

Abstract
Historically, the nature and extent of host damage by a microbe were considered highly dependent on virulence attributes of the microbe. However, it has become clear that disease is a complex outcome which can arise because of pathogen-mediated damage, host-mediated damage, or both, with active participation from the host microbiota. This awareness led to the formulation of the damage response framework (DRF), a revolutionary concept that defined microbial virulence as a function of host immunity. The DRF outlines six classifications of host damage outcomes based on the microbe and the strength of the immune response. In this review, we revisit this concept from the perspective of Candida albicans, a microbial pathogen uniquely adapted to its human host. This fungus commonly colonizes various anatomical sites without causing notable damage. However, depending on environmental conditions, a diverse array of diseases may occur, ranging from mucosal to invasive systemic infections resulting in microbe-mediated and/or host-mediated damage. Remarkably, C. albicans infections can fit into all six DRF classifications, depending on the anatomical site and associated host immune response. Here, we highlight some of these diverse and site-specific diseases and how they fit the DRF classifications, and we describe the animal models available to uncover pathogenic mechanisms and related host immune responses.
INTRODUCTION
Historically, the nature and extent of host damage by an opportunistic microbe were considered highly dependent on virulence attributes of the microbe. However, it is now quite clear that damage to the host during infection is also reflective of the immune status of the host and often mediated by host responses. Therefore, disease itself is a complex outcome which can arise because of pathogen-mediated damage, host-mediated damage, or both. Hence, in many interactions between pathogens and normal hosts, there is a continuum between pathogen-mediated and host-mediated damage, which results in disease only when the nature of the damage impairs the normal function of the host.
In this review, we revisit the concept of the damage response framework (DRF) initially introduced by Casadevall and Pirofski in 1999, which explains microbial pathogenesis as the outcome of an interaction between the host and microbe (1). The host-relevant outcome is microbe- and/or host-mediated damage, which provides a basis for a new pathogen classification scheme based on the amount of damage as a function of the host immune response (1). Here, we utilize the DRF to explore the pathogenesis of a ubiquitous fungal pathogen, Candida albicans, which is uniquely adapted to its human host, existing often as a harmless commensal at various mucosal sites (2, 3). As a pathogen, however, C. albicans is responsible for a wide range of infections, both mucosal and systemic, in both immunocompetent and immunocompromised individuals (4, 5). With no known environmental reservoir, C. albicans is acquired at or shortly after birth, often being transmitted from mother to child, and can remain a commensal or cause neonatal infections. Therefore, the DRF provides an excellent means to illustrate and explain all the various forms of candidiasis in the context of host response and host damage.
Much of what we know about C. albicans pathogenesis and host immune responses has come from animal model systems. The large panel of clinically relevant models available to study systemic and mucosal candidiasis highlights the diversity of niches in which this fungus can cause disease (4, 6). The rodent is ideal for studying C. albicans pathogenesis due to the demonstrated similarity to the human disease process and host immune responses. Experimental animal models of mucosal candidiasis, in particular, have been invaluable in elucidating the various compartmentalized host immune responses to C. albicans at the various mucosal sites. Similarly, rodent models have been instrumental in understanding host responses during the initiation and progression of systemic C. albicans infection. The main advantage of these models is that they allow for manipulations, genetically and pharmacologically, in order to mimic susceptible hosts, greatly contributing to our knowledge of factors leading to host susceptibility to Candida infections (7).
The versatility of C. albicans in its pathogenic potential and the diversity of its disease entities, depending on infection site, position it in a unique situation: based on the various clinical manifestations as a function of host immune status, this pathogen may arguably fit within each of the classes outlined by the DRF (1, 8). Accordingly, here we highlight some of the varied and site-specific diseases caused by this human pathogen (Fig. 1) in the context of the DRF. Within each class, disease pathogenesis is explored using both clinical data and data from animal models that support the DRF classification. Of note, while this perspective article focuses specifically on C. albicans, some of the literature cited to support the classifications of C. albicans within the DRF does not discriminate between individual Candida species. Accordingly, the classifications ultimately may be extended to other Candida species as well.
FIG 1.

Anatomical defenses and host damage associated with the various manifestations of candidiasis. The illustration shows the diverse and site-specific diseases caused by C. albicans, highlighting the disease pathogenesis in each case and site-specific host immune responses.
DAMAGE RESPONSE FRAMEWORK
The premise of the DRF is that damage to the host can be mediated by either the pathogen or the host, and therefore microbial virulence is a measure of the outcome of an interaction between a microbe and a susceptible host. This integrated view of pathogenesis in which the contributions of both the microbe and the host are incorporated became essential in light of the surge in infections caused by microbes once considered to be members of the commensal microbiota. It is well recognized that medical devices, immunosuppressive therapies, and diseases such as AIDS have created patient populations highly susceptible to infections. This awareness ultimately led to the development of the concept of microbial opportunism and opportunistic pathogens, viewed as microbes with pathogenic potential that becomes manifest in the setting of a weakened host immune system (8, 9).
The DRF is based on the core principle that there are no exclusive pathogens, commensals, or opportunists, but that microbial pathogenesis requires a microbe and a host to interact, with the relevant outcome being damage to the host. C. albicans pathogenesis has often been described using phrases based on the concept that C. albicans is the exclusive causative agent, such as “converts from commensal to pathogen” or exists as an “opportunistic pathogen.” Under the tenets of the DRF, these phrases or terminologies are invalid and should be avoided. More importantly, the ensuing damage results from microbial and/or host factors, with the host response contributing to microbe-mediated damage. Further, the host damage can stem from either weak or strong responses to microbes. This concept is illustrated in graphs depicting six different levels of damage to the host as a function of the range of host immune responses with the potential for considerable damage occurring at either or both extremes of the host response (1, 9). In a global context, the DRF shifted the focus away from exclusively microbe-mediated damage by emphasizing the role of the host as a contributor to damage. Importantly, in addition to host and microbe, this flexible conceptual framework also focuses on outcomes that are a function of multiple factors, which include the environment and time.
DRF CLASSIFICATION OF MICROBIAL SPECIES
Class 1: pathogens that cause damage only in the setting of weak immune responses.
Microorganisms placed in DRF class 1 are those usually considered opportunistic or commensal and are associated with disease only in individuals with impaired immune function.
Class 2: pathogens that cause damage either in hosts with weak immune responses or in the setting of normal immune responses.
In DRF category 2 are microorganisms that cause host damage by both host- and pathogen-mediated mechanisms and are viewed as opportunists because their prevalence is higher in groups with impaired immune function. However, the capacity of class 2 microorganisms to mediate disease in individuals with apparently normal immunity is indicative of the expression of microbial characteristics that promote their ability to evade normal host defenses that would otherwise eliminate them.
Class 3: pathogens that cause damage in the setting of appropriate immune responses and produce damage at both ends of the continuum of immune responses.
In DRF class 3, microorganisms can cause damage in normal hosts, which is amplified in the settings of either weak or strong immune responses.
Class 4: pathogens that cause damage primarily at the extremes of both weak and strong immune responses.
In normal hosts, microbes in DRF class 4 cause relatively little damage. However, a weak immune response can promote infection and pathogen-mediated damage, while a strong immune response can produce excessive inflammation.
Class 5: pathogens that cause damage across the spectrum of immune responses, but damage can be enhanced by strong immune responses.
In DRF class 5, microorganisms cause infections that result in pathogen-mediated damage but the damage is associated with protracted or chronic damage resulting from an excessive or inappropriate immune response.
Class 6: pathogens that can cause damage only as a result of strong immune responses.
DRF class 6 was first thought to be a largely theoretical category to describe a growing list of diseases that may have a microbial etiology not associated with impaired immune function. These organisms may also be members of the normal microbiota and confer a benefit to the host in settings of normal or weak responses.
According to the criteria for each class, C. albicans was initially categorized as a class 2 pathogen. However, unlike most pathogens, C. albicans is a human commensal with no known environmental reservoir, making it highly host adapted at various anatomical sites. Given its adaptability and the diversity of its pathogenic potential, classifying C. albicans within one of the DRF classes does not account for the full complexity of its pathogenesis. In our review of the various candidal diseases, it became clear that a select spectrum of C. albicans infections can be categorized within each of the six classifications, depending on the site and manifestation of infection and the nature of the host immune response.
CANDIDA ALBICANS: A VERSATILE FUNGAL PATHOGEN
Among fungal species, C. albicans is the most common human pathogen, causing diseases ranging from superficial mucosal to life-threatening systemic infections (2, 10–12). As part of the commensal human microbiota, C. albicans asymptomatically colonizes the gastrointestinal tract, oral cavity, and reproductive tract of healthy individuals (13, 14). At these various sites, its proliferation is controlled by the host immune system and colonization resistance is provided by other members of the microbiota. Therefore, its isolation from mucosal tissues does not necessarily indicate a diseased state (3, 4, 15, 16). However, disruptions in host immune status, barrier function, or local microenvironment (including microbiota composition) can lead to changes in C. albicans growth and physiology (gene/protein expression, metabolism, morphology), which can cause host damage, both microbe mediated and/or host mediated. Depending on the magnitude of damage, disease ensues, which can be acute and/or recurrent in nature (4, 17–19). As a result, C. albicans infections are recognized as a serious public health challenge with high clinical and socio-economic importance, representing one of the most prevalent agents identified in nosocomial infections (10, 20, 21).
As with other microbial pathogens, the ability of C. albicans to adhere to host surfaces is a prerequisite for both successful colonization and persistence during infection. Unsurprisingly, the majority of C. albicans infections are associated with its ability to form biofilms on host tissue or abiotic surfaces (6, 18, 19, 22–24). Biofilms are structured communities of surface-associated microbial populations embedded in a matrix of extracellular polysaccharides proposed to provide protection for biofilm cells (24–27). C. albicans biofilm-embedded cells are afforded a stable environment where they are protected from the host immune system and can tolerate extremely high concentrations of antimicrobials (28). The impact of these biofilms on public health is dramatic, as cells released from biofilms formed on implanted biomaterials, including vascular catheters, can potentiate systemic infections. Thus, C. albicans biofilm contamination of medical devices has been implicated as a risk factor for increased patient mortality (18, 19). The growing usage of and need for implanted medical devices and central venous catheters in managing patient care are important reasons why the incidence of C. albicans infections have steadily increased. In fact, C. albicans and other non-albicans Candida species are currently ranked by the Centers for Disease Control and Prevention as the third most commonly isolated bloodstream pathogens in hospitalized patients, with a mortality rate of up to 50% (6, 19, 24, 29). Overall, the annual cost of antifungal therapies for device-associated Candida species infections in the United States alone is estimated at $2.6 billion (30, 31).
A number of properties and virulence factors attributed to C. albicans are known to promote its biofilm-forming potential and its persistence within the host. First and foremost is the property of morphological transition, as the distinct morphological states (yeast, pseudohyphae, hyphae) of C. albicans dictate phases of colonization, growth, and dissemination; the yeast form has been associated with both initial attachment and dissemination, while the filamentous hyphal form enables C. albicans to form a biofilm and invade host tissue (24, 32). Importantly, mutants locked in either the yeast or pseudohyphal phase are generally less virulent and more easily cleared from tissues during intravenous infections than hyphal-phase-locked mutants (33). Another important virulence property is adhesion mediated by cell wall adhesins; most notable are the agglutinin-like sequence family (Als) and a hyphal wall protein (Hwp1) that is crucial for attachment of C. albicans to receptors on host tissues (30, 34). In addition, C. albicans also produces several extracellular secreted enzymes, such as lipases, esterases, and secreted aspartyl proteinases (Saps), as well as hemolysins, that are involved in host tissue invasion and nutrient acquisition (35). Compounded by the increase in resistance to the relatively limited number of available antifungal drugs, the continued increase in the incidence of C. albicans (including non-albicans Candida species) infections highlights the need for elucidating the fundamental pathogenic determinants of C. albicans and the reciprocal host protective mechanisms against this fungus (4).
