

Abstract
Colorectal cancer (CRC) is a genetic disease governed by clonal evolution1. Genotyping CRC tissue is employed for therapeutic purposes but this approach has significant limitations. A tissue sample represents a single snapshot in time, is subjected to selection bias due to tumor heterogeneity, and can be difficult to obtain. We exploited circulating DNA (ctDNA) to genotype colorectal tumors and track clonal evolution during therapies with the anti-EGFR antibodies cetuximab or panitumumab. We identified genomic alterations in KRAS, NRAS, MET, ERBB2, FLT3, EGFR and MAP2K1 in ctDNA of patients with primary or acquired resistance to EGFR blockade. Mutant RAS clones, which rise in blood during EGFR blockade, decline upon withdrawal of anti-EGFR antibodies indicating that clonal evolution continues beyond clinical progression. Pharmacogenomic analysis of CRC cells, which had acquired resistance to cetuximab, reveals that upon antibody withdrawal KRAS clones decay, while the population regains drug sensitivity. ctDNA profiles of patients who benefit from multiple challenging with anti-EGFR antibodies exhibit pulsatile levels of mutant KRAS. These results reveal that the CRC genome adapts dynamically to intermittent drug schedules and provide a molecular explanation for the efficacy of re-challenge therapies based on EGFR blockade.
Colorectal cancer tissue is used to define the molecular status of clinically relevant genes. For example, oncogenic mutations in KRAS and NRAS, which occur collectively in about 55% of the patients, are routinely assessed in tumor specimens as they predict lack of response to EGFR-_targeted monoclonal antibodies cetuximab and panitumumab2. Mutations in BRAF are also associated with lack of response to EGFR blockade and frequently evaluated in metastatic CRC patients3. In colorectal tumors lacking RAS pathway mutations, HER2 or MET gene amplifications are being explored as biomarkers of response to drugs inhibiting the corresponding oncoproteins4–6.
Tumor tissue genotyping has inherent limitations. It has been shown that the genomic profile of the primary tumor and the metastases are not always concordant due to the intrinsic molecular heterogeneity of the disease1,7,8. Furthermore, commonly used chemotherapeutic agents as well as _targeted drugs can alter the tumor molecular landscape1. To account for this, the genomic profiles of CRC patients should be evaluated repeatedly during the course of therapy. Several reasons prevent the reiterated use of tissue biopsies including the inherent risk of complications associated with these procedures 9. Moreover, biopsy samples are often limited and tissue processing might delay the initiation of treatment. To overcome these restrictions, we considered the analysis of circulating tumor DNA (ctDNA), a procedure known as liquid biopsy10–15.
We assessed whether blood-based molecular profiles could be used to identify actionable _targets, to monitor drug resistance and to track tumor dynamics in CRC patients. We started by evaluating whether the genotype of RAS pathway genes of clinical relevance could be determined in circulating tumor DNA. To analyze ctDNA, we exploited Droplet Digital PCR (ddPCR)16,17 and BEAMing18. Both technologies are based on micro compartmentalization of the PCR reaction and can detect mutant alleles with high sensitivity (0.01 to 0.001%)19.
We selected a total of 100 CRC cases (Supplementary Table 1 and Supplementary Figure 1) for which we collected matched pairs of tissue and blood samples. Droplet Digital PCR (ddPCR) was used to interrogate the mutational status of KRAS, BRAF and NRAS in plasma samples without prior knowledge of the tissue genotype. In 97/100 cases (97%) determinations of ‘RAS pathway mutations’ were concordant between tissue and blood (Supplementary Figure 1a and Supplementary Table 2). Two of the discordant cases (ONCG-CRC21 and ONCG-CRC55) were from patients with low tumor burden (lymph nodes-limited disease or lesions <1.5 cm in size) as assessed by imaging (data not shown).
In 8 cases, plasma analysis revealed mutations which were not detected in the matched tissue, thus denoting that the blood more comprehensively captures intra-patient disease heterogeneity (Supplementary Figure 1a and Supplementary Table 2). The mutational status did not correlate with the levels of circulating free DNA (genome equivalents, GE) indicating that the genotype does not overtly affect release of tumor DNA in the blood (Supplementary Figure 1b).
Presently, even when KRAS and NRAS mutant CRCs are excluded from treatment, only 20% of the patients benefit from monotherapy with EGFR _targeted agents20,21. Using a cohort of 10 patients who received anti-EGFR antibodies regimens without achieving clinical benefit, we asked whether the molecular bases of primary resistance could be ascertained in blood. We started by determining the mutational status of the RAS pathway in ctDNA using the approach described above. As expected, KRAS mutations were absent in the entire cohort. Two patients (MOLI-CRC02 and MOLI-CRC06) displayed NRAS mutations (Figure 1a and Supplementary Table 4a). Scrutiny of the clinical history of these patients revealed that anti-EGFR treatment had been initiated before the introduction of NRAS testing in this clinical setting2, thus explaining the findings.
Figure 1. Identification of genetic alterations associated with resistance to anti-EGFR antibodies in plasma samples.
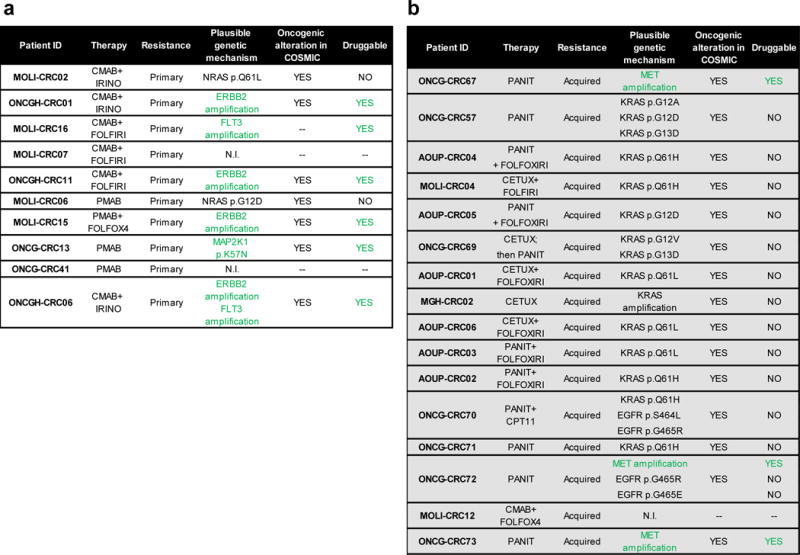
The tables list putative genetic mechanism of primary (a) and acquired (b) resistance to anti-EGFR antibodies which were identified in circulating tumor DNA of 26 patients in total. EGFR mutational analysis was not performed in AOUP-CRC04, AOUP-CRC05, AOUP-CRC01, AOUP-CRC06 and AOUP-CRC02 due to limited sample amount. Green colour highlights actionable _targets. CETUX: cetuximab; PANIT: panitumumab; IRINO: irinotecan. N.I. not identified.
