

Abstract
Comparative sequence analysis was carried out for the regions adjacent to experimentally validated transcriptional start sites (TSSs), using 3324 pairs of human and mouse genes. We aligned the upstream putative promoter sequences over the 1-kb proximal regions and found that the sequence conservation could not be further extended at, on average, 510 bp upstream positions of the TSSs. This discontinuous manner of the sequence conservation revealed a “block” structure in about one-third of the putative promoter regions. Consistently, we also observed that G+C content and CpG frequency were significantly different inside and outside the blocks. Within the blocks, the sequence identity was uniformly 65% regardless of their length. About 90% of the previously characterized transcription factor binding sites were located within those blocks. In 46% of the blocks, the 5′ ends were bounded by interspersed repetitive elements, some of which may have nucleated the genomic rearrangements. The length of the blocks was shortest in the promoters of genes encoding transcription factors and of genes whose expression patterns are brain specific, which suggests that the evolutional diversifications in the transcriptional modulations should be the most marked in these populations of genes.
As fellow mammals, humans share many physiological, anatomical, and metabolic parallels with mice (Nadeau and Taylor 1984). However, there are striking differences between the two species as well, that is, alterations in size, shape, and longevity. Above all, humans but not mice have developed highly complex neural systems in the brain. It has long been supposed that the genetic basis for these similarities/differences lies, at least in part, in alterations in the expression of genes rather than changes in the functions of their encoded protein products (King and Wilson 1975; Tautz 2000). Differential regulation of gene expression seems a likely explanation for many differences between humans and mice. Between humans and mice, many of the protein functions themselves have been shown to be comparable (Boguski 2002). To understand the molecular machinery that makes humans distinct from mice, the features in the transcriptional networks that are unique to humans should be identified. On the other hand, if the mechanisms that constitute the basic framework of the genetic network are to be delineated, the investigation should be focused on the features that are shared between humans and mice.
However, only limited knowledge has been accumulated about how and to what extent the transcriptional modulatory mechanisms are conserved or divergent between human and mouse genes. Although there are pioneering studies phylogenetically comparing the genomic sequences involved in transcriptional regulations (for reviews, see Ureta-Vidal et al. 2003; Wray et al. 2003), our understanding of the comprehensive systems of transcriptional regulation is still at a very primitive stage. To address this issue, it is essential to enrich our basic knowledge of the molecular mechanisms underlying the regulation of the transcription of each gene.
One of the most important regulatory steps for transcription is the initiation step. For many genes, it has been shown that the transcription level is regulated by controlling the efficiency of the formation of the RNA polymerase II pre-initiation complex (Mitchell and Tjian 1989; Roeder 1996). The DNA sequence just adjacent to the transcriptional start sites (TSSs) plays an important role in the regulation. This region is called the promoter, and several cis-regulatory sequence elements are embedded in it. These cis-acting elements are recognized by general transcription factors (GTFs), various kinds of transcription regulatory factors (TFs), or other protein factors. When these proteins are recruited to the promoter, they accelerate/inhibit the formation of the preinitiation complex through direct interaction or by changing the conformation of the docking platform (Novina and Roy 1996). To understand the molecular mechanisms of such transcriptional regulation, it is essential to identify and characterize what kinds of cis-elements are embedded within the promoters and what kinds of TFs are recruited onto the promoters (http://www.epd.isb-sib.ch; Eukaryotic Promoter Database; and http://www.gene-regulation.com/; TRANSFAC; Praz et al. 2002; Kel et al. 2003).
With the near completion of the human and mouse genome sequencing projects (http://genome.ucsc.edu/downloads.html; UCSC Genome Browser; Lander et al. 2001; Venter et al. 2001; Waterston et al. 2002), the basic materials to start genome-wide analyses of promoters have become available. Because the promoters are located proximal to or overlapping with the TSSs and because the 5′ ends of full-length cDNA sequences correspond to the TSS, it is possible to retrieve the putative promoter sequences (called “putative promoter regions” [PPRs] hereafter) from large volumes of genomic sequences by combining the information about genomic DNA and full-length cDNAs.
We previously developed a method to construct full-length cDNA libraries and have been collecting full-length cDNAs (Carninci and Hayashizaki 1999; Suzuki and Sugano 2003). So far, we have accumulated 400,225 human and 580,209 mouse cDNAs (http://fantom.gsc.riken.go.jp/; FANTOM), from a wide variety of tissues and cultured cells (Kawai et al. 2001; Okazaki et al. 2002; Waterston et al. 2002). Based on the data for these full-length cDNAs, in the present study we were able to determine the exact positions of their TSSs on the genomic sequences and retrieve the PPR sequences for 8793 human and 6875 mouse RefSeq genes (http://dbtss.hgc.jp/; DBTSS; and http://www.ncbi.nlm.nih.gov/RefSeq/; RefSeq). Of these, 3324 promoters could be paired with each other between mutually 1:1 homologous genes (Statistics of the data set used in the present study are summarized in Supplemental data Table 1; for further details refer to Suzuki et al. 2004). This collection of PPR sequences enabled us, for the first time, to precisely distinguish which parts of the genomic sequences correspond to the exonic regions, TSSs, and upstream regions. Here we report our first large-scale comparative sequence analyses of PPRs between human and mouse genes.