FITTING C. ALBICANS WITHIN THE DAMAGE RESPONSE FRAMEWORK
The ability of C. albicans to adapt to various and changing host environments, such as immune status and microbiota, is key to its ability to cause such a wide range of diseases. Here, we highlight some of the most common candidal diseases and, based on infection site and nature of host immune response, we propose their classification within the DRF (Fig. 2). Furthermore, host and pathogen factors mediating damage to the host and the animal models available to study the various diseases are described in Table 1.
FIG 2.

Damage response framework curves. Individual graphs are included for each class, with an associated Candida infection. (A) Class 1, pathogens that cause damage only in situations of weak immune responses (e.g., oral candidiasis). (B) Class 2, pathogens that cause damage either in hosts with weak immune responses or in the setting of normal immune responses (e.g., invasive candidiasis). (C) Class 3, pathogens that cause damage in the setting of appropriate immune responses and produce damage at both ends of the continuum of immune responses (e.g., intra-abdominal candidiasis). (D) Class 4, pathogens that cause damage primarily at the extremes of both weak and strong immune responses (e.g., gastrointestinal candidiasis). (E) Class 5, pathogens that cause damage across the spectrum of immune responses, but damage can be enhanced by strong immune responses (e.g., denture stomatitis). (F) Class 6, microorganisms that can cause damage only under conditions of strong immune responses (e.g., vaginal candidiasis). (Adapted from reference 1 with permission.)
TABLE 1.
Examples of C. albicans infections, host predisposing factors, and animal models for each DRF class
| DRF class | Candidal infection example | Primary predisposing factor(s) | Rodent model(s) |
|---|---|---|---|
| Class 1 | Oropharyngeal candidiasis | Immunocompromised state (AIDS, cancer patients) | Mouse oral infection model |
| Class 2 | Hematogenously disseminated candidiasis | GI tract mucosal disturbances; intravenous catheters; surgery; use of broad-spectrum antibiotics; neutropenia and other immunosuppressed conditions | Rodent intravenous, subcutaneous catheter and GI tract translocation models |
| Class 3 | Intra-abdominal candidiasis | Peritoneal dialysis; bowel surgery; GI tract perforation; hepatobiliary leaks | Mouse peritonitis and abscess models |
| Class 4 | Gastrointestinal candidiasis | Immune deficiency; cancer; antibiotic or immunosuppressive therapy | Mouse mucosal GI model |
| Class 5 | Denture stomatitis | Denture usage; poor oral hygiene | Rat denture model |
| Class 6 | Vulvovaginal candidiasis | Antibiotic or hormone therapy; estrogen replacement; vaginal microbiome dysbiosis | Rodent vaginal infection models |
Class 1 (damage occurs only in situations of weakened or compromised immune system): oropharyngeal candidiasis.
Oropharyngeal candidiasis (OPC), commonly known as thrush, encompasses infections of the tongue and other oral mucous membranes and may extend into the pharynx (Fig. 2A). OPC can present as white curd-like lesions (pseudomembranous) (Fig. 3A) or reddened patches (erythematous) (Fig. 3B). The diagnoses of both manifestations of OPC are essentially clinical and are based on the recognition of the lesions by a health care provider and can be confirmed by microscopic identification or culture of Candida (31, 36). OPC is rare in healthy adults, occurring almost exclusively in immunocompromised patients. In fact, OPC is the most common oral infection in HIV-positive individuals, although the incidence has been reduced significantly with antiretroviral therapies (ART) (16, 31, 37, 38). In addition to HIV infection, OPC occurs in 35% of cancer patients who have recently received chemotherapy, in the elderly and infants, and under conditions of malnutrition or local immune suppression (e.g., suppression resulting from use of steroid inhalers for asthma). Further, patients with Sjogren's syndrome, diabetes, or other metabolic or hormonal disorders or those on antibiotics are also predisposed to OPC (30, 37–39).
FIG 3.

Clinical manifestations of oral candidiasis. (A) Pseudomembranous candidiasis is characterized by white plaques formed on the tongue and the buccal mucosa. (B) Erythematous candidiasis example, showing the subtle red lesions on the tongue, which can also occur on the palate.
OPC is a biofilm-associated disease that results from the adherence of yeast cells to mucosal tissue, followed by hyphal invasion, which is associated with secreted proteolytic enzyme expression (40). Therefore, under conditions of weak oral immune responses, C. albicans invokes considerable damage upon the host, highlighting its class 1 designation as an opportunist. However, as part of the commensal microbiota in the oral cavity, C. albicans can exist asymptomatically in individuals with normal or strong protective host defenses. Under these conditions, C. albicans has the potential to develop a symbiotic relationship with the host, imparting a degree of protection against potentially harmful microbes that come in contact with the oral cavity (3). This potential for benefit in the context of strong host responses and for damage in the face of weak responses defines a DRF class 1 pathogen.
Experimental and clinical evidence has provided significant advances in terms of understanding more specifically what contributes to host defense against OPC. While it is clear that innate defenses (salivary flow, antimicrobial peptides) help limit C. albicans overgrowth in the oral cavity, it became quite clear during the HIV epidemic that CD4+ T cells were the primary protective host defense mechanism against OPC (37, 38, 41–46). An established mouse model of OPC has been widely used for investigating Candida virulence factors, immune mechanisms against candidiasis, and the efficacy of antifungal agents (4). In this relatively simple model, mice are rendered susceptible to oral infection by injection with cortisone acetate prior to sublingual inoculation with C. albicans (47). This process results in a reproducible level of infection, the histopathology (Fig. 4) of which mimics that of pseudomembranous OPC in patients (47, 48). The availability of a CD4/HIVMutA transgenic mouse model that develops AIDS-like disease was instrumental in demonstrating that HIV-mediated loss of CD4+ T cells underlies susceptibility to mucosal candidiasis (49). Originally, it was thought that the primary protective mechanism by CD4+ T cells was the Th1 phagocyte-dependent response (37). However, the discovery of the Th17 axis and subsequent in vivo studies using the OPC model identified the CD4+ Th17 response as the primary protective response (17, 50). Importantly, in the absence of interleukin-17 (IL-17) and related cytokines IL-12 and gamma interferon (IFN-γ), innate cell recruitment, activation, and phagocytosis of C. albicans cells fail to occur. Using knockout mice, defense against OPC was shown to be more dependent on Th17-type than Th1-type immunity, with Th17-deficient mice exhibiting impaired neutrophil recruitment and high fungal burdens (51). It is also noteworthy that Th17 cells enhance the expression of antimicrobial peptides, including defensins and histatins, which are produced by oral epithelial cells and salivary glands, respectively (52). Therefore, the Th1/Th17 immune response is central to combating and preventing oral candidal infections under immunocompetent conditions, providing a large-scale benefit with little to no damage from a strong immune response. Although the incidence of OPC can be high in the absence of ART, it is postulated that under CD4+ Th1/Th17 immunocompromised conditions, secondary host defense mechanisms can provide some protection against infection. These include oral epithelial cells via annexin-A1 and CD8+ T cells, although the mechanisms are not well understood (37, 53, 54).
FIG 4.

Mouse model of oral candidiasis. (A) An infected mouse exhibiting clinical signs of advanced candidiasis on the surface of the tongue 4 days post-sublingual infection with C. albicans. (B) Histopathology of an infected tongue tissue section, demonstrating the extensive presence of C. albicans around the periphery of the tongue. Hyphae can be seen penetrating the subepithelial tissue, along with a marked presence of host inflammatory cells. (C) Magnified image of tongue tissue, revealing the depth of hyphal invasion into the subepithelium (arrows). Bar, 20 μm. (D) Scanning electron micrograph of excised tongue showing the thick biofilm formed on the outer epithelial surface consisting of C. albicans hyphae invading the subepithelium. (E) Higher-magnification image of the outer surface of the tongue showing the epithelium spiny layer with hyphae penetrating through the surface. (F) Significant gap in the tissue caused by hyphae invading from the sublingual area as it emerges through the tongue surface. (Reprinted from reference 166.)
Class 2 (damage occurs in hosts with weak or normal immune responses): hematogenously disseminated candidiasis.
Hematogenously disseminated candidiasis (HDC) arises when C. albicans gains access to the bloodstream (candidemia), leading to deep-seated candidiasis (DSC), which is defined as C. albicans infection of internal organs (Fig. 2B) (55). Overall, C. albicans, along with other non-albicans Candida species, is the second leading cause of invasive infection in North American intensive care units (ICUs), causing approximately 18% of infections, inclusive of all origins of infection (56). HDC is a leading cause of mycosis-associated mortality, and C. albicans, along with other non-albicans species, is currently ranked the third leading cause of bloodstream infections (BSI) in hospitalized patients in the United States (6, 19, 29, 57). Once in the bloodstream, C. albicans can infect a wide range of _target organs, including the kidney, spleen, liver, heart, gastrointestinal (GI) tract, and lungs (58). Within _target organs, invasive infections lead to significant tissue damage (59, 60). One of the major problems in managing patients with HDC is the difficulty in establishing the diagnosis, because bloodstream Candida infections tend to present clinically with nonspecific symptoms similar to those seen with systemic bacterial infections (7). More importantly, the sensitivity of blood cultures, the current diagnostic gold standard, is only ∼50% (55). The nonspecific presentation and poor performance of blood culturing often lead to delays in the initiation of effective antifungal therapy, which contributes to the high mortality rates associated with Candida bloodstream infections (61).
HDC most commonly stems from translocation across a damaged GI tract mucosa into the systemic circulation or from infected vascular catheters or other access devices. GI tract translocation is often seen in neutropenic and other immunocompromised hosts, in particular cancer patients with mucositis stemming from chemotherapy (62, 63). Central line catheter-associated infections typically emerge from biofilms formed on catheters, providing a niche for microorganisms where they are protected from both the host immune system and antimicrobials (24). Successful therapy of these foreign body-associated infections relies on device removal in most instances. Catheter-associated biofilms provide the opportunity for direct inoculation of C. albicans into the bloodstream. If the device is not removed, continual seeding of the bloodstream can overwhelm intact host defenses in nonneutropenic patients (normal host response) and cause significant mortality despite antifungal therapy and often times an intact immune system (64–66). In central line catheter-associated bloodstream infections, the weak defense is related to a breach in the barrier protecting the systemic circulation, as opposed to a more specific immune deficiency. Damage is amplified in acutely ill patients with additional immune deficiencies who acquire HDC. Thus, the fact that HDC can occur in the presence of both weak and normal defenses, albeit disproportionately, highlights the DRF class 2 designation of C. albicans and its behavior as an “equal opportunist” under such circumstances. The ability of C. albicans to cause systemic disease under conditions other than immune deficiencies highlights the fact that the term opportunistic does not always apply to this pathogen.
