

Abstract
The p300/CBP‐associated factor (PCAF) and related GCN5 bromodomain‐containing lysine acetyl transferases are members of subfamily I of the bromodomain phylogenetic tree. Iterative cycles of rational inhibitor design and biophysical characterization led to the discovery of the triazolopthalazine‐based L‐45 (dubbed L‐Moses) as the first potent, selective, and cell‐active PCAF bromodomain (Brd) inhibitor. Synthesis from readily available (1R,2S)‐(−)‐norephedrine furnished L‐45 in enantiopure form. L‐45 was shown to disrupt PCAF‐Brd histone H3.3 interaction in cells using a nanoBRET assay, and a co‐crystal structure of L‐45 with the homologous Brd PfGCN5 from Plasmodium falciparum rationalizes the high selectivity for PCAF and GCN5 bromodomains. Compound L‐45 shows no observable cytotoxicity in peripheral blood mononuclear cells (PBMC), good cell‐permeability, and metabolic stability in human and mouse liver microsomes, supporting its potential for in vivo use.
Keywords: bromodomains, chemical probes, epigenetics, medicinal chemistry, structure-based design
Bromodomains proteins (Brds) bind to acetylated lysines (KAc) through the Brd acetyllysine‐binding site. Misregulation of these proteins is linked to the onset and progression of multiple disease states, such as cancer.1 Significant efforts have been made recently to interrogate the role of these _targets through the development of chemical probes and inhibitors.2 Considerable work has focused on the BET family (Brd sub‐family II),3 however non‐BET4 Brds are increasingly receiving the attention of small molecule intervention efforts, with the disclosure of more than 10 new chemical probes/inhibitors in 2016.5
The p300/CBP‐associated factor, PCAF (KAT2B), is a multi‐domain protein containing a single Brd, an N‐terminal domain, and a histone acetyltransferase (HAT) domain. Known to associate with CBP6 and p3006b during transcription, misregulation of PCAF has been linked to cancer,7 HIV infection,7a, 8 and neuroinflammation.7a, 9 Despite predictions of high druggability10 and links with inflammatory disease,7a, 11 the role of PCAF and, more specifically, contributions of the Brd in such disease states are poorly understood. The development of a small molecule modulator of PCAF Brd would provide a useful tool for interrogating this potential therapeutic _target and allow for dissociation of the roles of the Brd and enzymatic domains in disease. Initial reports of PCAF Brd inhibitors were focused on disrupting interactions between the HIV‐1 peptide TAT‐1 and PCAF Brd.8a,8d Wang et al. reported the first PCAF Brd inhibitor, compound 1 (PCAF IC50 1.6 μm, Figure 1), which was effective at disrupting HIV‐1 replication (EC50 2.8 μm).8c Further efforts made by Hu et al.12 towards more potent compounds such as 2 were described without significant increases in potency or indication of selectivity (PCAF IC50 0.93 μm, EC50 11.5 μm, Figure 1). Additional chemotypes have been disclosed from fragment‐based screening by Chaikuad et al.5l Concurrent to this work, Constellation/Genentech reported compound 3 13 and others, which are potent PCAF inhibitors (AlphaLISA IC50 13 nm) but lack reported selectivity over other Brds (Figure 1).7b,7c Despite recent developments of PCAF Brd inhibitors, a potent, selective, and cell‐active chemical probe has not been reported. The work herein describes the discovery of such a probe.
Figure 1.
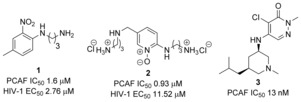
Reported PCAF bromodomain inhibitors.
Our first line of inquiry towards the first PCAF Brd chemical probe was focused on the core of non‐selective Brd inhibitors, bromosporine14 (PCAF isothermal titration calorimetry (ITC) K D: 5 μm) and [1,2,4]triazolo[4,3‐a]phthalazine15 derivatives as starting points. Small amine substituents, as in compounds 7–9 (Table 1), were designed to extend out of the narrow PCAF pocket and _target glutamic acid residues E750 and E756 at the edge of the KAc‐binding pocket through amine–acid salt bridge interactions (PDB: 5FE0).5l Commercially available 1,4‐dichlorophthalazine 4 underwent a scalable (up to 20 g) tandem SNAr/condensation reaction to furnish corresponding triazole intermediate 5 in good yields (Scheme 1). Significant efforts were employed to screen conditions using Pd‐catalyzed couplings of 5 with various amine nucleophiles; disappointing yields or lack of reactivity were observed in all of these cases. It was found that a KI/HCl‐catalyzed SNAr reaction allowed for a tractable divergent synthesis of various N‐linked derivatives (Scheme 1).