RESULTS
Sequence Comparison of Promoters Between Human and Mouse Genes
We aligned the PPR sequences of 3324 pairs of human and mouse genes over the regions proximal to the TSSs, from -1 kb to +200 bp (the TSS was designated as 0). The sequence identities calculated for these regions were 46% on average. Consistent with a previous report (Waterston et al. 2002), the average sequence identity was the highest in the -100-bp to +100-bp region, and it decreased as the distance from the TSSs increased (Fig. 1A).
Figure 1.

Sequence identity between human and mouse PPRs. Sequence alignments were calculated using LALIGN with the default parameters. The sequence identity was evaluated as the number of aligned nucleotides in the regions of -1000 to +200 (TSS: 0). The average sequence identities were calculated for each region (A). (B) The PPRs were separated into the 200-bp windows at the positions indicated in the inset. Sequence identity was calculated for each of the windows. Frequency as to which of the windows belong to which of the sequence identity groups represented on the horizontal axis is plotted.
For the alignment, we used the sequence alignment program LALIGN (Huang et al. 1992), because it is a relatively simple local alignment program that is robust against gaps (a typical example of the results is shown in Supplemental data Fig. 1). We also used CLUSTALW (Thompson et al. 1994), which is one of the most popular global alignment programs. However, CLUSTALW was inappropriate for our purpose. When CLUSTALW was used for the alignment, a relatively short gap disturbed the overall alignment in many cases (data not shown).
We further examined the sequence alignments and found that the aligned sequences did not always cover the entire 1-kb upstream region. Very frequently, the sequence alignments disappeared at particular positions within the 1-kb regions, which made the aligned parts look like “blocks” (a typical example is illustrated in Supplemental data Fig. 1). The boundary of the block was defined as the most distal aligned region according to the result of LALIGN. The observed patterns of gradually decreasing average identities mainly accounted for the difference in the frequency of the blocks covering the corresponding regions (Fig. 1B). The average length of the blocks was 510 bp (Fig. 2A). The sequence identity inside the blocks was uniformly around 65% irrespective of the block's length (Fig. 2B,C). The overall sequence similarities of the upstream sequences were mainly dependent on the length of the blocks. We performed similar analyses using different parameters for gap-opening penalties and gap-extension penalties. We observed essentially the similar results unless the effects parameter changes resulted in disruption of the alignments themselves (for further details, see Supplemental data Fig. 2).
Figure 2.

Sequence alignments of the block structure in PPRs and nongenic regions. (A) Frequency of the blocks belonging to each population is shown. (B) Relation between length of the block and the average sequence identity within it. (C) Relation between percentile position within the block and the average sequence identity. (D) Alignment of the nongenic sequences using LALIGN. The sequences ranging from -1 kb to + 200 bp of the putative syntenic regions located in nongenic regions as in UCSC genome browser were aligned and the frequencies of the aligned nucleotides were calculated at each of the positions. Vertical line represents the frequency of the nucleotide at the indicated position being located within the block. (Note that the vertical axis in Figure 1 represents the frequency of the sequence “identity”).
We also examined whether this discontinuous manner of the sequence conservation was specific to the PPRs using the sequences of the nongenic regions. Positional information of the putative syntenic regions of the human and mouse genomes were obtained from UCSC Genome Browser and those regions at least 100 kb apart from the so-called Ensembl regions were selected (http://genome.ucsc.edu/goldenPath/14nov2002/database/; Ensembl; and http://www.ensembl.org/; Ensembl). Using the distal sequences (-1 kb to 200 bp) of those putative “homologous” regions, a similar analysis was performed. As shown in Figure 2D, the discontinuity of the sequence conservation was also observed in the nongenic regions throughout the genome (for further details on these homologous regions in the nongenic regions, see Supplemental data Fig. 3).
Sequences Around the Distal Regions of the Block Structure
We examined why the sequence alignments could not be further extended at the edge of the blocks. It was rare that the alignments were terminated at the positions of sequence gaps (incompleteness in the genome sequencing) in either the human or mouse genomes. In humans, 31% of the boundaries were flanked by interspersed repetitive elements (Table 1). Of these, 16% corresponded to Alu elements, which are primate-specific repetitive elements (Mitchell and Tjian 1989; Deininger and Batzer 2002). Similarly, in mice, insertion of repetitive elements was observed for 20% of the boundaries, of which 8% were B1 elements, which are Alu superfamily elements in rodents. Taken together, in 46% of the blocks, repetitive elements were found at the boundaries in either the human or mouse genome. For this population, it is possible that the sequence alignments were disrupted because the repetitive elements were inserted into otherwise continuous regions. It was possible that LALIGN could not allocate “gaps” to them in the alignments. To address this issue, we excised the repetitive elements and generated the sequence alignments again. Still, we could not identify sequence similarity significantly greater than 30% in essentially any case. This is similar to the results obtained from the analyses of the remaining 54% of the edges of the blocks. In either case, the sequences outside of the blocks seemed completely lost from the corresponding parts of the counter genomes.
Table 1.