We next sought to systematically identify genetic mechanisms of primary resistance to anti-EGFR treatment in the 8 patients in which the ‘RAS pathway’ was confirmed wild-type in plasma. To this aim, we analyzed the coding region of 226 genes, which we selected as being involved in colorectal cancer progression, oncogenic signaling and sensitivity or resistance to therapy (Supplementary Table 3). The approach relies on massive parallel next generation sequencing (NGS) of genomic DNA, which we adapted to capture and sequence circulating tumor DNA. To confirm that the NGS-blood analysis could identify tumor exclusive (somatic) genetic alterations, we determined the mutational status of APC and TP53, two genes frequently mutated in CRC. The analysis identified APC and/or TP53 mutations in circulating DNA, which were not present in the patients’ germ-line DNA, (Supplementary Table 5).
The results of the analysis of the 226 genes were noteworthy: four cases (ONCGH-CRC01, ONCGH-CRC11, ONCGH-CRC06 and MOLI-CRC15) harbored ERBB2 amplification (Figure 1a and Supplementary Table 4b). We previously found that ERBB2 amplification is present in approximately 3% of CRC and correlate with lack of response to EGFR blockade5. These findings therefore validate the NGS-based experimental approach and increased our confidence in profiling plasma DNA to nominate molecular mechanisms of drug resistance. One of the ERBB2 positive samples also harbored FLT3 amplification; the latter has been previously detected in 3% of CRC (Kopetz, J Clin Oncol 32:5s, 2014; suppl; abstr 3509; www.cbioportal.org, TCGA Nature 2012). In one patient (ONCG-CRC13) we detected a somatic variant of the MAP2K1 gene (p.K57N) encoding the MEK1 protein (Figure 1a and Supplementary Table 4a). MAP2K1 mutations are present in 1.5% of colorectal tumors (www.cbioportal.org, TCGA Nature 2012) but have not yet been associated with resistance to EGFR blockade.
We next asked whether analysis of plasma DNA could be used to unveil mechanisms of acquired (secondary) resistance to cetuximab or panitumumab. To answer this question, we studied the blood of 16 patients who responded and then progressed upon anti-EGFR therapy (Figure 1b, Supplementary Table 4a and 4b). We and others previously reported that acquired resistance to anti-EGFR antibodies is associated with the emergence of RAS pathway mutations and that these mutations can be detected in the blood before disease progression is clinically manifest15,22–24. Accordingly, in most of the patients (11/16), KRAS mutated alleles were detected in blood samples obtained at progression (Figure 1b and Supplementary Table 4a). We also detected EGFR ectodomain mutations (in 2 cases) and KRAS or MET gene amplification in four instances (Figure 1b and Supplementary Table 4b). We previously reported that amplification of KRAS and MET genes, as well as mutations in the extracellular domain of EGFR, sustain resistance to anti-EGFR antibodies in CRC patients4,24–26.
The clinical management of patients who develop RAS pathway mutations, as a consequence of EGFR blockade, remains challenging. Most often these individuals receive additional lines of therapies based on chemotherapeutic agents alone or combined with anti-angiogenic drugs, and in some cases also monotherapy with the multikinase inhibitor regorafenib. It is unknown whether and how subsequent therapies affect RAS (resistant) clones, which were selected during treatment with anti-EGFR antibodies.
To address this issue we monitored the behavior of KRAS mutations and MET amplification during therapeutic schemes that are commonly administered at relapse after EGFR treatment. Resistant clones were tracked in the blood of patients who initially achieved benefit and then experienced disease progression after treatment with an anti-EGFR antibody, either in first-line in combination with chemotherapy or as monotherapy in third-line. For 5 patients who developed KRAS mutations and for one patient who acquired MET amplification, samples were available at baseline, at the time of first disease progression and at different time-points across subsequent lines of treatment (Figure 2 and Supplementary Table 6a).
Figure 2. KRAS mutant alleles emerge in circulating DNA during anti-EGFR therapy and decline when treatment is suspended.
