheless, their report stresses the need for exercising caution when drawing firm conclusions from cross-sectional biomarker studies.
The mechanism underlying the lower levels of circulating myeloperoxidase observed in persons with ischemic markers of CSVD, in contrast to higher levels in those with CMBs, is uncertain. If validated, in keeping with studies demonstrating more pronounced rotenone-induced neuronal injury in myeloperoxidase-deficient mice,29 and worsening cognitive decline in human participants with the myeloperoxidase AA genotype (leading to lower myeloperoxidase levels),30 myeloperoxidase deficiency may promote ischemic neuronal injury. An alternative explanation is that chronically progressing large volumes of ischemic neuronal and glial injury, and the ensuing inflammatory response, promotes microglial senescence (the dysfunction and dystrophy of microglia with advancing age).31
It is of interest that, irrespective of myeloperoxidase, we observed different inflammatory biomarker profiles between hemorrhagic and ischemic MRI markers of CSVD. Although this observation requires replication to ensure validity, if validated, it lends support to the involvement of different inflammatory pathways in the pathogenesis of ischemic vs hemorrhagic MRI markers or possibly the manner by which the 2 pathologies elicit cerebral tissue injury and activate the inflammatory cascade.
Our findings have a number of limitations. Our cross-sectional analysis does not allow for causal inferences, and we cannot exclude the possibility of residual confounding. Our secondary analysis relating markers to CMB burden was limited by small sample size. The predominant European descent of Framingham Heart Study participants limits generalization to other ethnic groups. We analyzed circulating biomarkers at only one point in time and we have not accounted for concurrent infection, rheumatologic disease, or malignancy that could have altered our results. It is uncertain whether systemic circulating biomarkers are adequate proxy measures of cerebrovascular inflammation and endothelial dysfunction. Although the accumulation of MRI markers of CSVD is considered an ongoing chronic process, they have been observed to occur rapidly within acute/subacute time periods32–34; accordingly, the time interval between serology and acquisition of MRI also limits our findings, despite our attempts to account for it as a covariate. Most notably, because all but one of our associations did not withstand Bonferroni correction, our findings could have occurred merely by chance, should be considered exploratory in nature, and require replication in an external sample to ensure validity. It is reassuring, however, that the association of some of these markers, such as TNFR2, withstood several models and analyses.
Our study further supports the view that inflammatory cytokines may be involved in the pathogenesis of CSVD and suggests the possibility of differing inflammatory pathways between ischemic and hemorrhagic manifestations of CSVD. Further research is required to ensure the validity of our findings and to determine whether any of these molecules might serve as prognostic markers or therapeutic _targets to combat CSVD and related neuronal injury.
Supplementary Material
GLOSSARY
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- CMB
cerebral microbleed
- CSVD
cerebral small vessel disease
- ICAM-1
intercellular adhesion molecule 1
- ln
natural logarithm
- Lp-PLA2
lipoprotein-associated phospholipase A2
- OR
odds ratio
- ROC
receiver operating characteristic
- SCI
silent cerebral infarct
- TNF-α
tumor necrosis factor α
- TNFR2
tumor necrosis factor receptor 2
- WMH
white matter hyperintensity
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study concept/design: Ashkan Shoamanesh, Sarah R. Preis, Alexa S. Beiser, Jose R. Romero, and Sudha Seshadri. Analysis and interpretation of data: Ashkan Shoamanesh, Sarah R. Preis, Alexa S. Beiser, Jose R. Romero, and Sudha Seshadri. Drafting/revising the manuscript for content: Ashkan Shoamanesh, Sarah R. Preis, Alexa S. Beiser, Ramachandran S. Vasan, Emelia J. Benjamin, Carlos S. Kase, Philip A. Wolf, Charles DeCarli, Jose R. Romero, and Sudha Seshadri. Acquisition of data: Sarah R. Preis, Alexa S. Beiser, Ramachandran S. Vasan, Emelia J. Benjamin, Carlos S. Kase, Philip A. Wolf, Charles DeCarli, Jose R. Romero, and Sudha Seshadri. Study supervision/coordination: Alexa S. Beiser, Jose R. Romero, and Sudha Seshadri. Obtained funding: Ramachandran S. Vasan, Emelia J. Benjamin, Philip A. Wolf, Jose R. Romero, and Sudha Seshadri.