Boundaries of the Blocks
| Human | Mouse | |||||
|---|---|---|---|---|---|---|
| Repeat | 31% | 20% | ||||
| Alu-type SINE | 16% | B1-type SINE | 8% | |||
| MIR-type SINE | 3% | B2-type SINE | 4% | |||
| LINE | 6% | LINE | 3% | |||
| LTR | 2% | LTR | 3% | |||
| MER | 2% | MER | 2% | |||
| others | 1% | others | 0% | |||
| Gap in genomic sequence | 0% | 4% | ||||
| Uncharacterized | 69% | 76% | ||||
| Total | 100% | 100% |
Indicated sequences were observed at the corresponding frequencies at the boundaries of the blocks.
Sequences Are Conserved in a Block Manner in the Promoters
To determine whether the observed block structures were derived from algorithmic artifacts of LALIGN, we aligned the PPR sequences using another type of sequence alignment program, SSEARCH (Smith and Waterman 1981; Pearson 1996). This program is based on the simple Smith-Waterman algorithm and gives the most precise alignments, though it is computationally expensive. Using the SSEARCH alignments, we again observed the similar block structures, and the sequence identities sharply dropped just outside the blocks. In these cases, the results were robust against changes of the parameters, as is the case for LALIGN (also see Supplemental data Fig. 4).
When a similar analysis was performed using the sequences around the 5′-end boundaries of the second exons (note that PPRs were defined as the regions upstream of the first exons), the SSEARCH scores dropped sharply at the 5′ ends of the exons (Fig. 3). Thus, the boundaries of the block structures were overlapped with the exon-intron boundaries in these cases. The boundaries between exonic and intronic sequences can be considered as transition points from the regions where most of the sequences play biologically significant roles to the regions where most of the sequences are biologically less relevant. Similarly, it can be suggested that, in the promoters, most of the biologically significant elements should be embedded inside rather than outside the blocks. It was also significant that such discontinuity in the sequence conservation has frequently been observed in the proximal regions of both the boundaries of the blocks in the PPRs and the exon-intron boundaries.
Figure 3.

Sequence alignments around the boundary of the block and that of the first intron and the second exon using SSEARCH. (A) Sequences of human and mouse PPRs were aligned using SSEARCH with a 50-bp moving window around the boundary of the block. The broken line represents the boundary of the block calculated using LALIGN. The vertical axis represents the average score of the SSEARCH calculated for the corresponding position. The horizontal axis represents the relative position to the boundary. (B) Result of an analysis similar to that shown in A, using the proximal sequences of the 5′ end of the second exons. The broken line represents the exon-intron boundary. The horizontal axis represents the relative position to the exon-intron boundary.
Differences in G+C Content and CpG Frequency Between the Sequences Inside and Outside the Blocks
We compared the G+C contents and the frequencies of the di-nucleotides, CpG, between the sequences inside and outside the blocks (Table 2). Promoters are frequently associated with the G+C-rich regions with increased frequency of the CpG (Cross and Bird 1995). For humans, when the sequences of 200 bp around the boundaries of the blocks were evaluated, the average G+C contents were 58% and 53% in the sequences inside (proximal sides to the TSSs) and outside (distal sides to the TSSs) of the blocks, respectively. The difference overall distributions of the G+C contents between them was statistically significant according to the standard t test (p < 1.0e-136), although the G+C contents vary between PPRs. The average frequencies of the di-nucleotide, CpG, were 12.7 sites/200 bp and 9.0 sites/200 bp for the regions inside and outside the blocks, respectively. Again, the difference in their distributions was statistically significant (p < 1.0e-105). As shown in Table 3, essentially the same results were obtained from mouse PPRs. This observation also supports our claim that the sequences inside and outside of the blocks are qualitatively distinct.
Table 2.
G+C Content and CpG Frequency Inside and Outside the Blocks
| Outside of −1 Kb to +200 bp | Within −1 kb to +200 bp | Within block | Outside of block | ||
|---|---|---|---|---|---|
| Human | Experimentally confirmed | 35 | 203 | 179 (88%) | 24 (12%) |
| Predicted | ND | 1898 | 1704 (90%) | 194 (10%) | |
| Mouse | Experimentally confirmed | 31 | 108 | 102 (94%) | 6 (6%) |
| Predicted | ND | 1853 | 1668 (90%) | 185 (10%) |
The sequences ±200 bp of the boundaries of the “blocks” were used for the calculation. ND = not determined.
Table 3.
TF Binding Sites Inside and Outside the Blocks
| Human
|
Mouse
|
|||
|---|---|---|---|---|
| Inside block | Outside block | Inside block | Outside block | |
| G+C content | 58%* | 53% | 56% | 48% |
| CpG frequency (sites/200 bp) | 12.7** | 9.0 | 11.0 | 6.0 |
The frequencies of the TF binding sites were calculated for each of the indicated regions. Statistical significance of the enrichment was *p < 1.0e-136 and **p < 1.0e-105.