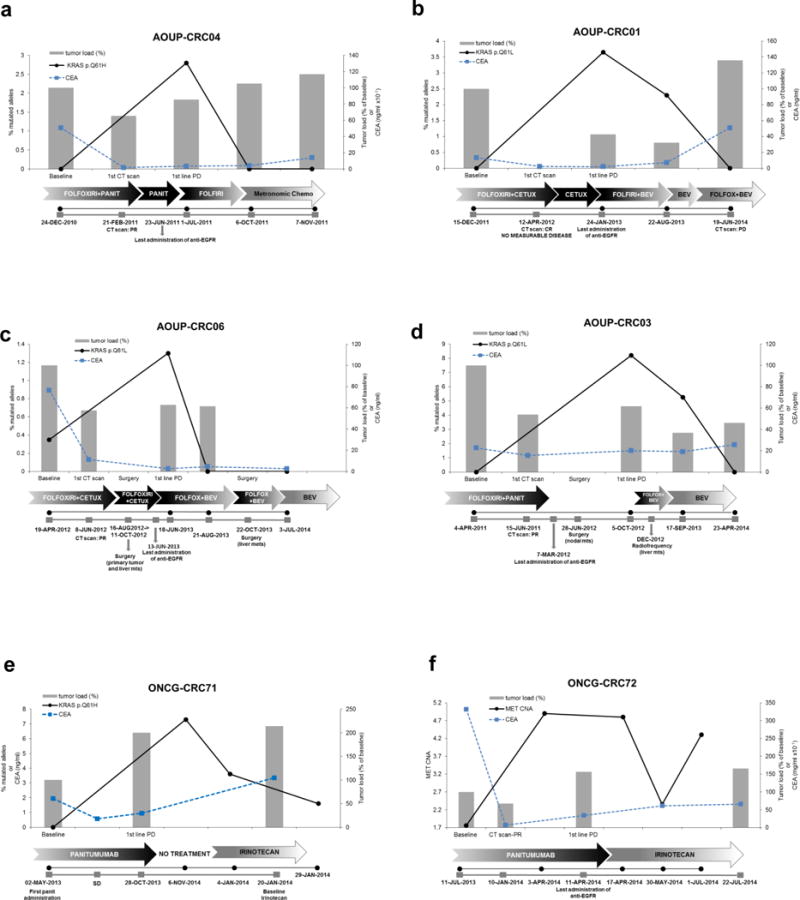
Detection of KRAS mutations (a–e) and MET amplification (f) in circulating DNA of patients who developed acquired resistance to first-line chemotherapy plus anti-EGFR and then received other lines of treatment. Grey bars represent the variation of tumor load, compared to baseline, during systemic treatments specified in arrows below the graphs. Tumor load is calculated as follows: measurable disease at the initiation of treatment (baseline) is assumed as 100%; responses or progression are calculated as % of tumor load as compared to baseline, as per RECIST criteria. Relevant clinical events are indicated in grey boxes below the graphs. Black lines indicate the frequency of KRAS mutation (% of alleles) or MET copy number alteration, detected in circulating DNA at the time points indicated below the graphs. Dotted blue line indicates CEA values.
CETUX: cetuximab; PANIT: panitumumab; BEV: bevacizumab; IRINO: irinotecan.
In patient AOUP-CRC01 the first CT scan revaluation assessed complete response (no measurable disease). KRAS mutational frequency is plotted on the primary axis while % of tumor load is plotted on the secondary axis.
In patient ONCG-CRC71 the first CT scan revaluation not available. CEA levels were used as a surrogate marker of response.
The analysis revealed that KRAS mutant alleles, which emerge at the time of disease progression, decline when treatment with anti-EGFR antibodies is suspended and remain below the limit of detection across subsequent lines of treatment (Figure 2). These findings led us postulate that clonal evolution of tumor cell populations that survive treatment with anti-EGFR antibodies continues beyond the point of clinically-established resistance.
To mechanistically evaluate this possibility, we studied populations of CRC cells (DiFi) in which acquisition of resistance to cetuximab is accompanied by amplification of the KRAS gene14. To parallel withdrawal of EGFR blockade, which occurs when patients develop cetuximab resistance, two populations of DiFi resistant cells were cultured for 160 days in the absence of the anti-EGFR antibody (Figure 3a). Analogously to what we had observed in the blood of patients, KRAS declined significantly in both cell models (Student’s t-Test p value <0.01) when the anti-EGFR antibody was suspended (Figure 2 and Figure 3a, top panel). The cell populations, which experienced antibody withdrawal, regained partial sensitivity to cetuximab (Student’s t-Test p value ≤0.001) as compared to the population in which the drug pressure was maintained (Figure 3a, bottom panel and Supplementary Figure 2).
Figure 3. Anti-EGFR antibodies re-challenge in colorectal cancer cells and patients.
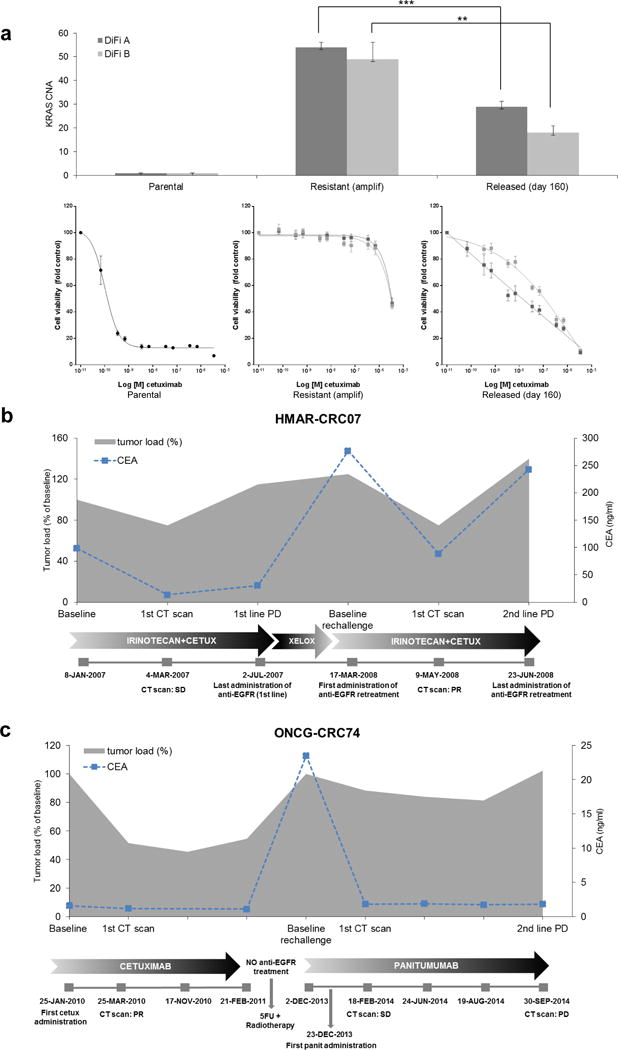
(a) Two colorectal cancer cell populations (DiFi A and DiFi B) that developed KRAS amplification as a resistance mechanism to cetuximab were allowed to replicate in the absence of the antibody for 160 days.
Top panel: KRAS amplification was assessed by qPCR in the indicated cell models (parental/sensitive, resistant derivatives and resistant cells after 160 days of antibody withdrawal). Grey bars indicate KRAS gene copy number (CNA). Statistical differences were calculated by Student’s t-Test. Data are expressed as means ± SD of three independent experiments. *** p value ≤0.001; ** p value ≤0.01.