Animal models of HDC have been essential for our understanding of disease initiation and progression and for development of more effective diagnostics and therapies. In the standard HDC model, mice are infected intravenously (i.v.) via lateral tail vein injection, which results in rapid dissemination of C. albicans in a reproducible fashion (67, 68). Although fungal growth is controlled in the spleen and to a lesser extent in the liver, due to the presence of resident phagocytes, C. albicans causes extensive disease in the kidneys that is accompanied by increased immune infiltrates (68, 69). Whereas with lower inocula tissue burdens and host responses within the kidney are controlled, with higher inocula sepsis might occur, leading to high fatality rates (67, 70). In another model that has been useful for studying C. albicans dissemination to kidneys and other organs, a central venous catheter (CVC) is used to study C. albicans biofilm formation in catheters implanted in the central venous system of rodents. The benefit of this model is that it provides a realistic model of the central venous catheter infection site, catheter-vascular flow, and host serum proteins and other blood components, as well as humoral immunity (71–73).
With the i.v. HDC model, studies have shown that immune detection of C. albicans is accomplished by pattern recognition receptors (PRRs) on innate immune cells, where host Toll-like receptors (TLRs) and dectin receptors recognize cell wall components of C. albicans, such as mannans and glucans (74). Recognition of C. albicans by these PRRs activates the expression and secretion of a bevy of inflammatory cytokines, which are pivotal for optimal activation of phagocytes. In addition, the CD4+ T cell-independent IL-17 response is also notable and is considered essential to an effective antifungal response during systemic infection. In fact, animal studies have shown that infection of IL-17Ra knockouts results in greater mortality, higher fungal burdens, and significantly decreased neutrophil recruitment in an HDC model of infection (75, 76). In addition, IL-17 can also stimulate the production of chemokines such as IL-8, CXCL1, CXCL2, CCL20, and CCL7, which recruit polymorphonuclear leukocytes (77). Non-CD4+ T cell sources of IL-17 include γδ T cells, which are expanded in HIV+ patients and produce IL-17A in response to C. albicans (78). In a murine model of HDC, the major source of IL-17A was lung γδ T cells, which were required for optimal neutrophil recruitment and control of infection (79). NK cells are not required for defense against intravenous infection in immunocompetent animals, but lack of NK cells in T/B-cell deficient SCID mice led to increased susceptibility to infection (80).
There are also several murine models of candidal dissemination via GI tract colonization that have been used to evaluate either host or fungal factors that promote disease (reviewed in reference 81). Most models rely upon antibiotic treatment to promote C. albicans GI tract colonization and upon immunosuppressive agents, such as 5-fluoruracil or cyclophosphamide. The liver is the most reliable _target organ for monitoring dissemination (82, 83), presumably because translocation from the GI mucosa can occur via the portal circulation or biliary tree. To more specifically address which facets of host immunity are required to prevent dissemination, investigators sequentially disrupted specific host defense elements. Surprisingly, the findings demonstrated that depletion of lymphocytes, neutrophils, or macrophages did not predispose to candidal dissemination (83). Similarly, disruption of enteric mucosal integrity with dextran sulfate was also not sufficient to induce disseminated candidiasis. However, when agents that ablated neutrophils and also caused gut barrier disruption were administered, lethal disseminated candidiasis developed (83). Using germfree animals to facilitate C. albicans colonization, another study found that mice lacking phagocyte oxidase (Phox−/−) and nitric oxide synthase 2 (NOS2−/−) were susceptible to not only GI mucosal colonization but also dissemination and mortality (84). Overall, these studies underscore the importance of an intact GI tract barrier and oxidative pathways of granulocytes to defend against both mucosal infection and the ability to disseminate from the GI tract.
Class 3 (damage occurs throughout the continuum of immune responses but is amplified at extremes of both weak and strong immune responses): intra-abdominal candidasis.
Intra-abdominal candidaisis (IAC) results from entry of C. albicans into the abdominal cavity through translocation across the GI mucosal barrier or by direct inoculation, which can occur via contaminated peritoneal dialysis catheters or as a result of perforation of the GI tract (85–88) (Fig. 2C). While HDC is a DRF class 2 disease, IAC is considered a class 3 disease. Even in otherwise-immune-sufficient patients, inoculation of the abdominal cavity can cause peritonitis, an inflammatory disease of the lining of the abdominal cavity, which could be amplified with uncontrolled host response, leading to host-mediated damage. The cardinal clinical signs and symptoms of peritonitis include fevers, chills, and abdominal pain, and complications of peritonitis include invasion of adjacent organs, such as the liver and spleen, and/or abscess formation (83, 89). Treatment usually entails both source control with drainage and surgical intervention as well as antifungal therapy. In rare instances, intra-abdominal candidiasis can disseminate via the bloodstream (HDC) and cause DSC. This may be exacerbated in immunocompromised patients who lack innate defenses that could help to contain or control C. albicans within the peritoneal cavity. It is estimated that secondary candidemia occurs in 5 to 20% of intra-abdominal candidiasis cases (90). IAC is distinct from HDC (class 2) in that host damage is equally severe during a strong response, due to the local nature of the peritoneal immune response, characterized by neutrophil recruitment and inflammatory cytokine production (90). While defects in innate defenses are associated with susceptibility to IAC, the robust response could ultimately compound the outcome of infection (91). The severity of these infections at either end of the host response continuum, with some damage occurring even under optimal host responsiveness, leads to C. albicans being classified in this scenario as class 3, with behavior as a “bipolar” pathogen. It is not clear, however, how pathogenesis and host responses in blood-borne C. albicans infections resulting from intra-abdominal candidiasis compare to those of bloodstream infections that originate through other mechanisms.
One unique feature of intra-abdominal infections involving C. albicans is that they are often polymicrobial, and fungal-bacterial mixed infections are associated with higher mortality rates than polymicrobial bacterial infections (92–95). Recently, several animal models were developed to study IAC in the context of bacterial contamination or coinfection (89, 96, 97). In one study, intraperitoneal inoculation of C. albicans along with sterile feces resulted in peritonitis within 6 h, characterized by a rise in pH and neutrophil influx into the peritoneal fluid. Organ invasion by hyphae and early abscess formation were evident 6 and 24 h after infection, respectively (89). It was postulated that the bacterial by-products acted synergistically with the fungi to cause disease. Another model using coinoculation of C. albicans with the bacterial pathogen Staphylococcus aureus showed similar synergistic effects on mortality compared with monomicrobial inoculation; the synergistic effects included dissemination of both species and sepsis (97). Intriguingly, however, mortality in the C. albicans-S. aureus coinfection model was associated with dramatic increases in inflammatory cytokines both locally and systemically very early postinoculation, but no increases in microbial burdens (96). These findings indicate that the host response is a key mediator of host damage, emphasizing the class 3 designation. Furthermore, a recent study examining the role of C. albicans morphogenesis in the disease process indicated that, unlike in the majority of C. albicans infections, hyphal formation is not a major contributor to the pathogenesis of C. albicans-bacterial pathogen peritonitis, although the signaling pathways governing morphogenesis are required (96, 98). Further, studies using a sublethal monomicrobial inoculum demonstrated a role for a secreted aspartyl protease (Sap6) in mediating peritoneal organ invasion and tissue damage independent of hyphal formation, supporting the concept that morphogenesis per se is not a virulence determinant during peritoneal infection (99). These mouse models provide powerful tools for measuring the relative virulence of infecting strains and evaluating in vivo gene expression. Additionally, they are also useful for understanding how C. albicans adapts to diverse host environments and for studying host responses within the peritoneal cavity. Importantly, the models are invaluable for identifying novel approaches for diagnosing, preventing, and treating intra-abdominal candidiasis and invasive candidiasis of intra-abdominal origin (89).
Class 4 (damage occurs primarily at the extremes of both weak and strong immune responses): gastrointestinal candidiasis.
Environmental factors such as antibiotic use and diet, which can alter bacterial microbiota levels and lower colonization resistance, are believed to lead to overgrowth of C. albicans in the GI tract in humans (100) (Fig. 2D). While this has been demonstrated using murine models (81), monitoring human C. albicans intestinal colonization levels is more problematic. However, more recent clinical studies examining fecal loads have demonstrated increased Candida levels with antibotic treatment (101). Overgrowth may facilitate localized mucosal infection and/or generalized GI tract disturbances, but the validity of GI candidiasis via overgrowth as a clinical entity is controversial (102, 103). This has been difficult to verify due to the lack of specific symptoms (belching, bloating, indigestion, nausea, diarrhea, and gas) and lack of diagnostic tests. Patients considered at risk for this type of candidiasis include cancer and transplant patients receiving immunosuppressive therapy or prolonged antibiotic prophylaxis, and therefore this represents a disease associated with impaired host immunity (104, 105). At the other end of the host response spectrum, experimental and clinical evidence also suggests that GI tract C. albicans colonization may be a cofactor for inflammatory diseases, which could be considered host-mediated damage. Therefore, based on current, albeit limited data, GI candidiasis fits within the class 4 designation, with C. albicans behaving in this case as an immunoreactive opportunist, causing host damage in the context of both strong and weak host responses.
It is clear from various animal models that the major defense against C. albicans overgrowth in the GI tract is the presence of the bacterial microbiota and normal gut peristalsis. Antibiotic treatment or the use of germfree gnotobiotic animals enhances consistent colonization of the gut (81). In terms of innate immunity, mice with congenital granulocytic cell deficiencies are susceptible to GI candidiasis but mount C. albicans-specific adaptive responses and eventually clear the infection (106). Treatment with a NOS inhibitor increased the severity of GI tract infection; however, the reactive chemical species responsible is likely peroxynitrite, as nitric oxide is not directly candidicidal in vitro (107). Studies using congenitally athymic T cell-deficient SCID mice or CD4+ T cell-depleted mice showed that T cells are critical for effective protection against GI candidiasis (108–112).
There are several experimental and clinical studies that support the notion that C. albicans overgrowth in the GI tract exacerbates inflammatory diseases, promoting local and systemic hyperreactivity. In one such murine model, inoculation of the GI tract with C. albicans was accompanied by dramatic increases in pulmonary allergic responses to experimental allergens (113, 114). These studies utilized immunocompetent mice and did not involve previous systemic antigen priming, as is typically used for inducing hypersensitivity to these allergens. There was also no evidence of microbial growth in the lungs or inflammation in the GI tract in this model. Combined, the findings from these studies demonstrate experimentally that disruption of the microbiota, including fungal GI tract colonization, can reduce tolerance to aeroallergens. Mechanistically, it has been suggested that aberrant immune responses to mucosal fungal colonization in the GI tract are responsible for disrupting normal tolerance mechanisms that control hyperreactivity. For example, mice deficient in dectin-1, which recognizes β-(1,3)-glucan in the fungal cell wall, are more susceptible to induction of colitis, which was associated with increases in intestinal fungi, including Candida spp. (115).
In humans, polymorphisms in dectin-1 are associated with severe medically refractive ulcerative colitis more strongly than in patients with less severe disease (115). Further, GI tract inflammation was shown to promote C. albicans colonization in chemically induced colitis in mice, which augments inflammatory responses via the PRR galectin-3 (116). C. albicans colonization also primed expansion of Th17 cells with commensal specificity, driving intestinal inflammation (117). Likewise, C. albicans colonization of the GI tract in germfree animals induced gastritis, demonstrating the inflammatory potential of this “benign” microbiota member (13). In humans, it has been noted that the fungal microbiota composition differs in patients with inflammatory bowel disease (IBD) from that in healthy individuals, with increases in fungal load and diversity (118). Therefore, PRRs that recognize fungal moieties may help promote responses to commensals in the GI tract and initiate damage. It is as yet unclear whether GI tract carriage of C. albicans in healthy hosts (up to 50% of individuals) imparts any benefit, but it is often asymptomatic and hence likely does not normally cause damage in the context of normal host responses (119).