Mapping of TF-Binding Sites
To study the relationship between the relative positions of the blocks and the TF-binding sites embedded in the upstream regions, we mapped previously determined TF-binding sites. For this, we used the information contained in TRANSFAC (version 7.4). This database is the most widely used database in which detailed information concerning TF-binding sites, which have been characterized by various experimental methods, is compiled (Kel et al. 2003). In the 3324 promoter pairs, there were 238 experimentally characterized TF-binding sites for human genes (further references about each of the TF-binding sites are recorded in TRANSFAC). Of these, 203 sites (85%) were located in regions proximal to the TSSs (within the -1 kb to +200 bp regions), which is consistent with previous observations that most TF-binding sites were located within this region (Praz et al. 2002; Liu et al. 2003). Among the TF-binding sites, 179 sites (88%) were located within the blocks. On the other hand, we also observed that 24 sites (12%) in human genes were located outside of the blocks, where no significant sequence similarities were found. For each of these sites, we both manually and computationally examined whether the same kind of TF-binding site could be identified in the corresponding regions of the promoter sequences of the mouse gene. All of these sites were completely missing from the corresponding regions of the mouse promoters, although there still remains a slight possibility that real TF-binding sites are located in regions distant from the TSSs, or that the TF binding sites were so diverged that they could not be identified using a computational method.
We performed similar analyses with regard to the computationally predicted TF-binding sites. Among the 1898 predicted TF-binding sites in human PPRs, 1704 (90%) were located within the blocks and 194 (10%) outside of the blocks. This corresponds well with the above results regarding the “experimentally characterized” TF sites. Essentially similar results were obtained from analyses from the mouse side, too (Table 3).
Correlation Between Sequence Conservation in the Promoters and Molecular Functions and Tissue Specificity of the Genes
We examined whether there is any correlation between sequence divergence of the promoters and molecular functions and expression patterns of the corresponding genes. We calculated the frequency of the PPRs in which blocks covered less than 50% (600 bp) of the sequences (designated as “encroached” PPRs) for each of the GO categories (Harris et al. 2004). Similarly, the frequency of those promoters was calculated for each population of the genes that showed tissue-specific patterns of gene expressions. For the expression profiles, we used the data obtained by iAFLP, which is an RT-PCR-mediated high-throughput method for detecting relative amounts of gene expression (Kawamoto et al. 1999; the iAFLP data used in this study are presented at http://cdna.ims.u-tokyo.ac.jp/iAFLP.xls). We tentatively defined the genes as “tissue specific” when more than 30% of the transcripts were attributed to a particular tissue.
As shown in Table 4A, the frequency of the encroached PPRs was significantly increased in the GO category of “transcription regulators”, which is the group of genes of TFs (p < 0.0002). In the 203 TF genes, the frequency of the genes with such promoters was 39%, which was higher than the frequency calculated for any other GO category. We also observed that encroached PPRs were enriched in genes whose expression patterns were “brain specific” (Table 4B). Although statistical significance in this case was not as clear as the case of the transcription regulators, the enrichment was higher than any other tissues (p < 0.05).
Table 4.
Correlation Between the Gene Ontology, Expression Profiles, and Sequence Conservation in the PPRs
| A. GO annotation | Total number of genes | Number of genes with encroached PPRs | Frequency (%) |
|---|---|---|---|
| Transcription regulator | 203 | 79 | 39* |
| Structural molecule | 125 | 35 | 28 |
| Enzyme | 871 | 225 | 26 |
| Enzyme regulator | 91 | 23 | 25 |
| Cell adhesion molecule | 56 | 14 | 25 |
| Defence/immunity | 29 | 7 | 24 |
| Transporter | 342 | 81 | 24 |
| Signal transducer | 362 | 78 | 22 |
| Total | 3324 | 921 | 28 |
| B. Tissue | Total number of genes with tissue-specific gene expression | Number of genes with encroached PPRs | Frequency (%) |
|---|---|---|---|
| Brain/neuron | 156 | 53 | 34** |
| Gastrointestinal | 121 | 35 | 29 |
| Immune | 98 | 29 | 30 |
| Reproductory | 137 | 34 | 25 |
| Endocrine | 17 | 4 | 24 |
| Circulatory/blood | 22 | 5 | 24 |
| Others | 148 | 42 | 29 |
| Total | 3324 | 921 | 28 |
The numbers and the frequencies of the genes were shown for each of the GO (A) and iAFLP (B) categories. Statistical significance of the enrichment was *p < 0.0002 and **p < 0.05, respectively (for further details on the procedure, see Methods).
DISCUSSION
Here we have described the first systematic and quantitative comparison of promoters regarding the manner in which and the extent to which promoter sequences are conserved between human and mouse genes. Using 3324 pairs of PPRs of human and mouse genes, we first demonstrated that the conserved parts frequently stood out against the nonconserved parts, forming blocks. The sequence similarities of around 65% in these blocks extended upstream of the TSSs and disappeared at particular points, on average, 510 bp upstream of the TSSs. This is inconsistent with the view generally held hitherto. The initial descriptions of the sequence similarity among promoters indicated that the independent alternations of the nucleotides are distributed in a gradually increasing manner in proportion to the distance from the TSSs (as shown in Fig. 1). Although the results of a previous study using 41 human-mouse promoter pairs suggested the block structure of the sequence conservation in the promoters, it was considered likely to be an artifact of the alignment program used (Jareborg et al. 1999). In the present study, we scrutinized the sequence alignments mainly using two alignment programs that are based on different algorithms and demonstrated that the block structures were observed regardless of the alignment programs in about one-third of the examined PPRs (Figs. 2, 3; for further details on the alignment programs, see Ureta-Vidal et al. 2003).