Table 1.
Amino‐substituted triazolophthalazine are potent PCAF Brd inhibitors.
| Compound | R1 | R2 | R3 | R4 | n | ΔT m [°C][a] | K D [μm] (ITC) |
|---|---|---|---|---|---|---|---|
| 7 | Me | H | H | Me | 1 | 8.5[b] | 8.0±0.65 |
| 8 | Me | H | H | Me | 2 | ND | >30 |
| 9 | Me | H | H | Me | 3 | ND | >30 |
| 10 | Me | H | Ph | Me | 1 | 1.7 | 1.0 |
| 11 | Me | Me | H | Me | 1 | 5.6 | 0.30±0.039 |
| (S)‐11 | Me | Me | H | Me | 1 | 7.4 | 0.28±0.029 |
| 12 | Me | Et | H | Me | 1 | 3.3 | 1.8±0.23 |
| 13 | Me | iBu | H | Me | 1 | 0.85 | >30 |
| 14 | Me | Me | H | Et | 1 | 0.0 | >30 |
| 15 | Me | H | Me | Me | 1 | 4.6 | 7.3±1.1 |
| 16 | Me | H | Et | Me | 1 | ND | 6.9±1.4 |
| (S)‐17 | CF3 | Me | H | Me | 1 | 0.65 | >30 |
[a] Compound concentration 10 μm, unless stated otherwise; [b] Compound concentration 100 μm; ND: not determined.
Scheme 1.
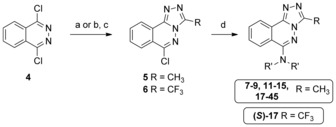
Synthesis of [1,2,4]triazolo[4,3‐a]phthalazine derivatives. Reagents and conditions: a) Acetohydrazide, DMF 120 °C 16 h, 62 %; b) N2H4 ⋅H2O, EtOH, 120 °C, 10 min, quant.; c) TFA, 100 °C, 2 h, 43 %; d) R′2NH (1.5–2.0 equiv) KI (0.1 equiv), HCl (0.05 equiv), EtOH or iPrOH, reflux, 3 days 8–94 %.
After the synthesis of a focused set of 20 compounds, screening conducted using a differential scanning fluorimetry (DSF) assay revealed two hits, dimethylamino compounds 7 and 10 (Table 1). It was found that compounds 8 and 9 featuring a longer amine chain were less potent. With the 2‐(dimethylamino)ethyl group of compounds 7 and 10 identified as optimal substituents, a virtual library of ∼12k compounds was constructed by in silico reaction of compound 5 with commercial compounds containing the 2‐(dimethylamino)ethyl motif.16 Over 60 compounds bearing a tethered 1,2‐diamine motif were chosen for synthesis based on docking score, diversity, and potential for new interactions with the PCAF Brd (Table 1, compounds 11–16 and Tables S1 and S2).
Table 2.
PCAF Brd‐binding affinity of compounds 39–45 measured by ITC.
| Compound | R | Configuration | K D (nm) (ITC) |
|---|---|---|---|
| 39 | F | (1S*, 2S*) | 195±23 |
| 40 | CO2Me | (1S*, 2S*) | 133±15 |
| 41 | Me | (1S*, 2S*) | 160±54 |
| 42 | Cl | (1S*, 2S*) | 223±78 |
| 43 | CF3 | (1S*, 2S*) | 163±117 |
| 44 | OMe | (1S*, 2S*) | 179±48 |
| 45 | H | (1S*, 2S*) | 168±27 |
| L ‐45/L‐Moses | H | (1S, 2S) | 126±15 |
| D ‐45 | H | (1R, 2R) | Inactive |
Derivatives were screened for PCAF Brd affinity by ITC, leading to the discovery of compound 11 (Table 1). By ITC, the stoichiometry of binding showed that all of the activity of the racemate lay in a single enantiomer, later found to have (S)‐configuration after synthesis using enantiopure building blocks (11 ITC K D 0.30 μm, Brd/11 2:1; (S)‐11 K D 0.28 μm, Brd/(S)‐11 1:1). Groups larger than a methyl substituent at R2 were detrimental to activity (compounds 12, 13) as was a bulkier N,N‐diethyl substituent (compound 14). Although a phenyl substituent at R3 conferred potency to compound 10, compounds 15 and 16 with smaller methyl and ethyl groups were less potent. Compound (S)‐17 featuring a trifluoromethyl group at position R1 caused a loss in activity consistent with previously reported Brd SAR of the [1,2,4]triazolo[4,3‐a]phthalazines.15
In a DSF panel of 48 human Brds, compound (S)‐11 showed binding to PCAF and GCN5 with no observable activity against other Brds (Figure S1). To improve the potency of (S)‐11, it was rationalized that a combination of appropriate substituents at R2/R3 might improve the avidity of binding interactions and addition of an aryl group at R3 would serve as a chemical handle for introduction of new functionality. The R2/R3‐substituted compounds would be a hybrid of the most potent analogues 10 and (S)‐11.