Bottom panel: cetuximab sensitivity assay. The indicated cell populations were treated for 1 week, with increasing concentrations of cetuximab. Cell viability was measured by the adenosine triphosphate (ATP) assay. Data points represent means ± SD of three independent experiments.
(b) Clinical synopsis of a patient treated with irinotecan plus cetuximab achieving SD in lung and bone metastases that lasted approximately 6 months. At progression, the patient began treatment with capecitabine plus oxaliplatin (XELOX) with progression of the disease after 3 months of treatment. The patient was subsequently re-treated with irinotecan plus cetuximab achieving a PR. Grey area represents tumor load (% of baseline); dotted blue line indicates CEA values.
(c) Clinical synopsis of a patient treated with cetuximab as third-line for pelvic relapse of rectal cancer, achieving partial response lasting 13 months; the patient then refused further therapy for skin toxicity. At disease progression, one year later, she was treated with radiotherapy and 5-fluorouracil with partial response and then progression after 6 months. She therefore received rechallenge with anti-EGFR (panitumumab monotherapy) achieving long-lasting SD (7 months). Grey area represents tumor load (% of baseline); dotted blue line indicates CEA values. CETUX: cetuximab.
The decline of KRAS alleles we observed in blood upon withdrawal of EGFR blockade and the cell based functional experiments suggest that re-challenge therapies may be worthy for CRC patients who, upon relapse, suspend EGFR-_targeted agents. The scrutiny of our clinical databases retrieved 2 patients who responded to anti-EGFR antibodies and upon relapse received additional lines of therapy before receiving again anti-EGFR treatment with cetuximab or panitumumab (Figure 3b and 3c). Notably, re-challenge with EGFR blockade was clinically effective and triggered partial responses or long lasting stabilization (Figure 3b and 3c). This is consistent with literature data showing that CRC patients can respond to anti-EGFR re-challenge27.
We next asked whether and to what extent intermittent treatment with anti-EGFR antibodies affected the genomic status of CRCs. To address this question we studied three additional patients who received multiple rounds of anti-EGFR antibodies and for which longitudinal tissue or plasma samples where available (Figure 4).
Figure 4. KRAS mutant clones dynamically evolve in response to pulsatile anti-EGFR antibodies therapy.
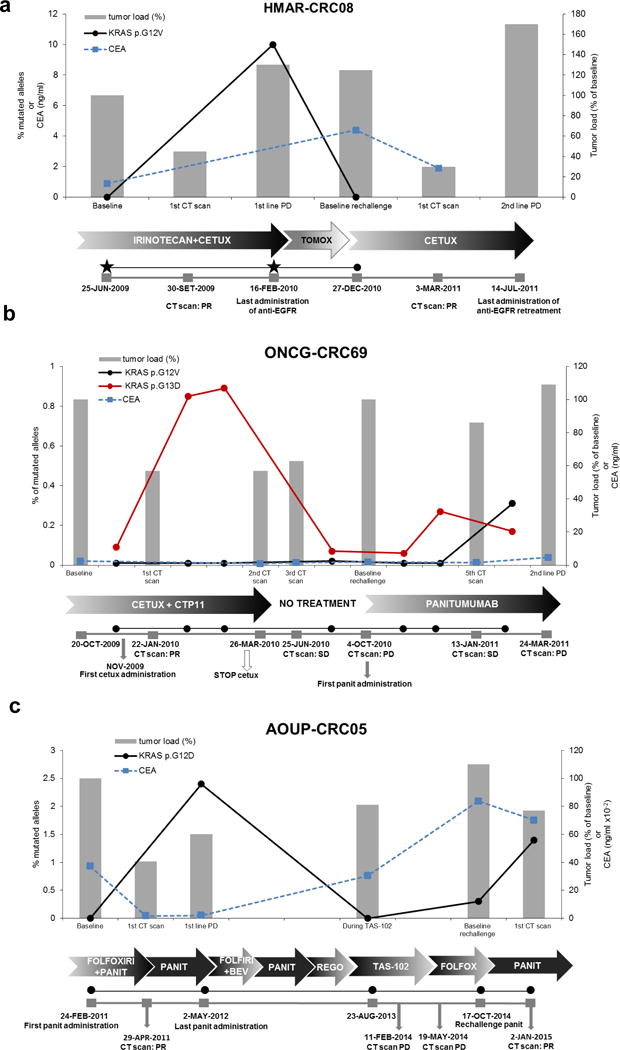
(a–c) Dynamics of KRAS mutant clones in plasma samples of three CRC patients receiving the indicated therapies. Grey bars represent variation of tumor load, compared to baseline, during treatments as specified below the graphs. Tumor load is calculated as follows: measurable disease at the initiation of treatment (baseline) is assumed as 100%; responses or progression are calculated as % of tumor load as compared to baseline, as per RECIST criteria.
Relevant clinical events are indicated in grey boxes below the individual graphs. Black and red lines indicate the frequency of KRAS mutation (% of alleles), detected in circulating DNA at the indicated time points. Black stars represent analyzed tissue samples. Dotted blue line indicates CEA values. CETUX: cetuximab; PANIT: panitumumab; REGO: regorafenib.
In the first patient (HMAR-CRC08) tumor tissue was available before the first administration of anti-EGFR, as well as at progression after a partial response of nearly 8 months. KRAS p.G12V was detected at resistance and declined when cetuximab plus irinotecan treatment was suspended, as assessed by plasma analysis (Figure 4a). The patient then received standard chemotherapy and when a new progression was detected, was re-challenged with cetuximab alone, achieving again a partial response, which lasted 18 months.
In the second patient (ONCG-CRC69) clonal tracking revealed that during the first round of EGFR blockade with cetuximab a KRAS p.G13D mutant clone raised in blood and declined when treatment was suspended as the patient refused further therapy (Figure 4b and Supplementary Table 6b). Subsequently, at disease progression, the patient received panitumumab-achieving SD with tumor shrinkage, which lasted for nearly six months (Figure 4b). Notably, during the second round of EGFR blockade, the KRAS p.G13D allele increased again in blood and was accompanied by the emergence of another KRAS mutant clone (p.G12V).