There still remains some possibility that the block structure observed in the present study was identified due to the inherent inability of the pre-existing alignment programs, most of which are designed for aligning sequences of genic (especially of protein-coding) regions. Also, we could not completely refute the possibility that alignment procedures employed here were not suitable for detecting relatively short motifs outside putative blocks separated by constitutive insertion or deletions of the nucleotides. However, we consider that such a possibility is low, because we selected relatively simple programs, LALIGN and SSEARCH, run by parameters for which no special “parameter tuning” was performed a priori. We also demonstrated that this observation was robust against the changes of the parameters (Supplementary data Figs. 2 and 3). Although it is possible further “optimization” of the programs and parameters may be useful for further precise determination of the boundaries of each of the blocks, we consider such perturbation would not greatly influence our conclusion that the segmentation occurred just around the TSSs very frequently.
It was also unlikely that our observations were obtained due to defects in our data set. Only rare data should represent spuriously identified promoter sequences resulting from erroneously cloned full-length cDNAs (truncated cDNAs), because, in most cases, the sequences could be aligned at least to some extent. If the promoters were spurious at all, they would not show any significant match against their counterparts. Mispairing of paralogs as orthologs could bring about the results observed here. As paralogs are generated by gene duplication (Frazer et al. 2003b), it is possible that there is some synteny just around the genic regions, which disappears at the boundaries of the duplication points. However, at least 80% of mouse genes have only a single identifiable homologous gene in the human genome, which should be an ortholog (Waterston et al. 2002). Also, we used the pairing information of the orthologs according to LocusLink information, in which 1:1 homologous genes are further inspected to pair orthologs (Wheeler et al. 2004). This should have excluded any remaining pseudo-orthologous pairs from our data set. Considering that the block structure was observed for more than one-third of the promoters, contamination by paralogs should not account much for our observations.
Based on all these facts and our findings, we concluded that the block structure is, in fact, a feature of the sequence conservation in about one-third of the PPRs examined here. We consider that this discontinuous manner of the sequence conservation should be a quite frequent feature of promoters throughout the human and mouse genomes. Although we could not show whether such discontinuous conservation would be observed in more distal regions from the TSSs in the gene of the remaining population, it is significant that such dynamic changes occurred just proximal regions of the TSSs at least one-third of the PPRs. In order to understand how the transcription modulation has evolved, this information should become the fundamental data.
Within the blocks, the sequence similarity was relatively uniform (Fig. 2) with an average identity of 65%. The overall sequence similarity between human and mouse at neutral sites has been estimated to be 53-54%, when assessed using relics of ancestral repeats (Waterston et al. 2002). If the regional variations of the neutral substitution rate are ignored (Hardison et al. 2003), the sequence identity is approximately 10% higher in the sites within the blocks. This difference implies that some parts of the promoters are subjected to selective pressure. Largely uniform sequence similarities within blocks were observed, maybe because the positions of the TF-binding sites are different between genes, allowing degeneracy within them to some extent. It is also possible that additional sequences as well as direct binding sites of TFs themselves should also be conserved, considering that the cognate sequences of the TFs are typically 6-10 bp long (Wray et al. 2003). Particular subregions of the promoter may not have been allowed to undergo free sequence divergence because the overall base composition or relative positions of TF-binding sites needed to be preserved. This could also explain the relatively flat patterns of sequence similarities within blocks. Extensive phylogenetic comparative analyses using forthcoming genomic sequences of other mammals (http://www.genome.gov/11007951) together with recently developed statistical methods (Elnitski et al. 2003) should lead to a more precise understanding of which sequences play a leading part, (serving as direct binding sites for TFs), and which play a supporting role.
We also observed that the sequence identity dropped just outside the blocks. It is possible that this is due to a discontinuous rate of random sequence substitution at the corresponding regions, despite the fact that the sequences themselves were continuous. However, the sequence identity outside the blocks was no more than 30%, even if the sequences were forced to be aligned (data not shown). This rate is somewhat lower than the conservation rate at neutral sites. It is unlikely that such extreme hot spots of random mutation are distributed within the regions 1 kb upstream of TSSs at such a frequency. It is more natural to suppose that totally unrelated sequences exist just outside the blocks. Consistently, the G+C content and CpG frequency were higher inside the blocks than outside (Table 3). This may also reflect that the sequences outside the blocks were foreign to the promoter sequences.
Genomic rearrangements, such as deletions, insertions, or recombination, may have taken place around the distal regions of the blocks. It is possible that the human genome has been rearranged significantly more in the course of evolution than previously thought. Although further confirmation is necessary, our result shown in Figure 2D also supports the idea that such segmentation prevails throughout the human and mouse genomes. Consistent with this possibility, recent publications have provided evidence that a large proportion of previously identified human-mouse syntenic regions contain multiple microrearrangements (Pevzner and Tesler 2003). Frazer et al. (2003a) observed genomic deletions, ranging from 0.2 to 8 kb in size, even between humans and chimpanzees. In particular, they observed integration of repetitive elements at the 3′-end boundaries of deletions in 23 out of 47 cases. In the present study, we showed that 46% of the 5′ ends of the blocks were bounded by interspersed repeats on either the human or mouse side (Table 1). Sometimes, the repetitive sequences may have acted as nucleation points for homologous recombination. In fact, it has been reported that this type of retroelement-mediated recombination has occasionally taken place in the human genome and is estimated to be responsible for at least 0.3% of human genetic disorders (Batzer and Deininger 2002).