STUDY FUNDING
This work was supported by the Framingham Heart Study's National Heart, Lung, and Blood Institute contract (N01-HC-25195) and by grants from the National Institute of Neurological Disorders and Stroke (R01 NS17950), the National Institute on Aging (R01 AG16495; AG08122; AG033193; AG031287; K23AG038444), and NIH grants (1RO1 HL64753; R01 HL076784; 1 R01 AG028321).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol 2013;12:483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koh SH, Park CY, Kim MK, et al. Microbleeds and free active MMP-9 are independent risk factors for neurological deterioration in acute lacunar stroke. Eur J Neurol 2011;18:158–164. [DOI] [PubMed] [Google Scholar]
- 3.Romero JR, Preis SR, Beiser AS, et al. Lipoprotein phospholipase A2 and cerebral microbleeds in the Framingham Heart Study. Stroke 2012;43:3091–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study: design and preliminary data. Prev Med 1975;4:518–525. [DOI] [PubMed] [Google Scholar]
- 5.Jefferson AL, Massaro JM, Wolf PA, et al. Inflammatory biomarkers are associated with total brain volume: the Framingham Heart Study. Neurology 2007;68:1032–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pikula A, Beiser AS, Chen TC, et al. Serum brain-derived neurotrophic factor and vascular endothelial growth factor levels are associated with risk of stroke and vascular brain injury: Framingham Study. Stroke 2013;44:2768–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeCarli C, Massaro J, Harvey D, et al. Measures of brain morphology and infarction in the Framingham Heart Study: establishing what is normal. Neurobiol Aging 2005;26:491–510. [DOI] [PubMed] [Google Scholar]
- 8.Pikula A, Beiser AS, DeCarli C, et al. Multiple biomarkers and risk of clinical and subclinical vascular brain injury: the Framingham Offspring Study. Circulation 2012;125:2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero JR, Preis SR, Beiser A, et al. Risk factors, stroke prevention treatments, and prevalence of cerebral microbleeds in the Framingham Heart Study. Stroke 2014;45:1492–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeerakathil T, Wolf PA, Beiser A, et al. Stroke risk profile predicts white matter hyperintensity volume: the Framingham study. Stroke 2004;35:1857–1861. [DOI] [PubMed] [Google Scholar]
- 12.Shoamanesh A, Kwok CS, Benavente O. Cerebral microbleeds: histopathological correlation of neuroimaging. Cerebrovasc Dis 2011;32:528–534. [DOI] [PubMed] [Google Scholar]
- 13.Faustman D, Davis M. TNF receptor 2 pathway: drug _target for autoimmune diseases. Nat Rev Drug Discov 2010;9:482–493. [DOI] [PubMed] [Google Scholar]
- 14.Pan W, Kastin AJ. Tumor necrosis factor and stroke: role of the blood-brain barrier. Prog Neurobiol 2007;83:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrero E, Zocchi MR, Magni E, et al. Roles of tumor necrosis factor p55 and p75 receptors in TNF-alpha-induced vascular permeability. Am J Physiol Cell Physiol 2001;281:C1173–C1179. [DOI] [PubMed] [Google Scholar]
- 16.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci 2002;22:RC216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breckwoldt MO, Chen JW, Stangenberg L, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci USA 2008;105:18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vita JA, Brennan ML, Gokce N, et al. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation 2004;110:1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada M, Itoh Y, Shintaku M, et al. Immune reactions associated with cerebral amyloid angiopathy. Stroke 1996;27:1155–1162. [DOI] [PubMed] [Google Scholar]
- 20.Weiss R, Lifshitz V, Frenkel D. TGF-beta1 affects endothelial cell interaction with macrophages and T cells leading to the development of cerebrovascular amyloidosis. Brain Behav Immun 2011;25:1017–1024. [DOI] [PubMed] [Google Scholar]
- 21.Townsend KP, Town T, Mori T, et al. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol 2005;35:901–910. [DOI] [PubMed] [Google Scholar]
- 22.Johnstone M, Gearing AJ, Miller KM. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol 1999;93:182–193. [DOI] [PubMed] [Google Scholar]
- 23.Ballantyne CM, Hoogeveen RC, Bang H, et al. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident ischemic stroke in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) Study. Arch Intern Med 2005;165:2479–2484. [DOI] [PubMed] [Google Scholar]
- 24.Wright CB, Moon Y, Paik MC, et al. Inflammatory biomarkers of vascular risk as correlates of leukoariosis. Stroke 2009;40:3466–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Campenhout A, Golledge J. Osteoprotegerin, vascular calcification and atherosclerosis. Atherosclerosis 2009;204:321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guldiken B, Guldiken S, Turgut B, et al. Serum osteoprotegerin levels in patients with acute atherothrombotic stroke and lacunar infarct. Thromb Res 2007;120:511–516. [DOI] [PubMed] [Google Scholar]
- 27.Castellanos M, Castillo J, Garcia MM, et al. Inflammation-mediated damage in progressing lacunar infarctions: a potential therapeutic _target. Stroke 2002;33:982–987. [DOI] [PubMed] [Google Scholar]
- 28.Giwa MO, Williams J, Elderfield K, et al. Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology 2012;78:167–174. [DOI] [PubMed] [Google Scholar]
- 29.Chang CY, Song MJ, Jeon SB, et al. Dual functionality of myeloperoxidase in rotenone-exposed brain-resident immune cells. Am J Pathol 2011;179:964–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pope SK, Kritchevsky SB, Ambrosone C, et al. Myeloperoxidase polymorphism and cognitive decline in older adults in the Health, Aging, and Body Composition Study. Am J Epidemiol 2006;163:1084–1090. [DOI] [PubMed] [Google Scholar]
- 31.Luo XG, Ding JQ, Chen SD. Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener 2010;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menon RS, Burgess RE, Wing JJ, et al. Predictors of highly prevalent brain ischemia in intracerebral hemorrhage. Ann Neurol 2012;71:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shoamanesh A, Catanese L, Sakai O, Pikula A, Kase CS. Diffusion-weighted imaging hyperintensities in intracerebral hemorrhage: microinfarcts or microbleeds? Ann Neurol 2013;73:795–796. [DOI] [PubMed] [Google Scholar]
- 34.Jeon SB, Kwon SU, Cho AH, Yun SC, Kim JS, Kang DW. Rapid appearance of new cerebral microbleeds after acute ischemic stroke. Neurology 2009;73:1638–1644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.