The third patient (AOUP-CRC05) developed a KRAS p.G12D clonal variant during the initial challenge with panitumumab which translated into a partial response. Upon progression the patient received several salvage therapies during which a single plasma samples was collected and again displayed a decline of the KRAS allele that emerged through the initial EGFR blockade. More than a year from the initial response to the anti-EGFR antibody the patient was treated again with panitumumab achieving again a partial response. During panitumumab re-challenge the levels of the KRAS p.G12D raised again in plasma (Figure 4c and Supplementary Table 6b).
Variations of KRAS clones detected in blood and cell lines suggest that colorectal cancers treated with anti-EGFR antibodies display remarkable plasticity. This likely reflects dynamic clonal competition and provides molecular evidence that re-challenge with EGFR blockade after a withdrawal period is effective as it exerts genomic selection in metastatic colorectal cancers.
Currently, CRC patients are treated according to mutational landscapes ascertained using a small fragment of the primary tumor or an individual metastatic lesion. Essentially, a single molecular snapshot is taken for each patient and based on this knowledge several rounds of therapy are administered, often over many years. Rather than being based on out-of-date ‘molecular’ pictures, each round of therapy should ideally be instituted on a recent genetic profile as comprehensive as possible of the overall disease.
In this work we evaluated whether ctDNA can be used throughout the clinical management of CRC patients to gather real time updates on the molecular landscape of the disease. We find that mutational profiles determined in blood and tissues are highly concordant. In a relevant fraction of the cases (8%), analysis of circulating DNA identified KRAS or NRAS mutated alleles, which were not detected in tissue samples, underscoring the ability of liquid biopsies to better capture the heterogeneity of the overall disease. Our findings suggest that blood-based tests may more comprehensively interrogate the ‘RAS pathway’ mutational status than tissue-derived genotypes.
Only a small fraction (20%) of patients who receive anti-EGFR therapies benefit from treatment. The molecular bases of the lack of response in the remaining patients are largely unknown. We find that ctDNA analyses can often identify genetic alterations likely responsible for resistance to EGFR blockade including amplification of the HER2 and MET genes or mutations in MAP2K1. We also observe that CRC patients refractory to anti-EGFR antibodies, who do not carry RAS pathway mutations, frequently display clinically-actionable oncogenic events. This approach may therefore be used to rapidly identify rare population of patients likely to respond to _targeted agents.
In accordance with our previous findings and other reports, KRAS or NRAS mutated clones often emerge in the blood of CRC patients who respond to cetuximab or panitumumab therapies14,28–30. At present, these individuals discontinue anti-EGFR antibodies treatment and receive salvage therapies based mainly on chemotherapeutic agents. We discovered that KRAS mutant clones decline in blood when anti-EGFR antibodies are withdrawn, suggesting that resistant cell populations are highly dynamic. To functionally substantiate these discoveries, we examined preclinical models of acquired resistance to cetuximab. Analogously to what observed in patients, we noted that KRAS clones decay in resistant populations upon antibody suspension.
These results, together with the clinical cases in which we documented effectiveness of re-challenge with anti-EGFR antibodies paralleled by rise and fall of KRAS mutant clones, provide the rationale for adaptive therapy strategies. In this regard discontinuous dosing strategy with BRAF inhibitors has been successfully attempted in preclinical models of BRAF driven melanomas31. Furthermore there is evidence that re-challenge with _targeted therapies can be effective in patients with different tumors types27,32–34.
Our data will likely trigger further study to assess whether CRC cells who developed resistance to EGFR may display fitness disadvantage in the absence of the drug and whether this can be exploited to forestall the onset of lethal drug-resistant disease. Drug schedules, aimed at maintaining a stable population of drug-sensitive cells to suppress the growth of resistant clones through intratumoral competition, could be explored in ad-hoc clinical trials. Accordingly, we plan to investigate the re-introduction of anti-EGFR antibodies in patients who achieve benefit from cetuximab or panitumumab and subsequently display a decline of KRAS mutant clones in circulating tumor DNA.
In conclusion, our data indicate that blood, rather than tissue, can be employed to monitor -virtually continuously- the molecular evolution of metastatic colorectal tumors. The findings that the genome of CRC adapts dynamically to pulsatile drug schedules provide rationale for additional lines of therapy for patients who benefit from an initial challenge with anti EGFR antibodies.
Material and Methods
Patients and samples
Tumor specimens (formalin fixed paraffin embedded sections) and plasma samples were collected from histologically confirmed metastatic colorectal cancer (CRC) patients with clinical annotation as detailed in Supplementary Table 1, and treated at Ospedale Niguarda Ca’ Granda, Milano, Italy, Azienda Ospedaliera Universitaria Pisana (Pisa, Italy), Candiolo Cancer Institute (Candiolo, Italy), San Giovanni Battista Hospital (Turin, Italy), Istituto Nazionale Tumori Fondazione G. Pascale – IRCCS (Naples, Italy), Massachusetts General Hospital Cancer Center (Boston, MA) and Hospital del Mar (Barcelona, Spain). Samples were obtained through protocols approved by the local Ethical Committees. Availability of tumor sample qualitatively and quantitatively suitable for molecular analyses was a requirement for being considered in the present study. The study was conducted according to the provisions of the Declaration of Helsinki, and all patients signed their informed consent before sample collection. Disease extension and response were assessed by CT scans and clinical response was evaluated according to RECIST 1.1 criteria. Two independent oncologists and radiologists verified in a blinded manner the clinical response for all patients. For a subset of the patients, plasma samples were also collected at selected timepoints (baseline, during EGFR-_targeted therapy with cetuximab or panitumumab, at progression and during subsequent lines of treatment).
Mutational analysis of tissue specimens
Mutation analysis in tissue (primary tumor or metastasis) was carried out in the context of the standard management care of patients with mCRC considered for anti-EGFR treatment by the Institutions that participated in the study. The methodologies used in the study to determine mutations in tissue samples include High Resolution Melting Analysis (HMRA), Pyrosequencing and Sanger sequencing. These analyses were performed in pathology labs under diagnostic guidelines35–38. The sensitivity for mutations detection across all platforms was at least 15%.