Deletion of TF-binding sites could have accompanied some of the rearrangements. However, alterations that occurred inside the transcriptional regulatory modules in the promoters would mostly have been unfavorable for proper biological functions, and thus, would have been deleted from the population. The “block” structure we identified in the present study seemed to have formed as a consequence of such selective pressure. We observed that most of the previously characterized TF-binding sites were located within the blocks (Table 2). For these TF-binding sites, the cognate sequences as well as the relative positions of the TF-binding sites and distances to the TSSs were preserved.
Alterations that occurred outside blocks may generally have been tolerated. Some might have led to the acquisition of altered modes of transcriptional modulation. It has been reported that polymorphisms that cause an approximately twofold difference in transcription activation activities frequently occur without showing organismal phenotypes within human populations (Rockman and Wray 2002). Repetitive elements at the boundaries of the blocks could contribute to such modifications. There are a number of examples in which retroelements integrated in the vicinity of TSSs became involved in transcriptional regulation via changes in their sequences (Norris et al. 1995; Vansant and Reynolds 1995; Hamdi et al. 2000). It is likely that such variations have accumulated during evolution and have laid the genetic background to drive speciation during certain periods of time.
Intriguingly, we observed that the blocks in the PPRs were most encroached in the genes encoding transcription factors and genes whose expression patterns are brain specific (Fig. 3). This suggests that alterations within the proximal regions of the TSSs have been accumulated for these gene populations. It is possible that evolutionary diversification between humans and mice has been caused by slight changes in the regulation by TFs, which are located at the apexes of the regulatory hierarchy of transcriptional networks, rather than changes of the downstream proteins. Moreover, the evolutional changes may be the most significant in the genes expressed and functioning in the brain, which is the most distinctly different organ between humans and mice. Further characterization of the TF-binding sites that are similar to or distinctive in mice and humans as well as cross-validation of expression analyses should help to elucidate the molecular mechanisms underlying the alterations in transcriptional modulation responsible for the speciation of humans and mice. To this end, the present work has provided a first glimpse of how the modulation of transcriptional networks is likely to have differentially evolved between humans and mice.
MATERIALS AND METHODS
Promoter Data Set
The putative promoter regions were extracted by computational mapping of the 5′ ends of the human and mouse full-length cDNA sequences onto the corresponding genomic sequences obtained from UCSC Genome Browser (human: hg13; mouse: mm2). In total, 400,225 human and 580,209 mouse cDNAs were used to retrieve 8793 human and 6875 mouse promoters by the sequential use of BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat?command=start; BLAT) and SIM4 (http://pbil.univ-lyon1.fr/sim4.php; SIM4). The identified promoters were located about 4 kb upstream of the 5′ ends of the previously registered public cDNA sequences on average. Among the retrieved promoters, 3324 were correlated with each other as putative mutually orthologous genes using the table obtained from ftp://ftp.ncbi.nih.gov/pub/HomoloGene/. The statistics of the generated promoter data set are provided as Supplemental data Table 1. Details of the procedures for cDNA mapping and promoter pairing are described in Suzuki et al. (2004). Further information on the gene definitions used for the present study is also available in Supplemental data Table 1. As described there, at least two-thirds of the promoters were supported by three independently isolated full-length cDNAs. Considering that the average frequency of the full-length cDNAs (full-length-ness) in each of the libraries is >70%, there should be little chance that all of them are truncated. Also, we discarded all of the CDS-minus cDNAs, which increased the full-length-ness even more (for further discussion of this issue, please refer to Suzuki et al. 2001).
Sequence Alignment of the Promoters
LALIGN was obtained from http://www.ch.embnet.org/software/LALIGN_form.html and used for aligning sequences of the promoters with the default settings in the main text. The results of similar analyses using different parameter sets are shown in Supplemental data Figure 2. When LALIGN results split the sequence alignments allowing a large gap(s), most distal positions were recognized as the boundaries of the blocks. A graphical view of the sequence alignment and calculated sequence identities are shown in Supplemental data Figure 1.
For aligning nongenic regions, the putative syntenic regions were obtained according to the information from the UCSC genome alignment map (http://genome.cse.ucsc.edu/goldenPath/14nov2002/vsMm2/axtTight/). The alignments located within 100 kb for the Ensembl regions (http://genome.ucsc.edu/goldenPath/14nov2002/database/) were excluded and the 183,733 boundary sequences ranging from -1 kb to +200 bp were retrieved. Using these sequences, the alignments were generated using LALIGN.
SSEARCH was obtained from ftp://ftp.virginia.edu/pub/fasta/ as FASTA package programs. SSEARCH was used with default parameters for the detailed alignment of the sequences at the distal regions of the blocks and the proximal regions of the 5′ ends of the second exons. The results of a similar analysis using different parameter sets are shown in Supplemental data Figure 3.
Search for the Repetitive Sequences in the Promoters
The positions of the boundaries of the blocks were compared with those of annotated repetitive sequences. For positional information about the repetitive sequences, http://genome.ucsc.edu/goldenPath/14nov2002/bigZips/chromOut.zip and http://genome.ucsc.edu/goldenPath/mmFeb2002/bigZips/chromOut.zip were used for the human and mouse genomes, respectively. Classification of the repetitive sequences was also as described there.