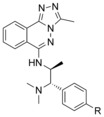
Synthesis of aryl substituted compounds was achieved through a non‐selective aza‐Henry reaction with p‐substituted benzaldehydes (Scheme 2). p‐Substituted benzaldehydes were chosen as provisional in silico scoring of potential inhibitors suggested that o‐ or m‐substitutions would be less tolerated for binding. Highly unstable olefins 18–24 were telescoped through a diastereoselective (d.r. 4.6:1–33:1) nitro–olefin conjugate addition furnishing racemic (S*,S*)‐configured17 compounds 25–31, then reduced to corresponding amines, 32–38, using either Pd/C‐ or Raney/Ni‐catalyzed hydrogenation. Compounds 32–38 were isolated as single diastereomers and submitted to the aforementioned KI‐catalyzed SNAr reaction (Scheme 2) to produce compounds 39–45 in low to good yields (16–79 %). Racemic compounds were screened by ITC for PCAF‐binding affinity (Table 2). All of the compounds showed an increase in potency compared to compound (S)‐11, with the simple unsubstituted derivative 45 having highest potency.
Scheme 2.
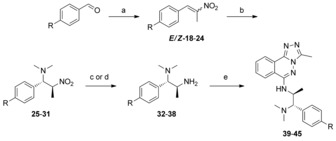
Synthesis of threo‐substituted derivatives 39–45. Reagents and conditions: a) NH4OAc (0.2 equiv), EtNO2, reflux, 1:1 E/Z, quant.; b) Me2NH (5 equiv), THF, RT, 16 h, d.r. 4.6:1–33:1; c) H2 (1 atm), Pd/C (10 %), MeOH, RT, 16 h, 11–15 % over two steps, single diastereomer; d) H2 (1 atm), Ra/Ni (0.3 equiv), MeOH, RT, 16 h, 25–28 %, over two steps, single diastereomer; e) 5 (0.8 equiv) KI (0.1 equiv), HCl (0.05 equiv), EtOH or iPrOH, reflux, 3 days 16–79 %.
Pleasingly, it was found following resolution by preparative chiral stationary phase HPLC, that active enantiomer L ‐45, which was dubbed L ‐Moses, showed good binding affinity for PCAF Brd (PCAF KD 126 nm, ITC). The other enantiomer D ‐45 showed no observable binding, implying its utility as an inactive control compound. Having achieved good potency against PCAF Brd, L ‐45 was then screened for selectivity against the panel of 48 human bromodomains using DSF (Figure 2 B). Homologous Brd of GCN5 was the only other Brd that showed any affinity for L ‐45, confirmed by ITC (ΔT m+3.6 °C, KD 0.55 μm). L ‐45 competitively displaced a biotinylated tool derivative, compound 46 (Supporting Information) in a homogeneous time‐resolved resonance fluorescence (HTRF) assay (PCAF Ki 47 nm), corresponding to exquisite selectivity over BRD4 (>4500‐fold selective).
Figure 2.
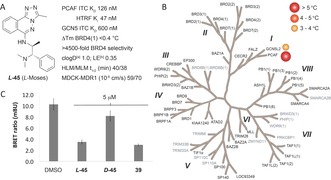
A) Profile of L ‐45. B) L ‐45 is selective in a DSF assay panel of 48 Brds (black text). C) Displacement of PCAF‐Brd from H3.3‐nanoLuc in live HEK‐293 cells using the nanoBRET assay. [a] clogD was calculated using ChemAxon.18 [b] Ligand efficiency.19
In a cellular context, L ‐45 was shown to displace nanoLuciferase‐tagged PCAF‐Brd from halo‐tagged‐H3.3 in a nanoBRET _target engagement assay at a single digit μm concentration (Figure 2 C).20 Inactive enantiomer D ‐45 had no effect in the same assay.