Plasma Samples Collection
At least 10 mL of whole blood were collected by blood draw using EDTA as anticoagulant. Plasma was separated within 5 hours through 2 different centrifugation steps (the first at room temperature for 10 minutes at 1,600 × g and the second at 3,000 × g for the same time and temperature), obtaining up to 3 mL of plasma. Plasma was stored at −80°C until ctDNA extraction.
ctDNA isolation and genome equivalents quantification (GE/ml plasma) and CEA assessment
ctDNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (QIAGEN) according to the manufacturer’s instructions. 6 μl of ctDNA were used as template for each reaction. All samples were analyzed in triplicate. PCR reactions were performed using 10 μl final volume containing 5 μl GoTaq® qPCR Master Mix, 2× with CXR Reference Dye) (Promega) and LINE-1 [12,5 μmol] forward and reverse primers. DNA at known concentrations was also used to build the standard curve. Primer sequences are available upon request.
CEA levels were assessed by Cobas 8000 modular analyzer system immunoassay (Roche Diagnostics) as for good clinical practice.
BEAMing
BEAMing was performed as described previously 14,28 with further optimizations in our laboratory. Primers and probes sequences are available upon request.
Droplet digital PCR analysis
Isolated circulating free DNA was amplified using ddPCR™ Supermix for Probes (Bio-Rad) using KRAS, NRAS, BRAF (PrimePCR™ ddPCR™ Mutation Assay, Bio-Rad), EGFR (custom designed) ddPCR assays for point mutations and and KRAS, MET and EIF2C1 (reference) for gene copy number variations (PrimePCR™ ddPCR™ Copy Number Assay, Bio-Rad). ddPCR was then performed according to manufacturer’s protocol and the results reported as percentage or fractional abundance of mutant DNA alleles to total (mutant plus wild type) DNA alleles. 8 to 10 μl of DNA template was added to 10 μl of ddPCR™ Supermix for Probes (Bio-Rad) and 2 μl of the primer/probe mixture. This reaction mix was added to a DG8 cartridge together with 60 μl of Droplet Generation Oil for Probes (Bio-Rad) and used for droplet generation. Droplets were then transferred to a 96 well plate (Eppendorf) and then thermal cycled with the following conditions: 5 minutes at 95°C, 40 cycles of 94°C for 30s, 55°C for 1 minute followed by 98°C for 10 minutes (Ramp Rate 2°C/sec). Droplets were analyzed with the QX200™ Droplet Reader (Bio-Rad) for fluorescent measurement of FAM and HEX probes. Gating was performed based on positive and negative controls, and mutant populations were identified. The ddPCR data were analyzed with QuantaSoft analysis software (Bio-Rad) to obtain Fractional Abundance and Copy Number Variations of the mutant/amplified DNA alleles in the wild-type/normal background. The quantification of the _target molecule was presented as number of total copies (mutant plus WT) per sample in each reaction. Fractional Abundance is calculated as follows: F.A. % = (Nmut/(Nmut+Nwt))*100), where Nmut is number of mutant events and Nwt is number of WT events per reaction. The number of positive and negative droplets is used to calculate the concentration of the _target and reference DNA sequences and their Poisson-based 95% confidence intervals, as previously shown39. ddPCR analysis of normal control plasma DNA (from cell lines) and no DNA template controls were always included. Samples with too low positive events were repeated at least twice in independent experiments to validate the obtained results.
Next Generation Sequencing analysis
Libraries were prepared with Nextera Rapid Capture Custom Enrichment Kit (Illumina Inc., San Diego, CA, USA), according to the manufacturer’s protocol. Preparation of libraries was performed starting from up to 10–150 ng of plasma ctDNA and 50–100 ng of gDNA from PBMCs (as corresponding normal reference, or hg19 when germ-line DNA from the patient was not available). gDNA was fragmented using transposones, adding simultaneously adapter sequences. For ctDNA libraries preparation was used NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England BioLabs Inc., Ipswich MA). Purified tagmented gDNA and ctDNA was used as template for subsequent PCR to introduce unique sample barcodes. Fragments’ size distribution of the DNA was assessed using the 2100 Bioanalyzer with a High Sensitivity DNA assay kit (Agilent Technologies, Santa Clara, CA). Equal amount of DNA libraries were pooled and subjected to _targeted panel hybridization capture. Libraries were then sequenced using Illumina MiSeq or NextSeq500 sequencer (Illumina Inc., San Diego, CA, USA).
Bioinformatic analysis
We built a bioinformatic pipeline for NGS data analysis in order to compare normal and tumor samples and to identify three kind of variations: somatic mutations, insertions or deletions (INDEL) and gene copy number alterations. To this end, we combined open-source tools and custom scripts into a complete analysis workflow. Initially, FastQ sequences files generated by Illumina sequencer for each sample were mapped to the human reference genome (assembly version hg19) using BWA mapper version 0.7.640 with default parameters. The reads’ alignment was saved as BAM files; these were subsequently sorted depending on their mapping position with the “sort” command in the software package SAMtools version 0.1.19 41. PCR duplicates were removed from BAM files using the command “rmdup” of SAMtools package41. By making use of the “mpileup” command41 output, we calculated the alleles’ counts using a custom script: for each position in the chromosome, we compared normal and tumor samples allele by allele. Somatic variations were then called only when supported by a 5% significance level obtained with a Fisher’s Test. Mutations were annotated by a custom script printing out gene information, number of normal/mutated reads, the allelic frequencies and the variation effect (synonymous, non-synonymous, stop-loss/gain). Each of these entries was supplemented with the corresponding number of occurrences in the COSMIC database http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/42, the world’s largest resource for exploring the impact of somatic mutations in human cancer. To find possible INDELs, we further analyzed the alignment files by comparing normal/tumor samples using Pindel software43 with human genome as reference. We retained a subset of hits that was present only in tumor sample with a minimum support of 10% altered reads. INDELS in homopolymeric context were filtered and discarded. Gene copy number (GCN) was addressed starting from “mpileup” output, by calculating the ratio of the median gene read depth to the median read depth of all coding regions _targeted by IRCC panel. Alterations of tumor sample with respect to normal were obtained by considering the ratio of the corresponding GCNs. For each gene, we reported its copy number (CN) in the normal and tumor samples and GCN ratio. In every plot, we added the 2N line, representing the ratio of the corresponding median reads depth of each sample.