Computational Prediction of the Putative TF-Binding Sites in the Promoters
For information about previously experimentally characterized TF-binding sites, TRANSFAC Professional 74 was used. For the computational prediction of the putative TF-binding sites, the promoter sequences were surveyed using MATCH. For the predictions, the cutoff value set of minFP.prf, which has been demonstrated to minimize “false positives”, were used.
Relating GO Criteria and Expression Profiles With the Sequence Divergence in the Promoters
The correlation tables between GO terms and RefSeq IDs were obtained from http://www.geneontology.org/. For each GO term, the frequencies of the promoters whose block lengths were greater or less than 600 bp were determined. As for the expression profiles, for those genes whose relative expression level was limited to a particular organ by more than 0.3, a similar calculation was performed. Classification of the organs is shown together with the iAFLP data file (http://cdna.ims.u-tokyo.ac.jp/iAFLP.xls). A detailed characterization of the iAFLP data will be published elsewhere.
Statistical significance of the difference in the frequencies of the encroached PPRs was evaluated by calculating hypergeometric distribution using the following equation:
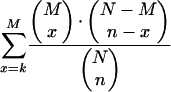 |
where N = 3324, n = 921, M = 203, k = 79 (“transcriptional regulators”) in the case of GO terms and N = 3324, n = 921, M = 156, k = 53 (“brain specific”) in the case of expression profiles.
Acknowledgments
We thank T. Hasui for his support with computational analyses of the promoters. We are grateful to K. Abe, M. Morinaga, M. Ishizawa, M. Kawamura, T. Mizuno, A. Kanai, and H. Hata for their excellent technical support. We are thankful to K. Koyanagi, M. Nakao, and E. Nakajima for helpful discussions and critical reading of the manuscript. This study was supported by a Grant-in-Aid for Scientific Research on Priority Areas and by Special Coordination Funds for Promoting Science and Technology (SCF), both from the Ministry of Education, Culture, Sports, Science, and Technology in Japan. This study was also supported by a Research Grant for the RIKEN Genome Exploration Research Project from the Ministry of Education, Culture, Sports, Science, and Technology of the Japanese Government to Y.H.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
References
- Batzer, M.A. and Deininger, P.L. 2002. Alu repeats and human genomic diversity. Nat. Rev. Genet. 3: 370-379. [DOI] [PubMed] [Google Scholar]
- Boguski, M.S. 2002. Comparative genomics: The mouse that roared. Nature 420: 515-516. [DOI] [PubMed] [Google Scholar]
- Carninci, P. and Hayashizaki, Y. 1999. High-efficiency full-length cDNA cloning. Methods Enzymol. 303: 19-44. [DOI] [PubMed] [Google Scholar]
- Cross, S.H. and Bird, A.P. 1995. CpG islands and genes. Curr. Opin. Genet. Dev. 5: 309-314. [DOI] [PubMed] [Google Scholar]
- Deininger, P.L. and Batzer, M.A. 2002. Mammalian retroelements. Genome Res. 12: 1455-1465. [DOI] [PubMed] [Google Scholar]
- Elnitski, L., Hardison, R.C., Li, J., Yang, S., Kolbe, D., Eswara, P., O'Connor, M.J., Schwartz, S., Miller, W., and Chiaromonte, F. 2003. Distinguishing regulatory DNA from neutral sites. Genome Res. 13: 64-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K.A., Chen, X., Hinds, D.A., Pant, P.V., Patil, N., and Cox, D.R. 2003a. Genomic DNA insertions and deletions occur frequently between humans and nonhuman primates. Genome Res. 13: 341-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K.A., Elnitski, L., Church, D.M., Dubchak, I., and Hardison, R.C. 2003b. Cross-species sequence comparisons: A review of methods and available resources. Genome Res. 13: 1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi, H.K., Nishio, H., Tavis, J., Zielinski, R., and Dugaiczyk, A. 2000. Alu-mediated phylogenetic novelties in gene regulation and development. J. Mol. Biol. 299: 931-939. [DOI] [PubMed] [Google Scholar]
- Hardison, R.C., Roskin, K.M., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13: 13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M.A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., et al. 2004. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32: D258-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X., Miller, W., Schwartz, S., and Hardison, R.C. 1992. Parallelization of a local similarity algorithm. Comput. Appl. Biosci. 8: 155-165. [DOI] [PubMed] [Google Scholar]
- Jareborg, N., Birney, E., and Durbin, R. 1999. Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Res. 9: 815-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai, J., Shinagawa, A., Shibata, K., Yoshino, M., Itoh, M., Ishii, Y., Arakawa, T., Hara, A., Fukunishi, Y., Konno, H., et al. 2001. Functional annotation of a full-length mouse cDNA collection. Nature 409: 685-690. [DOI] [PubMed] [Google Scholar]