Class 5 (damage occurs across the spectrum of immune responses, but damage is enhanced by strong immune responses): denture stomatitis.
Candida-associated denture stomatitis (DS) is the most common form of oral candidal infections in otherwise-healthy individuals and is primarily caused by C. albicans (120, 121) (Fig. 2E). Denture stomatitis is a chronic disease characterized by localized or generalized inflammation of the denture-bearing mucosa and affects patients wearing removable partial or complete dental prostheses (Fig. 5). Symptoms of Candida-associated DS range from mild to severe, including palatal edema, painful inflammation, and papillary hyperplasia (small pebble-like sores) (120). This condition is prevalent in approximately 70% of denture wearers, with very high recurrence rates despite antifungal therapy (120–122). C. albicans readily adheres to the acrylic denture material and forms biofilms, which could result in continuous seeding of biofilm-associated organisms on the palatal tissue. This is particularly exacerbated in situations where an ill-fitting denture and frictional irritation damage the normally protective mucosal barrier, allowing infiltration of C. albicans into the tissue (30). Under these conditions, a strong immune response, likely chronic in nature, will invoke considerable damage to the host, whereas a moderate response results in less damage, highlighting the DRF class 5 designation of C. albicans in DS as an immunoreactive pathogen. We recognize that assigning this class may be somewhat premature based on available data; clarification/confirmation of this assignment should be forthcoming, pending further research.
FIG 5.
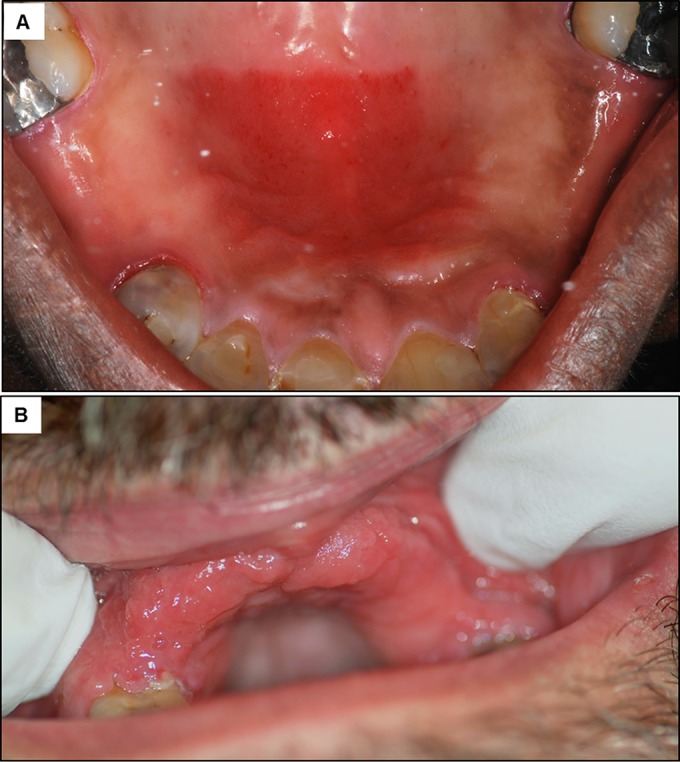
Clinical manifestations of denture stomatitis. (A) Red inflammatory lesions formed on the denture-associated palatal tissue in a patient with a partial denture. (B) Inflammation of the gingival tissue in a patient with a full denture.
To study Candida-associated DS in vivo, a rat acrylic denture model that recapitulates features of DS was developed (123, 124) (Fig. 6). In this model, a custom-fitted fixed and removable denture system (U.S. patent 8,753,113 [125]) is installed in the rat palate, followed by inoculation with C. albicans (123). This system allows longitudinal analysis of rats, which become chronically and stably infected while the denture is in place. Microscopic analysis demonstrated biofilm formation on both the palate and denture by 4 weeks postinoculation with no obvious tissue invasion (superficial infection). Rats also exhibited gradual worsening of erythematous palatal inflammation, while histopathology of the palate tissue revealed neutrophil infiltration, indicative of a strong inflammatory response (123). Using a similar rat model of DS, a separate study indicated that C. albicans SAP expression correlated with histological changes, suggesting that specific fungal virulence factors promote tissue damage (126).
FIG 6.

Rat model of Candida-associated denture stomatitis. Individual impressions were made for each rat, using vinyl polysiloxane impression material. Molds were made from the impressions, which were then used to construct the fixed and removable portions of the denture system. (B) Scanning electron and confocal fluorescence microscopy analysis images of a C. albicans biofilm formed in vivo on the denture and palate of rats 4 and 8 weeks postinfection with C. albicans. (Reprinted from reference 166.)
Clinically, even in DS patients without obvious clinical symptoms (limited inflammation/damage; normal immune response), there may be detrimental sequelae from a contaminated denture. Chronic denture infection could lead to seeding of the GI tract, which serves as a major portal for systemic infection in immunosuppressed or hospitalized patients (127). In fact, DS patients have higher rates of GI tract carriage of C. albicans, with similar species isolated from the oral cavity and feces (128). This could become clinically important in the elderly population, which has high rates of denture wearing and increased risk of developing immunosuppressive diseases. Clinical studies have also demonstrated that elderly denture patients have lower expression and activity levels of salivary innate defenses, which may promote oral C. albicans levels (129).
Class 6 (damage occurs only under conditions of strong immune responses): vulvovaginal candidiasis.
Similar to the oral cavity, C. albicans is a resident of the normal vaginal microbiota and the leading causative agent of vulvovaginal candidiasis (VVC) (14, 130–135) (Fig. 2F). Vulvovaginal candidiasis caused by Candida spp. is characterized by itching, burning, pain, and redness of the vulva and vaginal mucosa and is often accompanied by vaginal discharge. It is estimated that 75% of all otherwise-healthy immunocompetent women of childbearing age will be affected by VVC at least once in their lifetime (135, 136). Although VVC is easily treated with antifungals and the infection is normally cleared, approximately 5 to 8% of afflicted women will suffer from recurring episodes characterized by having four or more episodes per year requiring continual antifungal therapy (14, 53, 131, 136, 137). Predisposing factors for primary VVC include high-estrogen oral contraceptive use, hormone replacement therapy, antibiotic usage, and underlying diabetes mellitus. Importantly, disruption of the vaginal microbiota is also considered an important contributor to this complex disease (14, 133, 138). Recurrent VVC (RVVC) is considered idiopathic with no known predisposing factors, although the mechanisms of VVC and RVVC pathogenesis are likely identical. Importantly, VVC and RVVC are not associated with immunodeficiency but are instead associated with a vigorous local inflammatory response, qualifying C. albicans in the context of this infection as a DRF class 6 designation, as it behaves as an immunoreactive commensal.
For study of VVC, a well-established estrogen-dependent mouse model is available (139). Unlike humans, laboratory rodents do not naturally harbor C. albicans as a commensal; however, the experimental infection model closely parallels the human infection, and findings from the animal model are translatable to the human host (4). This includes the host immunopathological response and drug efficacy. In addition, and in contrast to humans, mice maintain a neutral vaginal pH that favors hyphal formation, thus making them a robust model system for C. albicans vaginitis (reviewed in reference 4). Several properties of C. albicans have been proposed to play major roles in causing VVC, most of which have been investigated using the animal model. Most notably, strains of C. albicans defective in hypha formation displayed significantly reduced vaginitis symptomatology, indicating a requirement for hyphae in the pathogenesis of VVC (130, 137). C. albicans tissue penetration is mediated by the invasive filamentous hyphae and is aided by candidal secreted proteolytic enzymes, which degrade epithelial cell barriers and facilitate hyphal penetration (140–143). While not a focus of this article, it should be noted that C. glabrata, which does not form hyphae, is the second most common cause of symptomatic VVC (144). Although one may argue that this contradicts a role for hyphae in the disease pathogenesis, a recent study employing an animal model of C. glabrata VVC showed no evidence of an immunopathologic response despite consistent long-term vaginal colonization (145). Hence, it is unclear how symptoms occur in clinical cases of C. glabrata VVC.
Although these fungal virulence factors are important for the initial onset of VVC, following initial insult from C. albicans the propagation of disease is largely mediated by the host immune system (130, 146). In fact, the mucosal damage in symptomatic VVC is associated with an aggressive neutrophil migration into the vagina and a subsequent acute host inflammatory response (134, 147). This neutrophil response is initiated by the interaction of C. albicans with vaginal epithelial cells, which have no apparent ability to clear C. albicans (131, 134). Therefore, neutrophils contribute more to the symptoms associated with vaginitis (i.e., damage) than they provide protection against disease (130, 135, 147).
It is postulated that the neutrophil response is triggered by the sensitivity of the vaginal epithelium to C. albicans, with epithelial cells of women with RVVC considered sensitive to C. albicans resulting in a response, whereas cells of women with no history of VVC are resistant to such responses (146). Epithelial cell triggers are considered ultimately dependent on a threshold level of C. albicans, such that under sensitive conditions, C. albicans will stimulate the epithelial cells to produce alarmins and proinflammatory cytokines that ultimately lead to neutrophil migration and the inflammatory symptomatic condition (147, 148). On the other hand, under resistant conditions, the epithelial cells fail to elicit the alarmin response and instead inhibit C. albicans growth in a noninflammatory manner mediated by annexin-A1 (147, 149). Interestingly, adaptive cell-mediated or humoral immunity appears to play no role in protection against infection, although certain epitope-specific antibodies characterized as protective antibodies can be used therapeutically against infection (134, 135, 150–152). Another interesting caveat is that while Th17 responses are critical to neutrophil responses at other anatomical sites (oral, bloodstream), the role for Th17 responses in the vaginal neutrophil response is controversial (139, 153). The strongest evidence to date has shown that mice deficient in several cytokines encompassing the Th17 axis showed no change in the C. albicans-induced neutrophil response, suggesting little to no role for the Th17 response in VVC (139).
In women with asymptomatic vaginal carriage of C. albicans, damage is limited. C. albicans, as a member of the normal vaginal microbiota, colonizes approximately 70% of healthy women (154). Thus, it is tempting to speculate that there is evolutionary pressure to maintain C. albicans in the majority of healthy women, with colonization imparting a benefit to the host. Carriage is not associated with other common vaginal infections, such as bacterial vaginosis (BV) (154). Molecular characterization of the vaginal microbiota has revealed that lactobacilli dominate in healthy women, while BV is associated with vaginal dysbiosis and decreased diversity (155). In contrast, vaginal colonization with C. albicans is more common in women with a lactobacillus-dominated microbiota (healthy composition) than in women with dysbiosis (156). Therefore, C. albicans may support bacterial homeostasis as a benefit to the host, and C. albicans adherence to the vaginal epithelium may also provide colonization resistance against sexually transmitted pathogens. Such benefits to the host can be considered to be within the properties of the theoretical DRF class 6 designation.