Cell culture
DiFi A and DiFi B models of acquired resistance to cetuximab were generated as previously reported14. DiFi cells were cultured in F12 medium (Invitrogen) supplemented with 5% FBS. The genetic identity of the cell lines used in this study was confirmed by STR profiling (Cell ID, Promega). Cell models were allowed to replicate for 160 days in the absence of cetuximab and their mutational status was assessed by qPCR. All cell lines were tested and resulted negative for mycoplasma contamination with Venor® GeM Classic kit (Minerva biolabs).
Drug assays
Cetuximab was obtained from the Pharmacy at Niguarda Ca’ Granda Hospital, Milan, Italy. DiFi cells were seeded in 100 μL medium at 2×103 density in 96-well culture plates. After serial dilutions, drugs in serum-free medium were added to cells, and medium-only wells were included as controls. Plates were incubated at 37°C in 5% CO2 for 6 days, after which cell viability was assessed by ATP content using the CellTiter-Glo® Luminescent Assay (Promega). All assays were performed independently at least three times.
Gene copy number analysis (qPCR)
Real time PCR was performed with 150 ng of DNA per single reaction using GoTaq QPCR Master Mix (Promega) and determined by real time PCR using an ABI PRISM®7900HT apparatus (Applied Biosytems). All primer sequences are available upon request.
Supplementary Material
(a) The mutational status of KRAS, BRAF and NRAS was assessed in plasma by ddPCR and matched tissue sample of 100 colorectal cancer patients. Green indicates that mutations identified in plasma and tissues are concordant; yellow denoted cases in which mutations were identified only in plasma; grey highlights mutations identified only in tissue.
(b) Levels of genome equivalents (GE, log scale, ascending order) per milliliter of plasma in the same colorectal cancer patients reported in panel (a). The colors of the bars identify genotype (grey: RAS/BRAF WT-negative; lilac: KRAS mutant; light blue: NRAS mutant; light red: KRAS and BRAF mutant; green: BRAF mutant).
Supplementary Figure 2: Bar graph shows cell viability at clinically relevant concentration of 10 μg/ml of cetuximab in the resistant cell populations (DiFi A and DiFi B) and in the cells after 160 days of antibody withdrawal. Statistical differences were calculated by Student’s t-Test. Data are expressed as means ± SD of three independent experiments. ***p value ≤0.001.
Supplementary Table 1: Clinical characteristics of patients included in the plasma-tissue concordance analysis (n=100).
Supplementary Table 2: The table lists the ddPCR events (mutant cases only) obtained analyzing ctDNA of patients included in the concordance study (Supplementary Figure 1). Poisson corrected (95% C.I.) and total number of copies per sample analyzed is also shown.
Supplementary Table 3: (a) The table shows ddPCR measurements. Plasma samples were initially tested for KRAS, BRAF, NRAS and EGFR mutations, and for KRAS and MET copy number alterations. Negative cases were subjected to NGS analysis.
(b) Description of the IRCC-_target (Therapeutically Applicable Genes for Effective Treatment) panel. The table indicates gene name, exons and bases analyzed by next generation sequencing (Illumina MiSeq). The panel is designed to capture the entire coding region of listed genes using plasma DNA as a source.
Supplementary Table 4: Alterations in plasma DNA identified in patients resistant to anti-EGFR treatment. Raw data obtained by digital-based PCR and IRCC _target panel (mutated and wild type reads, respectively). N.I. not identified. Poisson corrected (95% C.I.) and total number of copies per sample analyzed is also shown.
Supplementary Table 5: The table shows APC and TP53 alterations found in patients through Next Generation Sequencing of plasma DNA (raw data). N.I. not identified.
Supplementary Table 6: KRAS mutations identified by ddPCR and BEAMing at indicated time points in plasma of patients who developed acquired resistance to first-line chemotherapy plus anti-EGFR and then received other lines of treatment (a) or were re-challenged with anti-EGFR drugs (b). Poisson corrected (95% C.I.) and total number of copies per sample analyzed is also shown. cetux: cetuximab; panit: panitumumab. N.A. not available.
Acknowledgments
The authors would like to thank all the patients that participated in the study and their families. We are grateful to the members of the laboratory of Molecular Genetics for critical reading and editing of this manuscript.
Supported by The European Community‘s Seventh Framework Programme under grant agreement no. 259015 COLTHERES (A.B.); Associazione Italiana per la Ricerca sul Cancro (AIRC) IG grant no. 12812 (A.B.); AIRC MFAG no. 11349 (F.D.N.); grant —Farmacogenomicall—5 per mille 2009 MIUR—Fondazione Piemontese per la Ricerca sul Cancro—ONLUS (F.D.N.); AIRC 2010 Special Program Molecular Clinical Oncology 5 per mille, project no. 9970 (A.B.); FPRC 5 per mille 2010 and 2011 Ministero della Salute (A.B.); Ministero dell‘Istruzione, dell‘Università e della Ricerca, progetto PRIN (A.B.); Progetti di Ateneo-2011, Università di Torino (ORTO11RKTW to A.B.). J.A. is a recipient of intensification program ISCIII/FEDER. Partially supported by a grant to A. Avallone from Italian Ministry of Health (RF2009-1539464). This work was supported by RD12/0036/0051, PI12/00989, PI12/00680, PT13/0010/0005 and 2014 SGR 740 grants and by the Xarxa de Banc de Tumors de Catalunya (C.M.)