- Kawamoto, S., Ohnishi, T., Kita, H., Chisaka, O., and Okubo, K. 1999. Expression profiling by iAFLP: A PCR-based method for genome-wide gene expression profiling. Genome Res. 9: 1305-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kel, A.E., Gossling, E., Reuter, I., Cheremushkin, E., Kel-Margoulis, O.V., and Wingender, E. 2003. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31: 3576-3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, M.C. and Wilson, A.C. 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107-116. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Liu, R., McEachin, R.C., and States, D.J. 2003. Computationally identifying novel NF-κB-regulated immune genes in the human genome. Genome Res. 13: 654-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, P.J. and Tjian, R. 1989. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 245: 371-378. [DOI] [PubMed] [Google Scholar]
- Nadeau, J.H. and Taylor, B.A. 1984. Lengths of chromosomal segments conserved since divergence of man and mouse. Proc. Natl. Acad. Sci. 81: 814-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, J., Fan, D., Aleman, C., Marks, J.R., Futreal, P.A., Wiseman, R.W., Iglehart, J.D., Deininger, P.L., and McDonnell, D.P. 1995. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J. Biol. Chem. 270: 22777-22782. [DOI] [PubMed] [Google Scholar]
- Novina, C.D. and Roy, A.L. 1996. Core promoters and transcriptional control. Trends Genet. 12: 351-355. [PubMed] [Google Scholar]
- Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., Nikaido, I., Osato, N., Saito, R., Suzuki, H., et al. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563-573. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. 1996. Effective protein sequence comparison. Methods Enzymol. 266: 227-258. [DOI] [PubMed] [Google Scholar]
- Pevzner, P. and Tesler, G. 2003. Genome rearrangements in mammalian evolution: Lessons from human and mouse genomes. Genome Res. 13: 37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praz, V., Perier, R., Bonnard, C., and Bucher, P. 2002. The Eukaryotic Promoter Database, EPD: New entry types and links to gene expression data. Nucleic Acids Res. 30: 322-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman, M.V. and Wray, G.A. 2002. Abundant raw material for cis-regulatory evolution in humans. Mol. Biol. Evol. 19: 1991-2004. [DOI] [PubMed] [Google Scholar]
- Roeder, R.G. 1996. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 21: 327-335. [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147: 195-197. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y. and Sugano, S. 2003. Construction of a full-length enriched and a 5′-end enriched cDNA library using the oligo-capping method. Methods Mol. Biol. 221: 73-91. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., Taira, H., Tsunoda, T., Mizushima-Sugano, J., Sese, J., Hata, H., Ota, T., Isogai, T., Tanaka, T., Morishita, S., et al. 2001. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep. 2,5: 388-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, Y., Yamashita, R., Shirota, M., Sakakibara, Y., Chiba, J., Mizushima-Sugano, J., Kel, A.E., Arakawa, T., Carninci, P., Kawai, J., et al. 2004. Large-scale collection and characterization of promoters of human and mouse genes. In Silico Biol. 4: 0036. [PubMed] [Google Scholar]
- Tautz, D. 2000. Evolution of transcriptional regulation. Curr. Opin. Genet. Dev. 10: 575-579. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ureta-Vidal, A., Ettwiller, L., and Birney, E. 2003. Comparative genomics: Genome-wide analysis in metazoan eukaryotes. Nat. Rev. Genet. 4: 251-262. [DOI] [PubMed] [Google Scholar]
- Vansant, G. and Reynolds, W.F. 1995. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc. Natl. Acad. Sci. 92: 8229-8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Wheeler, D.L., Church, D.M., Edgar, R., Federhen, S., Helmberg, W., Madden, T.L., Pontius, J.U., Schuler, G.D., Schriml, L.M., Sequeira, E., et al. 2004. Database resources of the National Center for Biotechnology Information: Update. Nucleic Acids Res. 32: D35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray, G.A., Hahn, M.W., Abouheif, E., Balhoff, J.P., Pizer, M., Rockman, M.V., and Romano, L.A. 2003. The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20: 1377-1419. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://dbtss.hgc.jp/; DBTSS.
- http://fantom.gsc.riken.go.jp/; FANTOM.
- ftp://ftp.virginia.edu/pub/fasta/; SSEARCH.
- http://genome.ucsc.edu/cgi-bin/hgBlat?command=start; BLAT.
- http://genome.ucsc.edu/downloads.html; UCSC Genome Browser.
- http://pbil.univ-lyon1.fr/sim4.php; SIM4.
- http://www.ch.embnet.org/software/LALIGN_form.html; LALIGN.
- http://www.ensembl.org/; Ensembl.
- http://www.epd.isb-sib.ch; Eukaryotic Promoter Database.
- http://www.gene-regulation.com/; TRANSFAC.
- http://www.geneontology.org/; GO.
- http://www.ncbi.nlm.nih.gov/RefSeq/; RefSeq.
- http://cdna.ims.u-tokyo.ac.jp/iAFLP.xls; iAFLP Expression Data.
- http://www.genome.gov/11007951; NHGRI Genome Projects.
- ftp://ftp.ncbi.nih.gov/pub/HomoloGene/; HomoloGene.
- http://genome.ucsc.edu/goldenPath/14nov2002/database/; Ensembl at UCSC.
- http://genome.ucsc.edu/goldenPath/14nov2002/bigZips/chromOut.zip; Human Genome.
- http://genome.ucsc.edu/goldenPath/mmFeb2002/bigZips/chromOut.zip; Mouse Genome.
- http://cdna.ims.u-tokyo.ac.jp/iAFLP.xls; iAFLP expression data.
- http://genome.cse.ucsc.edu/goldenPath/14nov2002/vsMm2/axtTight/; Human-Mouse Alignment.