Compounds DL‐ 45 and p‐fluoro derivative 39 were then tested for liver microsomal stability in vitro. DL‐ 45 showed good metabolic stability in both human (t 1/2 40 min) and mouse (t 1/2 38 min) liver microsome preparations. para‐F derivative 39 showed a slightly increased metabolic stability in both human (t 1/2 48 min) and mouse (t 1/2 65 min) liver enzymes, likely due to metabolic protection of the para‐substituted aryl ring. DL‐ 45 showed good kinetic solubility (>200 μm) and permeability in MDCK‐MDR1 cells with low efflux (Figure 2 A). L ‐45 was also tested in peripheral blood mononuclear cells and showed no observable cytotoxicity after treatment at 10 μm for 24 hours.
Although attempts to obtain a co‐crystal structure of recombinant PCAF with L ‐45 were unsuccessful, which was surprising given that numerous structures of less potent PCAF fragments have been reported recently.5l A structure using highly homologous (64 % identity) Brd from Plasmodium falciparum, PfGCN5, of which L ‐45 is also a potent ligand (ITC KD 280 nm), was successfully obtained (PDB: 5TPX, Figure 3). L ‐45 bound as expected in the KAc‐binding site of PfGCN5 with key interactions that include a salt bridge between E1389 (conserved in PCAF as E756) and the dimethylamino motif of L ‐45 (Figure 3 A). Additional contacts are also observed in the form of an edge‐to‐face π‐π stacking interaction between W1379 (conserved in PCAF as W746) and the phenyl substituent of L ‐45 (average distance 4.5 Å); a π–π stacking interaction between Y1442 (conserved in PCAF as Y809) and pyridazo ring of the triazolophthalazine motif (average distance 3.7 Å); and characteristic H‐bonds from the triazolophthalazine group and N1436 residue (conserved in PCAF as N803) and a water molecule. Intolerance of substitution of L ‐45 in R2 and R3 positions (compounds 12–16, Table 1) was rationalized by the tight fit of the alkyl amine chain of L ‐45 (Figure 3 B). Interestingly, K1383 in PfGCN5 is substituted with E750 in human PCAF, and as such the Plasmodium homologue features a slightly open KAc‐binding site (Figure 3 B). _targeting this difference may allow for design of Plasmodium‐selective Brd inhibitors. As previously supported by SAR, the absolute configuration of L ‐45 was confirmed to be (1S,2S).
Figure 3.
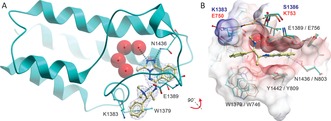
Co‐crystal structure of L ‐45 with PfGCN5 (PDB ID 5TPX). A) L ‐45 (pale sticks) binds in the KAc‐binding pocket of PfGCN (blue ribbon and sticks) and makes H‐bonds (dotted lines) through the triazole to N1436 and the first of a network of four water molecules (red spheres). The dimethylamino group forms a salt bridge with E1389. Blue mesh: 2 F o F c omitted map contoured at 2.5 σ. B) Surface view of complex of PfGCN5 (surface, blue sticks) and L ‐45 (pale sticks). The phenyl group of L ‐45 lies in a hydrophobic groove between W1379 and the alkyl linker of K1383. The structure of PCAF (orange sticks, PDB ID 5FTZ) is superimposed to show key residue similarities (black text PfGCN5/PCAF) and differences (blue text PfGCN5, red text PCAF).
For the asymmetric synthesis of L ‐45, commercially available (1R,2S)‐(−)‐norephedrine was Boc‐protected and cyclized to a sulfamidite and then directly oxidized using sodium periodate to boc‐protected sulfamidate 46 in reasonable yields (Scheme 3). Subsequent treatment with dimethylamine facilitated regio‐selective ring opening of sulfamidate 46,21 extruding SO3 and furnishing protected diamine 47 as a single diastereoisomer with inversion of configuration at the benzylic centre. Following a deprotection of 47 to the free amine and SNAr with aryl chloride 5, L ‐45 was furnished in six steps as a single stereoisomer.
Scheme 3.
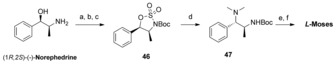
Asymmetric synthesis of L ‐45. Reagents and conditions: a) Boc2O, DIPEA, CH2Cl2, RT, 16 h, 51 % b) SOCl2, Pyridine, MeCN, 2 h, −40 °C to 0 °C; c) NaIO4 (1.5 equiv), RuCl3⋅3 H2O (0.05 equiv), MeCN, 1 h, 0 °C, 48 % (over two steps); d) Me2NH (3 equiv), THF, RT, 16 h, 63 %; e) TFA, CH2Cl2, quant.; f) 5 (0.8 equiv) KI (0.1 equiv), HCl (0.05 equiv), iPrOH, reflux, 3 days, 30 %.