THE HOST AND THE HOST MICROBIOTA
Scientists have estimated that there are at least as many microbial cells living in and on the human body as human cells in the body (157). The host-inhabiting microbes, defined as the microbiota, are an intricate mixture of microorganisms that have coevolved with their human host (158). Therefore, from the microbial perspective, the host is considered a complex environment. However, these host-microbe interactions influence various aspects of host physiology, and when in homeostasis, the microbiota contributes significantly to maintaining host health. Importantly, the microbiota has profound effects on host immunity and susceptibility to microbial diseases and, therefore, alterations to the microbiota could lead to host-microbe interactions that can produce host damage (8). Both extrinsic and intrinsic factors can cause perturbations to the system, which could lead to alterations in host physiology with potential adverse effects on host health (159). Consistent with the coevolved symbiotic relationship between microbes and humans, the indigenous microbiota provides many crucial functions to the host. In fact, the origin of the concept of colonization resistance dates back to 1965, when the role of the microbiota in antagonizing colonization with a potential pathogen was demonstrated (160). The introduction of broad-spectrum antimicrobial agents, in particular, provided the first evidence of the impact of loss of constituents of the microbiota (161). This was particularly applicable to C. albicans, for which it was first noted in the 1950s that the use of antimicrobial agents was linked to the increase in oral candidiasis in otherwise-healthy people (162).
It is well-established that the bacterial microbiome of the GI tract, including lactobacilli, plays a vital role in preventing fungal colonization, as indicated by the enhanced susceptibility of germfree mice to C. albicans colonization (163). Although ample studies have focused on the ability of the GI microbiota to influence C. albicans levels, surprisingly little is known about the role of C. albicans in shaping the bacterial microbiota, particularly during antibiotic recovery (13). Similarly, VVC is a common side effect of antibiotic treatment, indicating that the vaginal microbiota might modulate colonization of C. albicans (14). However, the role of the vaginal microbiota in VVC is controversial in the literature; where one study comparing the Lactobacillus species cultured from the vaginal secretions of women with or without VVC showed no significant differences, in another study Lactobacillus colonization was associated with a ≥4-fold increase in symptomatic VVC (132, 133). A subsequent comprehensive study of the vaginal microbiota found no altered or unusual bacterial communities in women with VVC, suggesting that commensal vaginal bacterial species might be incapable of preventing VVC (138). Therefore, to understand the interactions of fungi and bacteria within the human GI and vaginal microbiota and their impact on health and disease states, more studies using high-throughput sequencing techniques with longitudinal samples are warranted (14).
As the microbiota is known to be critical for proper immunological function, it can be viewed as an active participant in the outcome of host-microbe interactions (164). The original formulation of the DRF did not incorporate a role for the microbiota; however, one of the utilities of the DRF is its flexibility to accommodate advances in knowledge in the field of microbial pathogenesis. Therefore, in 2015, the DRF was reformulated to incorporate the rapidly accumulating new information emerging from human microbiome studies. This reconciliation redefined the host as “the entity that houses its associated microbiome/microbiota, interacts with microbes, and responds to them in a way that results in damage, benefit, or indifference, thus resulting in the states of symbiosis, colonization, commensalism, latency, and disease” (8).
Researchers have only just begun to describe the microbial communities that are associated with humans and the extent of the interactions between a host and its microbiota. However, dissecting the host immune system responses to perturbations in microbiota remains a considerable challenge, primarily due to the enormous and diverse microbial communities that colonize various parts of the body. Understanding the mechanisms by which host homeostasis is restored is critical for future therapies aimed at manipulating the microbiota. In advancing our immunological insights into diseases, it has become clear that we also need to identify the mechanisms that allow specific members of the microbiota to modulate the host's immune health. Only with an integrated approach will it be possible to make new connections between the microbiota and the immune system. Such insights may allow for prediction of disease development and will unveil the therapeutic potential for restoration of the microbiota in order to reestablish microbial homeostasis. Therefore, improved mouse models, such as those that contain a humanized microbiota, would allow for malleable therapeutic manipulations that would assist the testing and translation of potential therapeutic interventions (162, 165).
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
C. albicans is a highly adaptable microbial species able to cause infection at various anatomical sites with equally diverse host responses. Remarkably, this diversity allows C. albicans to fit into all six of the DRF categories, highlighting the complexity of the pathogenesis associated with C. albicans infections and, importantly, emphasizing the flexibility or plasticity of the parabolas within the DRF. This obliges us to take into account the DRF as much as the virulence attributes or host responses when uncovering the mechanisms associated with each infection. Similarly, animal models that have been instrumental in identifying host response mechanisms and the requirements of the organism for pathogenesis at each anatomical site, as well as for the development of new therapeutic approaches, should now be considered within the context of the DRF for future investigations. It is important to note, however, that we have considered only a subset of C. albicans infections relative to the DRF classifications. Therefore, it is likely that as additional C. albicans infections are considered, they will also be integrated into the diverse classifications of the DRF. Finally, as the nuances of the roles of both host and pathogen continue to be revealed through better understanding of the DRF, it has become critical to consider therapeutic strategies that _target the host in an effort to modulate immune responses in order to more effectively minimize damage to the host.
In conclusion, over the past 2 decades, we have come far in our understanding of the complex host-C. albicans interactions. Nevertheless, there remain considerable gaps in our knowledge of C. albicans pathogenicity, host immune responses and, significantly, the importance of the role of C. albicans as a constituent of the human microbiota. Incorporation of the DRF into studies addressing those gaps should strengthen the impact of findings, provide better global perspectives on various diseases, and identify appropriate host _targets for therapeutics.
ACKNOWLEDGMENTS
We thank Arturo Casadevall for critical reading of the manuscript. We also thank Timothy Meiller for providing us with clinical images of patients with oral candidiasis attending the University of Maryland Dental School.
The work was supported by the National Institutes of Health, grants DE14424 (M.A.J.-R.), DE022069 (M.N.), and AI116025 (M.N.).
We declare no conflicts of interest.
REFERENCES
- 1.Casadevall A, Pirofski L-A. 1999. Host-pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun 67:3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calderone RA. 2012. Candida and candidiasis. ASM Press, Washington, DC. [Google Scholar]
- 3.Williams D, Jordan R, Wei X-Q, Alves C, Wise M, Wilson M, Lewis M. 21 October 2013. Interactions of Candida albicans with host epithelial surfaces. J Oral Microbiol doi: 10.3402/jom.v5i0.22434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naglik JR, Fidel PL, Odds FC. 2008. Animal models of mucosal Candida infection. FEMS Microbiol Lett 283:129–139. doi: 10.1111/j.1574-6968.2008.01160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naglik JR, Challacombe SJ, Hube B. 2003. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 67:400–428. doi: 10.1128/MMBR.67.3.400-428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tournu H, Serneels J, Van Dijck P. 2012. Candida biofilms and the host: models and new concepts for eradication. Int J Microbiol 2012:845352. doi: 10.1155/2012/845352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szabo EK, MacCallum DM. 2011. The contribution of mouse models to our understanding of systemic candidiasis. FEMS Microbiol Lett 320:1–8. doi: 10.1111/j.1574-6968.2011.02262.x. [DOI] [PubMed] [Google Scholar]
- 8.Casadevall A, Pirofski L-A. 2015. What is a host? Incorporating the microbiota into the damage-response framework. Infect Immun 83:2–7. doi: 10.1128/IAI.02627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casadevall A, Pirofski L-A. 2003. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol 1:17–24. doi: 10.1038/nrmicro732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganguly S, Mitchell A. 2011. Mucosal biofilms of Candida albicans. Curr Opin Microbiol 14:380–385. doi: 10.1016/j.mib.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellis M. 2002. Invasive fungal infections: evolving challenges for diagnosis and therapeutics. Mol Immunol 38:947–957. doi: 10.1016/S0161-5890(02)00022-6. [DOI] [PubMed] [Google Scholar]
- 13.Mason K, Erb-Downward J, Mason K, Falkowski N, Eaton K, Kao J, Young V, Huffnagle G. 2012. Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect Immun 80:3371–3380. doi: 10.1128/IAI.00449-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu M-B, Xu S-R, He Y, Deng G-H, Sheng H-F, Huang X-M, Ouyang C-Y, Zhou H-W. 2013. Diverse vaginal microbiomes in reproductive-age women with vulvovaginal candidiasis. PLoS One 8:e79812. doi: 10.1371/journal.pone.0079812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Southern P, Horbul J, Maher D, Davis DA. 2008. C. albicans colonization of human mucosal surfaces. PLoS One 3:e2067. doi: 10.1371/journal.pone.0002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fidel P., Jr 2002. Immunity to Candida. Oral Dis 8(Suppl 2):69–75. doi: 10.1034/j.1601-0825.2002.00015.x. [DOI] [PubMed] [Google Scholar]
- 17.Pirofski L-A, Casadevall A. 2009. Rethinking T cell immunity in oropharyngeal candidiasis. J Exp Med 206:269. doi: 10.1084/jem.20090093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finkel J, Mitchell A. 2011. Genetic control of Candida albicans biofilm development. Nat Rev Microbiol 9:109–118. doi: 10.1038/nrmicro2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathe L, Van Dijck P. 2013. Recent insights into Candida albicans biofilm resistance. Curr Genet 59:251–264. doi: 10.1007/s00294-013-0400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perlroth J, Choi B, Spellberg B. 2007. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med Mycol 45:321–346. doi: 10.1080/13693780701218689. [DOI] [PubMed] [Google Scholar]
- 21.Filioti J, Spiroglou K, Panteliadis CP, Roilides E. 2007. Invasive candidiasis in pediatric intensive care patients: epidemiology, risk factors, mangement, and outcome. Intensive Care Med 33:1272–1283. doi: 10.1007/s00134-007-0672-5. [DOI] [PubMed] [Google Scholar]
- 22.Nett J, Andes D. 2006. Candida albicans biofilm development, modeling a host-pathogen interaction. Curr Opin Microbiol 9:340–345. doi: 10.1016/j.mib.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Jabra-Rizk MA, Falkler WA Jr, Meiller TF. 2004. Fungal biofilms and drug resistance. Emerg Infect Dis 10:14–19. doi: 10.3201/eid1001.030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsui CKE, Jabra-Rizk MA. 2016. Pathogenesis of Candida albicans biofilm. Pathog Dis 74:ftw018. doi: 10.1093/femspd/ftw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costerton JW, Montanaro L, Arciola CR. 2005. Biofilm in implant infections: its production and regulation. Int J Artif Organs 28:1062–1068. [DOI] [PubMed] [Google Scholar]