References
- 1.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Douillard JY, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 3.Di Nicolantonio F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 4.Bardelli A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertotti A, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic _target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 6.Yonesaka K, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Mattos-Arruda L, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25:1729–1735. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overman MJ, et al. Use of research biopsies in clinical trials: are risks and benefits adequately discussed? J Clin Oncol. 2013;31:17–22. doi: 10.1200/JCO.2012.43.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siravegna G, Bardelli A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014;15:449. doi: 10.1186/s13059-014-0449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer–a survey. Biochim Biophys Acta. 2007;1775:181–232. doi: 10.1016/j.bbcan.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 14.Misale S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diaz LA, et al. The molecular evolution of acquired resistance to _targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinert T, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2015 doi: 10.1136/gutjnl-2014-308859. [DOI] [PubMed] [Google Scholar]
- 17.Sanmamed MF, et al. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin Chem. 2015;61:297–304. doi: 10.1373/clinchem.2014.230235. [DOI] [PubMed] [Google Scholar]
- 18.Diehl F, et al. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3:551–559. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 19.Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov. 2014;4:1269–1280. doi: 10.1158/2159-8290.CD-14-0462. [DOI] [PubMed] [Google Scholar]
- 21.Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- 22.Beaver JA, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014;20:2643–2650. doi: 10.1158/1078-0432.CCR-13-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montagut C, et al. Evolution of heterogeneous mechanisms of acquired resistance to cetuximab-based therapy in colorectal cancer. ASCO. Vol abstr 3526 (Journal of Clinical Oncology, 2014) [Google Scholar]
- 24.Arena S, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 25.Mohan S, et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 2014;10:e1004271. doi: 10.1371/journal.pgen.1004271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valtorta E, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-_targeted therapy. Int J Cancer. 2013;133:1259–1265. doi: 10.1002/ijc.28106. [DOI] [PubMed] [Google Scholar]
- 27.Hata A, et al. Panitumumab rechallenge in chemorefractory patients with metastatic colorectal cancer. J Gastrointest Cancer. 2013;44:456–459. doi: 10.1007/s12029-012-9453-7. [DOI] [PubMed] [Google Scholar]
- 28.Misale S, et al. Blockade of EGFR and MEK Intercepts Heterogeneous Mechanisms of Acquired Resistance to Anti-EGFR Therapies in Colorectal Cancer. Sci Transl Med. 2014;6:224ra226. doi: 10.1126/scitranslmed.3007947. [DOI] [PubMed] [Google Scholar]
- 29.Morelli MP, et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann Oncol. 2015 doi: 10.1093/annonc/mdv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das Thakur M, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seghers AC, Wilgenhof S, Lebbé C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012;22:466–472. doi: 10.1097/CMR.0b013e3283541541. [DOI] [PubMed] [Google Scholar]
- 33.Hata A, Katakami N, Kaji R, Fujita S, Imai Y. Does T790M disappear? Successful gefitinib rechallenge after T790M disappearance in a patient with EGFR-mutant non-small-cell lung cancer. J Thorac Oncol. 2013;8:e27–29. doi: 10.1097/JTO.0b013e318282e047. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura T, et al. Application of a highly sensitive detection system for epidermal growth factor receptor mutations in plasma DNA. J Thorac Oncol. 2012;7:1369–1381. doi: 10.1097/JTO.0b013e31825f2821. [DOI] [PubMed] [Google Scholar]
- 35.Ma ES, Wong CL, Law FB, Chan WK, Siu D. Detection of KRAS mutations in colorectal cancer by high-resolution melting analysis. J Clin Pathol. 2009;62:886–891. doi: 10.1136/jcp.2008.063677. [DOI] [PubMed] [Google Scholar]
- 36.Tiacci E, et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood. 2012;119:192–195. doi: 10.1182/blood-2011-08-371179. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez de Castro D, et al. A comparison of three methods for detecting KRAS mutations in formalin-fixed colorectal cancer specimens. Br J Cancer. 2012;107:345–351. doi: 10.1038/bjc.2012.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sundström M, et al. KRAS analysis in colorectal carcinoma: analytical aspects of Pyrosequencing and allele-specific PCR in clinical practice. BMC Cancer. 2010;10:660. doi: 10.1186/1471-2407-10-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayden RT, et al. Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J Clin Microbiol. 2013;51:540–546. doi: 10.1128/JCM.02620-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forbes SA, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) The mutational status of KRAS, BRAF and NRAS was assessed in plasma by ddPCR and matched tissue sample of 100 colorectal cancer patients. Green indicates that mutations identified in plasma and tissues are concordant; yellow denoted cases in which mutations were identified only in plasma; grey highlights mutations identified only in tissue.
(b) Levels of genome equivalents (GE, log scale, ascending order) per milliliter of plasma in the same colorectal cancer patients reported in panel (a). The colors of the bars identify genotype (grey: RAS/BRAF WT-negative; lilac: KRAS mutant; light blue: NRAS mutant; light red: KRAS and BRAF mutant; green: BRAF mutant).
Supplementary Figure 2: Bar graph shows cell viability at clinically relevant concentration of 10 μg/ml of cetuximab in the resistant cell populations (DiFi A and DiFi B) and in the cells after 160 days of antibody withdrawal. Statistical differences were calculated by Student’s t-Test. Data are expressed as means ± SD of three independent experiments. ***p value ≤0.001.
Supplementary Table 1: Clinical characteristics of patients included in the plasma-tissue concordance analysis (n=100).
Supplementary Table 2: The table lists the ddPCR events (mutant cases only) obtained analyzing ctDNA of patients included in the concordance study (Supplementary Figure 1). Poisson corrected (95% C.I.) and total number of copies per sample analyzed is also shown.
Supplementary Table 3: (a) The table shows ddPCR measurements. Plasma samples were initially tested for KRAS, BRAF, NRAS and EGFR mutations, and for KRAS and MET copy number alterations. Negative cases were subjected to NGS analysis.
(b) Description of the IRCC-_target (Therapeutically Applicable Genes for Effective Treatment) panel. The table indicates gene name, exons and bases analyzed by next generation sequencing (Illumina MiSeq). The panel is designed to capture the entire coding region of listed genes using plasma DNA as a source.
Supplementary Table 4: Alterations in plasma DNA identified in patients resistant to anti-EGFR treatment. Raw data obtained by digital-based PCR and IRCC _target panel (mutated and wild type reads, respectively). N.I. not identified. Poisson corrected (95% C.I.) and total number of copies per sample analyzed is also shown.
Supplementary Table 5: The table shows APC and TP53 alterations found in patients through Next Generation Sequencing of plasma DNA (raw data). N.I. not identified.
Supplementary Table 6: KRAS mutations identified by ddPCR and BEAMing at indicated time points in plasma of patients who developed acquired resistance to first-line chemotherapy plus anti-EGFR and then received other lines of treatment (a) or were re-challenged with anti-EGFR drugs (b). Poisson corrected (95% C.I.) and total number of copies per sample analyzed is also shown. cetux: cetuximab; panit: panitumumab. N.A. not available.