- 26.Lewis K. 2001. Riddle of biofilm resistance. Antimicrob Agents Chemother 45:999–1007. doi: 10.1128/AAC.45.4.999-1007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghannoum M, Roilides E, Katragkou A, Petraitis V, Walsh T. 2015. The role of echinocandins in Candida biofilm-related vascular catheter infections: in vitro and in vivo model systems. Clin Infect Dis 61(Suppl 6):S618–S621. doi: 10.1093/cid/civ815. [DOI] [PubMed] [Google Scholar]
- 28.Taff HT, Mitchell KF, Edward JA, Andes DR. 2013. Mechanisms of Candida biofilm drug resistance. Future Microbiol 8:1325–1337. doi: 10.2217/fmb.13.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in U.S. hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 30.Williams D, Lewis M. 28 January 2011. Pathogenesis and treatment of oral candidosis. J Oral Microbiol doi: 10.3402/jom.v3i0.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Cuesta C, Sarrion-Pérez M, Bagán J. 2014. Current treatment of oral candidiasis: a literature review. J Clin Exp Dent 6:e576–e582. doi: 10.4317/jced.51798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo H-L, Kohler J, DiDomenico D, Loebenberg D, Cacciapuoti A, Fink G. 1997. Nonfilamentous C. albicans mutants are avirulent. Cell 90:939–949. doi: 10.1016/S0092-8674(00)80358-X. [DOI] [PubMed] [Google Scholar]
- 33.Cleary IA, Reinhard SM, Lazzell AL, Monteagudo C, Thomas DP, Lopez-Ribot JL, Saville SP. 2016. Examination of the pathogenic potential of Candida albicans filamentous cells in an animal model of haematogenously disseminated candidiasis. FEMS Yeast Res 16:fow011. doi: 10.1093/femsyr/fow011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoyer LL. 2001. The ALS gene family of Candida albicans. Trends Microbiol 9:176–180. doi: 10.1016/S0966-842X(01)01984-9. [DOI] [PubMed] [Google Scholar]
- 35.Naglik JR, Albrecht A, Bader O, Hube B. 2004. Candida albicans proteinases and host/pathogen interactions. Cell Microbiol 6:915–926. doi: 10.1111/j.1462-5822.2004.00439.x. [DOI] [PubMed] [Google Scholar]
- 36.Peters ES Jr, Eisenberg E. 1990. Oral candidiasis: the clinical diagnostic spectrum. J Conn State Dent Assoc 66:34–37. [PubMed] [Google Scholar]
- 37.Fidel PL., Jr 2011. Candida-host interactions in HIV disease: implications for oropharyngeal candidiasis. Adv Dent Res 23:45–49. doi: 10.1177/0022034511399284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fidel PL., Jr 2006. Candida-host interactions in HIV disease: relationships in oropharyngeal candidiasis. Adv Dent Res 19:80–84. doi: 10.1177/154407370601900116. [DOI] [PubMed] [Google Scholar]
- 39.Redding SW, Zellars RC, Kirkpatrick WR, McAtee RK, Caceres MA, Fothergill AG, Lopez-Ribot JL, Bailey CW, Rinaldi MG, Paterson TF. 1999. Epidemiology of oropharyngeal Candida colonization and infection in patients receiving radiation for head and neck cancer. J Clin Microbiol 37:3896–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naglik JR, Newport G, White TC, Fernandes-Naglik LL, Greenspan JS, Greenspan D, Sweet SP, Challacombe SJ, Agabian N. 1999. In vivo analysis of secreted aspartyl proteinase expression in human oral candidiasis. Infect Immun 67:2482–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong EF TC, Boyce H, Ibrahim A, Hoag SW, Karlsson AJ, Meiller TF, Jabra-Rizk MA. 2015. Development and in vivo evaluation of a novel histatin-5 bioadhesive hydrogel formulation against oral candidiasis. Antimicrob Agents Chemother 60:881–889. doi: 10.1128/AAC.02624-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan S, Fidel P Jr, Al Thunayyan A, Meiller T, Jabra-Rizk M. 2013. Impaired histatin-5 level and salivary antimicrobial activity against C. albicans in HIV-infected individuals. J Acquir Immune Defic Syndr Clin Res 4:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgerton M, Koshlukova SE, Lo TE, Chrzan BG, Straubinger RM, Raj PA. 1998. Candidacidal activity of salivary histatins. J Biol Chem 272:20438–20447. [DOI] [PubMed] [Google Scholar]
- 44.Peters B, Shirtliff M, Jabra-Rizk M. 2010. Antimicrobial peptides: primeval molecules or future drugs? PLoS Pathog 6:e1001067. doi: 10.1371/journal.ppat.1001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashman R, Farah C, Wanasaengsakul S, Hu V, Pang G, Clancy R. 2004. Innate versus adaptive immunity in Candida albicans infection. Immunol Cell Biol 82:196–204. doi: 10.1046/j.0818-9641.2004.01217.x. [DOI] [PubMed] [Google Scholar]
- 46.Farah C, Elahi S, Drysdale K, Pang G, Gotjamanos T, Seymour G, Clancy R, Ashman R. 2002. Primary role for CD4(+) T lymphocytes in recovery from oropharyngeal candidiasis. Infect Immun 70:724–731. doi: 10.1128/IAI.70.2.724-731.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solis N, Filler S. 2012. Mouse model of oropharyngeal candidiasis. Nat Protoc 7:637–642. doi: 10.1038/nprot.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kong E, Kucharíková S, Van Dijck P, Peters B, Shirtliff M, Jabra-Rizk M. 2015. Clinical implications of oral candidiasis: host tissue damage and disseminated bacterial disease. Infect Immun 83:604–613. doi: 10.1128/IAI.02843-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Repentigny L, Aumont F, Ripeau J-S, Fiorillo M, Kay D, Hanna Z, Jolicoeur P. 2002. Mucosal candidiasis in transgenic mice expressing human immunodeficiency virus type 1. J Infect Dis 185:1103–1114. doi: 10.1086/340036. [DOI] [PubMed] [Google Scholar]
- 50.Gaffen SL, Hernandez-Santos N, Peterson A. 2011. IL-17 signaling in host defense against Candida albicans. Immunol Res 50:181–187. doi: 10.1007/s12026-011-8226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conti H, Shen F, Nayyar N, Stocum E, Sun J, Lindemann M, Ho A, Hai J, Yu J, Jung J, Filler S, Masso-Welch P, Edgerton M, Gaffen S. 2009. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang S, Tan X, Luxenberg D, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser L. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harriott M, Lilly E, Rodriguez T, Fidel P Jr, Noverr M. 2010. Candida albicans forms biofilms on the vaginal mucosa. Microbiology 156:3635–3644. doi: 10.1099/mic.0.039354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hernández-Santos N, Huppler A, Peterson A, Khader S, McKenna K, Gaffen S. 2013. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol 6:900–910. doi: 10.1038/mi.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clancy CJ, Nguyen MH. 2013. Finding the “missing 50%” of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care. Clin Infect Dis 56:1284–1292. doi: 10.1093/cid/cit006. [DOI] [PubMed] [Google Scholar]
- 56.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K. 2009. International study of the prevalence and outcomes of infection in intensive care units. JAMA 302:2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 57.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections. Annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 58.Lewis RE, Cahyame-Zuniga L, Leventakos K, Chamilos G, Ben-Ami R, Tamboli P, Tarrand J, Bodey GP, Luna M, Kontoyiannis DP. 2013. Epidemiology and sites of involvement of invasive fungal infections in patients with haematological malignancies: a 20-year autopsy study. Mycoses 56:638–645. doi: 10.1111/myc.12081. [DOI] [PubMed] [Google Scholar]
- 59.Parker JC Jr, McCloskey JJ, Knauer KA. 1976. Pathobiologic features of human candidiasis. A common deep mycosis of the brain, heart and kidney in the altered host. Am J Clin Pathol 65:991–1000. [DOI] [PubMed] [Google Scholar]
- 60.Louria D, Stiff D, Bennett B. 1962. Disseminated moniliasis in the adult. Medicine 41:307–338. doi: 10.1097/00005792-196212000-00002. [DOI] [Google Scholar]
- 61.Morrell M, Fraser V, Kollef M. 2005. Delaying the empiric treatment of candida bloodstream infection until positive blood culture results are obtained: a potential risk factor for hospital mortality. Antimicrob Agents Chemother 49:3640–3645. doi: 10.1128/AAC.49.9.3640-3645.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasqualotto AC, Nedel WL, Machado TS, Severo LC. 2006. Risk factors and outcome for nosocomial breakthrough candidaemia. J Infect 52:216–222. doi: 10.1016/j.jinf.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 63.Eggimann P, Pittet D. 2014. Candida colonization index and subsequent infection in critically ill surgical patients: 20 years later. Intensive Care Med 40:1429–1448. doi: 10.1007/s00134-014-3355-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spellberg B. 2008. Novel insights into disseminated candidiasis: pathogenesis research and clinical experience converge. PLoS Pathog 4:e38. doi: 10.1371/journal.ppat.0040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vonk A, Netea M, van der Meer J, Kullberg B. 2006. Host defence against disseminated Candida albicans infection and implications for antifungal immunotherapy. Expert Opin Biol Ther 6:891–903. doi: 10.1517/14712598.6.9.891. [DOI] [PubMed] [Google Scholar]
- 66.Pappas PG. 2006. Invasive candidiasis. Infect Dis Clin North Am 20:485–506. doi: 10.1016/j.idc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 67.MacCallum D, Odds F. 2005. Temporal events in the intravenous challenge model for experimental Candida albicans infections in female mice. Mycoses 48:151–161. doi: 10.1111/j.1439-0507.2005.01121.x. [DOI] [PubMed] [Google Scholar]
- 68.MacCallum D. 2009. Massive induction of innate immune response to Candida albicans in the kidney in a murine intravenous challenge model. FEMS Yeast Res 9:1111–1122. doi: 10.1111/j.1567-1364.2009.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castillo LMD, Brown AJ, Gow NA, Odds FC. 2011. Differential regulation of kidney and spleen cytokine responses in mice challenged with pathology-standardized doses of Candida albicans mannosylation mutants. Infect Immun 79:146–152. doi: 10.1128/IAI.01004-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spellberg B, Ibrahim A, Edwards J Jr, Filler S. 2005. Mice with disseminated candidiasis die of progressive sepsis. J Infect Dis 192:336–343. doi: 10.1086/430952. [DOI] [PubMed] [Google Scholar]
- 71.Lazzell A, Chaturvedi A, Pierce C, Prasad D, Uppuluri P, Lopez-Ribot J. 2009. Treatment and prevention of Candida albicans biofilms with caspofungin in a novel central venous catheter murine model of candidiasis. J Antimicrob Chemother 64:567–570. doi: 10.1093/jac/dkp242. [DOI] [PubMed] [Google Scholar]
- 72.Schinabeck MK, Long LA, Hossain MA, Chandra J, Mukherjee PK, Mohamed S, Ghannoum MA. 2004. Rapid model of Candida albicans biofilm infection: liposomal amphotericin B antifungal lock therapy. Antimicrob Agents Chemother 48:1727–1732. doi: 10.1128/AAC.48.5.1727-1732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andes D, Nett J, Pschel P, Albrecht R, Marchillo K, Pitula A. 2004. Development and characterization of an in vivo central venous catheter Candida albicans biofilm model. Infect Immun 72:6023–6031. doi: 10.1128/IAI.72.10.6023-6031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Netea M, Gow N, Munro C, Bates S, Collins C, Ferwerda G, Hobson R, Bertram G, Hughes H, Jansen T, Jacobs L, Buurman E, Gijzen K, Williams D, Torensma R, McKinnon A, MacCallum D, Odds F, Van der Meer J, Brown A, Kullberg B. 2006. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest 116:1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van de Veerdonk F, Kullberg B, Verschueren I, Hendriks T, van der Meer J, Joosten L, Netea M. 2010. Differential effects of IL-17 pathway in disseminated candidiasis and zymosan-induced multiple organ failure. Shock 34:407–411. doi: 10.1097/SHK.0b013e3181d67041. [DOI] [PubMed] [Google Scholar]
- 76.Huang W, Na L, Fidel P Jr, Schwarzenberger P. 2004. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 77.Onishi R, Gaffen S. 2010. Interleukin-17 and its _target genes: mechanisms of interleukin-17 function in disease. Immunology 129:311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maher CO, Dunne K, Comerford R, O'Dea S, Loy A, Woo J, Rogers TR, Mulcahy F, Dunne PJ, Doherty DG. 2015. Candida albicans stimulates IL-23 release by human dendritic cells and downstream IL-17 secretion by Vδ1 T cells. J Immunol 194:5953–5960. doi: 10.4049/jimmunol.1403066. [DOI] [PubMed] [Google Scholar]
- 79.Dejima T, Shibata K, Yamada H, Hara H, Iwakura Y, Naito S, Yoshikai Y. 2011. Protective role of naturally occurring interleukin-17A-producing γδ T cells in the lung at the early stage of systemic candidiasis in mice. Infect Immun 79:4503–4510. doi: 10.1128/IAI.05799-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quintin J, Voigt J, van der Voort R, Jacobsen ID, Verschueren I, Hube B, Giamarellos-Bourboulis EJ, van der Meer JW, Joosten LA, Kurzai O, Netea MG. 2014. Differential role of NK cells against Candida albicans infection in immunocompetent or immunocompromised mice. Eur J Immunol 44:2405–2414. doi: 10.1002/eji.201343828. [DOI] [PubMed] [Google Scholar]
- 81.Koh A. 2013. Murine models of Candida gastrointestinal colonization and dissemination. Eukaryot Cell 12:1416–1422. doi: 10.1128/EC.00196-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clemons K, Gonzalez G, Singh G, Imai J, Espiritu M, Parmar R, Stevens D. 2006. Development of an orogastrointestinal mucosal model of candidiasis with dissemination to visceral organs. Antimicrob Agents Chemother 50:2650–2657. doi: 10.1128/AAC.00530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koh A, Köhler J, Coggshall K, Van Rooijen N, Pier G. 2008. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog 8:e35. doi: 10.1371/journal.ppat.0040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Balish E, Warner T, Nicholas P, Paulling E, Westwater C, Schofield D. 2005. Susceptibility of germfree phagocyte oxidase- and nitric oxide synthase 2-deficient mice, defective in the production of reactive metabolites of both oxygen and nitrogen, to mucosal and systemic candidiasis of endogenous origin. Infect Immun 73:1313–1320. doi: 10.1128/IAI.73.3.1313-1320.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lionakis M. 2014. New insights into innate immune control of systemic candidiasis. Med Mycol 52:555–564. doi: 10.1093/mmy/myu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raaijmakers R, Schroder C, Monnens L, Cornelissen E, Warris A. 2007. Fungal periotonitis in children on peritoneal dialysis. Pediatr Nephrol 22:288–293. doi: 10.1007/s00467-006-0289-x. [DOI] [PubMed] [Google Scholar]
- 87.Rebolledo M, Sarria J. 2013. Candida peritonitis intra-abdominal fungal infections. Curr Opin Infect Dis 26:441–446. doi: 10.1097/01.qco.0000433309.21148.f7. [DOI] [PubMed] [Google Scholar]
- 88.Montravers P, Dupont H, Eggimann P. 2013. Intra-abdominal candidiasis: the guidelines—forgotten non-candidemic invasive candidiasis. Intensive Care Med 39:2226–2230. doi: 10.1007/s00134-013-3134-2. [DOI] [PubMed] [Google Scholar]
- 89.Cheng S, Clancy C, Xu W, Schneider F, Hao B, Mitchell A, Nguyen M-H. 2013. Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J Infect Dis 208:1529–1537. doi: 10.1093/infdis/jit335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vergidis P, Clancy CJ, Shields RK, Park SY, Wildfeuer BN, Simmons RL, Nguyen MH. 2016. Intra-abdominal candidiasis: the importance of early source control and antifungal treatment. PLoS One 11:e0153247. doi: 10.1371/journal.pone.0153247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wojtowicz A, Tissot F, Lamoth F, Orasch C, Eggimann P, Siegemund M, Zimmerli S, Flueckiger UM, Bille J, Calandra T, Marchetti O, Bochud PY, Fungal Infection Network of Switzerland . 2014. Polymorphisms in tumor necrosis factor-alpha increase susceptibility to intra-abdominal Candida infection in high-risk surgical ICU patients. Crit Care Med 42:e304–e308. doi: 10.1097/CCM.0000000000000208. [DOI] [PubMed] [Google Scholar]
- 92.Dupont H, Paugam-Burtz C, Muller-Serieys C, Fierobe L, Chosidow D, Marmuse JP, Mantz J, Desmonts JM. 2002. Predictive factors of mortality due to polymicrobial peritonitis with Candida isolation in peritoneal fluid in critically ill patients. Arch Surg 137:1341–1346. doi: 10.1001/archsurg.137.12.1341. [DOI] [PubMed] [Google Scholar]
- 93.Montravers P, Gauzit R, Muller C, Marmuse JP, Fichelle A, Desmonts JM. 1996. Emergence of antibiotic-resistant bacteria in cases of peritonitis after intraabdominal surgery affects the efficacy of empirical antimicrobial therapy. Clin Infect Dis 23:486–494. doi: 10.1093/clinids/23.3.486. [DOI] [PubMed] [Google Scholar]
- 94.Calandra T, Bille J, Schneider R, Mosimann F, Francioli P. 1989. Clinical significance of Candida isolated from peritoneum in surgical patients. Lancet ii:1437–1440. [DOI] [PubMed] [Google Scholar]
- 95.Blot SI, Vandewoude KH, De Waele JJ. 2007. Candida peritonitis. Curr Opin Crit Care 13:195–199. doi: 10.1097/MCC.0b013e328028fd92. [DOI] [PubMed] [Google Scholar]
- 96.Nash E, Peters B, Palmer G, Fidel P Jr, Noverr M. 2014. Morphogenesis is not required for Candida albicans-Staphylococcus aureus intra-abdominal infection-mediated dissemination and lethal sepsis. Infect Immun 82:3426–3435. doi: 10.1128/IAI.01746-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Peters B, Noverr M. 2013. Candida albicans-Staphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect Immun 81:2178–2189. doi: 10.1128/IAI.00265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nash E, Peters B, Fidel P Jr, Noverr M. 2015. Morphology-independent virulence of Candida species during polymicrobial intra-abdominal infections with Staphylococcus aureus. Infect Immun 84:90–98. doi: 10.1128/IAI.01059-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Felk A, Kretschmar M, Albrecht A, Schaller M, Beinhauser S, Nichterlein T, Sanglard D, Korting H, Schäfer W, Hube B. 2002. Candida albicans hyphal formation and the expression of the Efg1-regulated proteinases Sap4 to Sap6 are required for the invasion of parenchymal organs. Infect Immun 70:3689–3700. doi: 10.1128/IAI.70.7.3689-3700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koh A. 2013. Gastrointestinal colonization of fungi. Curr Fungal Infect Rep 7:144–151. doi: 10.1007/s12281-013-0133-2. [DOI] [Google Scholar]
- 101.Rashid MU, Rosenborg S, Panagiotidis G, Soderberg-Lofdal K, Weintraub A, Nord CE. 2015. Ecological effect of ceftaroline-avibactam on the normal human intestinal microbiota. Antimicrob Agents Chemother 59:4504–4509. doi: 10.1128/AAC.00530-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lacour M, Zunder T, Huber R, Sander A, Daschner F, Frank U. 2002. The pathogenetic significance of intestinal Candida colonization: a systematic review from an interdisciplinary and environmental medical point of view. Int J Hyg Environ Health 205:257–268. doi: 10.1078/1438-4639-00159. [DOI] [PubMed] [Google Scholar]
- 103.Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, Brown J, Funari VA, Wang HL, Crother TR, Arditi M, Underhill DM, Iliev ID. 2016. Immunological consequences of intestinal fungal dysbiosis. Cell Host Microbe 19:865–873. doi: 10.1016/j.chom.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Erdogan A, Rao S. 2015. Small intestinal fungal overgrowth. Curr Gastroenterol Rep 17:16. doi: 10.1007/s11894-015-0436-2. [DOI] [PubMed] [Google Scholar]
- 105.Schulze J, Sonnenborn U. 2009. Yeasts in the gut: from commensals to infectious agents. Dtsch Arztebl Int 106:837–842. doi: 10.3238/arztebl.2009.0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cantorna M, Balish E. 1991. Acquired immunity to systemic candidiasis in immunodeficient mice. J Infect Dis 164:936–943. doi: 10.1093/infdis/164.5.936. [DOI] [PubMed] [Google Scholar]
- 107.Vazquez-Torres A, Jones-Carson J, Warner T, Balish E. 1995. Nitric oxide enhances resistance of SCID mice to mucosal candidiasis. J Infect Dis 172:192–198. doi: 10.1093/infdis/172.1.192. [DOI] [PubMed] [Google Scholar]
- 108.Cantorna M, Balish E. 1990. Mucosal and systemic candidiasis in congenitally immunodeficient mice. Infect Immun 58:1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Balish E, Jensen J, Warner T, Brekke J, Leonard B. 1993. Mucosal and disseminated candidiasis in gnotobiotic SCID mice. J Med Vet Mycol 31:143–154. doi: 10.1080/02681219380000161. [DOI] [PubMed] [Google Scholar]
- 110.Balish E, Balish M, Salkowski C, Lee K, Bartizal K. 1984. Colonization of congenitally athymic, gnotobiotic mice by Candida albicans. Appl Environ Microbiol 47:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cenci E, Mencacci A, Spaccapelo R, Tonnetti L, Mosci P, Enssle K, Puccetti P, Romani L, Bistoni F. 1995. T helper cell type 1 (Th1)- and Th2-like responses are present in mice with gastric candidiasis but protective immunity is associated with Th1 development. J Infect Dis 171:1279–1288. doi: 10.1093/infdis/171.5.1279. [DOI] [PubMed] [Google Scholar]
- 112.Jones-Carson J, Vazquez-Torres A, Warner T, Balish E. 2000. Disparate requirement for T cells in resistance to mucosal and acute systemic candidiasis. Infect Immun 68:2363–2365. doi: 10.1128/IAI.68.4.2363-2365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Noverr M, Noggle R, Toews G, Huffnagle G. 2004. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect Immun 72:4996–5003. doi: 10.1128/IAI.72.9.4996-5003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Noverr M, Falkowski N, McDonald R, McKenzie A, Huffnagle G. 2005. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect Immun 73:30–38. doi: 10.1128/IAI.73.1.30-38.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, Rotter JI, Wang HL, McGovern DP, Brown GD, Underhill DM. 2012. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jawhara S, Thuru X, Standaert-Vitse A, Jouault T, Mordon S, Sendid B, Desreumaux P, Poulain D. 2008. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J Infect Dis 197:972–980. doi: 10.1086/528990. [DOI] [PubMed] [Google Scholar]
- 117.Xin L, Jiang T, Chaturvedi V, Kinder J, Ertelt J, Rowe J, Steinbrecher K, Way S. 2014. Commensal microbes drive intestinal inflammation by IL-17-producing CD4+ T cells through ICOSL and OX40L costimulation in the absence of B7-1 and B7-2. Proc Natl Acad Sci U S A 111:10672–10677. doi: 10.1073/pnas.1402336111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ott S, Kuhbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, Drews O, Weichert W, Timmis K, Schreiber S. 2008. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol 43:831–841. doi: 10.1080/00365520801935434. [DOI] [PubMed] [Google Scholar]
- 119.Enache-Angoulvant A, Bourget M, Brisse S, Stockman-Pannier C, Diancourt L, Francois N, Rimek D, Fairhead C, Poulain D, Hennequin C. 2010. Multilocus microsatellite markers for molecular typing of Candida glabrata: application to analysis of genetic relationships between bloodstream and digestive system isolates. J Clin Microbiol 48:4028–4034. doi: 10.1128/JCM.02140-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Webb BC, Thomas CJ, Willcox MDP, Harty DWS, Knox KW. 1998. Candida-associated denture stomatitis. Aetiology and management: a review. Part 3. Treatment of oral candidosis. Austr Dent J 43:244–249. [DOI] [PubMed] [Google Scholar]
- 121.Ramage G, Tomsett K, Wickes BL, Lopez-Ribot JL, Redding SW. 2004. Denture stomatitis: a role for Candida biofilms. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 98:53–59. doi: 10.1016/j.tripleo.2003.04.002. [DOI] [PubMed] [Google Scholar]
- 122.Budtz-Jörgensen E. 1974. The significance of Candida albicans in denture stomatitis. Eur J Oral Sci 82:151–190. doi: 10.1111/j.1600-0722.1974.tb00378.x. [DOI] [PubMed] [Google Scholar]
- 123.Johnson C, Yu A, Lee H, Fidel PL, Noverr MC. 2012. Development of a contemporary animal model of Candida albicans-associated denture stomatitis using a novel intraoral denture system. Infect Immun 80:1736–1743. doi: 10.1128/IAI.00019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee H, Yu A, Johnson CC, Lilly EA, Noverr MC, Fidel PL Jr. 2011. Fabrication of a multi-applicable removable intraoral denture system for rodent research. J Oral Rehabil 38:686–690. doi: 10.1111/j.1365-2842.2011.02206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee H, Alika Y, Fidel P. June 2014. Removable rodent intraoral device. US Patent 8,753,113 B2.
- 126.Tobouti PL, Casaroto AR, de Almeida RS, de Paula Ramos S, Dionísio TJ, Porto VC, Santos CF, Lara VS. 2016. Expression of secreted aspartyl proteinases in an experimental model of Candida albicans-associated denture stomatitis. J Prosthodont 25:127–134. doi: 10.1111/jopr.12285. [DOI] [PubMed] [Google Scholar]
- 127.Miranda LN, van der Heijden IM, Costa SF, Sousa AP, Sienra RA, Gobara S, Santos CR, Lobo RD, Pessoa VP Jr, Levin AS. 2009. Candida colonisation as a source for candidaemia. J Hosp Infect 72:9–16. doi: 10.1016/j.jhin.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 128.Bergendal T, Holmberg K, Nord C-E. 1979. Yeast colonization in the oral cavity and feces in patients with denture stomatitis. Acta Odontol Scand 37:37–45. doi: 10.3109/00016357909004683. [DOI] [PubMed] [Google Scholar]
- 129.Gasparoto T, de Oliveira C, Vieira N, Porto V, Gasparoto C, Campanelli A, Lara V. 2012. The pattern recognition receptors expressed on neutrophils and the associated cytokine profile from different aged patients with Candida-related denture stomatitis. Exp Gerontol 47:741–748. doi: 10.1016/j.exger.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 130.Peters B, Yano J, Noverr M, Fidel P. 2014. Candida vaginitis: when opportunism knocks, the host responds. PLoS Pathog 10:e1003965. doi: 10.1371/journal.ppat.1003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sobel J, Faro S, Foxman B, Ledge W, Nyirjesy P, Reed B, Summers P. 1998. Vulvovaginal candidiasis: epidemiologic, diagnostic, and therapeutic considerations. Am J Obstet Gynecol 178:203–211. doi: 10.1016/S0002-9378(98)80001-X. [DOI] [PubMed] [Google Scholar]
- 132.Sobel J, Chaim W. 1996. Vaginal microbiology of women with acute recurrent vulvovaginal candidiasis. J Clin Microbiol 34:2497–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McClelland R, Richardson B, Hassan W, Graham S, Kiarie J, Baeten J, Mandaliya K, Jaoko W, Ndinya-Achola J, Holmes K. 2009. Prospective study of vaginal bacterial flora and other risk factors for vulvovaginal candidiasis. J Infect Dis 199:1883–1890. doi: 10.1086/599213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fidel PL Jr, Barousse M, Espinosa T, Ficarra M, Sturtevant J, Martin DH, Quayle AJ, Dunlap K. 2004. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect Immun 72:2939–2946. doi: 10.1128/IAI.72.5.2939-2946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fidel P., Jr 2004. History and new insights into host defense against vaginal candidiasis. Trends Microbiol 12:220–227. doi: 10.1016/j.tim.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 136.Sobel J. 1992. Pathogenesis and treatment of recurrent vulvovagnial candidiasis. Clin Infect Dis 14:S148–S153. doi: 10.1093/clinids/14.Supplement_1.S148. [DOI] [PubMed] [Google Scholar]
- 137.Sobel JD, Muller G, Buckley HR. 1984. Critical role of germ tube formation in the pathogenesis of candidal vaginitis. Infect Immun 44:576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou X, Westman R, Hickey R, Hansmann M, Kennedy C, Osborn T, Forney L. 2009. Vaginal microbiota of women with frequent vulvovaginal candidiasis. Infect Immun 77:4130–4135. doi: 10.1128/IAI.00436-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yano J, Fidel P Jr. 2011. Protocols for vaginal inoculation and sample collection in the experimental mouse model of Candida vaginitis. J Vis Exp 58:3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.de Bernardis F, Arancia S, Morelli L, Hube B, Sanglard D, Schafer W, Cassone A. 1990. Evidence that members of the secretory aspartyl proteinases gene family (SAP), in particular SAP2, are virulence factors for Candida vaginitis. J Infect Dis 179:201–208. [DOI] [PubMed] [Google Scholar]
- 141.de Bernardis F, Cassone A, Sturtevant J, Calderone R. 1995. Expression of Candida albicans SAP1 and SAP2 in experimental vaginitis. Infect Immun 63:1887–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lian CH, Liu WD. 2007. Differential expression of Candida albicans secreted aspartyl proteinase in human vulvovaginal candidiasis. Mycoses 50:383–390. doi: 10.1111/j.1439-0507.2007.01384.x. [DOI] [PubMed] [Google Scholar]
- 143.Naglik JR, Moyes D, Makwana J, Kanzaria P, Tsichlaki E, Weindl G, Tappuni AR, Rodgers CA, Woodman AJ, Challacombe SJ, Schaller M, Hube B. 2008. Quantitative expression of the Candida albicans secreted aspartyl proteinase gene family in human oral and vaginal candidiasis. Microbiology 154:3266–3280. doi: 10.1099/mic.0.2008/022293-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fidel PL Jr, Vazquez JA, Sobel JD. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev 12:80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nash EE, Peters BM, Lilly EA, Noverr MC, Fidel PL Jr. 2016. A murine model of Candida glabrata vaginitis shows no evidence of an inflammatory immunopathogenic response. PLoS One 11:e0147969. doi: 10.1371/journal.pone.0147969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yano J, Noverr MC, Fidel PL Jr. 2012. Cytokines in the host response to Candida vaginitis: identifying a role for non-classical immune mediators, S100 alarmins. Cytokine 58:118–128. doi: 10.1016/j.cyto.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yano J, Lilly E, Barousse M, Fidel PL Jr. 2010. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun 78:5126–5137. doi: 10.1128/IAI.00388-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bruno VM, Wang Z, Marjani SL, Euskirchen GM, Martin J, Sherlock G, Snyder M. 2010. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res 20:1451–1458. doi: 10.1101/gr.109553.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Steele C, Fidel P Jr. 2002. Cytokine and chemokine production by human oral and vaginal epithelial cells in response to Candida albicans. Infect Immun 70:577–583. doi: 10.1128/IAI.70.2.577-583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fidel PJ. 2003. Immune regulation and its role in the pathogenesis of candida vaginitis. Curr Infect Dis Rep 5:488–493. doi: 10.1007/s11908-003-0092-9. [DOI] [PubMed] [Google Scholar]
- 151.De Bernardis F, Arancia S, Sandini S, Graziani S, Norelli S. 2015. Studies of immune responses in Candida vaginitis. Pathogens 4:697–707. doi: 10.3390/pathogens4040697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cassone A. 2015. Vulvovaginal Candida albicans infections: pathogenesis, immunity and vaccine prospects. BJOG 122:785–794. doi: 10.1111/1471-0528.12994. [DOI] [PubMed] [Google Scholar]
- 153.Pietrella D, Rachini A, Pines M, Pandey N, Mosci P, Bistoni F, d'Enfert C, Vecchiarelli A. 2011. Th17 cells and IL-17 in protective immunity to vaginal candidiasis. PLoS One 6:e22770. doi: 10.1371/journal.pone.0022770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Drell T, Lillsaar T, Tummeleht L, Simm J, Aaspollu A, Vain E, Saarma I, Salumets A, Donders G, Metsis M. 2013. Characterization of the vaginal micro- and mycobiome in asymptomatic reproductive-age Estonian women. PLoS One 8:e54379. doi: 10.1371/journal.pone.0054379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vitali B, Pugliese C, Biagi E, Candela M, Turroni S, Bellen G, Donders G, Brigidi P. 2007. Dynamics of vaginal bacterial communities in women developing bacterial vaginosis, candidiasis, or no infection, analyzed by PCR-denaturing gradient gel electrophoresis and real-time PCR. Appl Environ Microbiol 73:5731–5741. doi: 10.1128/AEM.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.van de Wijgert J, Borgdorff H, Verhelst R, Crucitti T, Francis S, Verstraelen H, Jespers V. 2014. The vaginal microbiota: what have we learned after a decade of molecular characterization? PLoS One 9:e105998. doi: 10.1371/journal.pone.0105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sender R, Fuchs S, Milo R. 2016. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164:337–340. doi: 10.1016/j.cell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 158.Ursell L, Metcalf J, Parfrey L, Knight R. 2012. Defining the human microbiome. Nutr Rev 70:S38–S44. doi: 10.1111/j.1753-4887.2012.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Belizario JE, Napolitano M. 2015. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol 6:1050. doi: 10.3389/fmicb.2015.01050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mushin R, Dubos R. 1965. Colonization of the mouse intestine with Escherichia coli. J Exp Med 122:745. doi: 10.1084/jem.122.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Francino MP. 2015. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol 6:1543. doi: 10.3389/fmicb.2015.01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dethlefsen L, McFall-Ngai M, Relman DA. 2007. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wagner RD, Pierson C, Warner T, Dohnalek M, Hilty M, Balish E. 2000. Probiotic effects of feeding heat-killed Lactobacillus acidophilus and Lactobacillus casei to Candida albicans-colonized immunodeficient mice. J Food Prot 63:638–644. [DOI] [PubMed] [Google Scholar]
- 164.Rooks MG, Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16:341–352. doi: 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Blumberg R, Powrie F. 2012. Microbiota, disease, and back to health: a metastable journey. Sci Transl Med 4:137rv137. doi: 10.1126/scitranslmed.3004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kong EF, Fidel P, Jabra-Rizk MA. 2015. Candida albicans: love-hate relationship with its human host. Microbe Mag 10:413–418. doi: 10.1128/microbe.10.413.1. [DOI] [Google Scholar]