In conclusion, we report the discovery of L ‐45, the first nanomolar, selective, and cell‐active chemical probe of the PCAF bromodomain. Iterative cycles of rational inhibitor design, in silico docking studies, and synthesis furnished L ‐45 after generation of a focused PCAF inhibitor library. L ‐45 shows a clean toxicity profile in primary PBMCs, and disrupts interactions between PCAF Brd and H3.3 in HEK293 cells, indicating cellular _target engagement.
Good cell permeability in a MDCK‐MDR1 assay and stability to metabolism in both human and mouse liver microsomes indicate that L ‐45, dubbed L ‐Moses, may also have utility in vivo. L ‐Moses will allow for robust interrogation of PCAF Brd inhibition and pharmacological effects in relevant diseases models. Future work will investigate the use of L ‐Moses in functional assays pertaining to PCAF‐associated diseases.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge Cyprotex for providing in vitro metabolism data and Charles Rivers Laboratories for providing MDCK‐MDR1 cell permeability data. Authors thank S. Velupillai for aiding the crystallographic analysis. SGC is a registered charity (number 109773 7) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA [ULTRA‐DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation‐FAPESP, Takeda, and Wellcome Trust [092809/Z/10/Z]. M.M. is supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1). P.G.K.C. gratefully acknowledges the Woolf Fisher Trust. We would also like to extend our gratitude to Tiger M. Frystone and Maximilian H. K. Brennan for preparing compounds 8 and 9.
M. Moustakim, P. G. K. Clark, L. Trulli, A. L. Fuentes de Arriba, M. T. Ehebauer, A. Chaikuad, E. J. Murphy, J. Mendez-Johnson, D. Daniels, C.-F. D. Hou, Y.-H. Lin, J. R. Walker, R. Hui, H. Yang, L. Dorrell, C. M. Rogers, O. P. Monteiro, O. Fedorov, K. V. M. Huber, S. Knapp, J. Heer, D. J. Dixon, P. E. Brennan, Angew. Chem. Int. Ed. 2017, 56, 827.
Contributor Information
Prof. Dr. Darren J. Dixon, Email: darren.dixon@chem.ox.ac.uk
Prof. Dr. Paul E. Brennan, Email: paul.brennan@sgc.ox.ac.uk.
References
- 1.
- 1a. Arrowsmith C. H., Audia J. E., Austin C., et al., Nat. Chem. Biol. 2015, 11, 536–541; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Arrowsmith C. H., Bountra C., Fish P. V., et al., Nat. Rev. Drug Discovery 2012, 11, 384–400; [DOI] [PubMed] [Google Scholar]
- 1c. Filippakopoulos P., Knapp S., Nat. Rev. Drug Discovery 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- 2. http://www.thesgc.org/chemical-probes.
- 3.
- 3a. Filippakopoulos P., Qi J., Picaud S., et al., Nature 2010, 468, 1067–1073; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Garnier J. M., Sharp P. P., Burns C. J., Expert Opin. Ther. Pat. 2014, 24, 185–199. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Theodoulou N. H., Tomkinson N. C. O., Prinjha R. K., et al., ChemMedChem 2016, 11, 477–487; [DOI] [PubMed] [Google Scholar]
- 4b. Moustakim M., Clark P. G. K., Hay D. A., et al., Med. Chem. Commun. 2016, 7, 2246–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Gerstenberger B. S., Trzupek J. D., Tallant C., et al., J. Med. Chem. 2016, 59, 4800–4811; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Bamborough P., Barnett H. A., Becher I., et al., ACS Med. Chem. Lett. 2016, 7, 552–557; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Bennett J., Fedorov O., Tallant C., et al., J. Med. Chem. 2016, 59, 1642–1647; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Unzue A., Xu M., Dong J., et al., J. Med. Chem. 2016, 59, 1350–1356; [DOI] [PubMed] [Google Scholar]
- 5e. Sutherell C. L., Tallant C., Monteiro O. P., et al., J. Med. Chem. 2016, 59, 5095–5101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Palmer W. S., Poncet-Montange G., Liu G., et al., J. Med. Chem. 2016, 59, 1440–1454; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5g.W. Palmer, P. Jones, G. Liu, et al., University of Texas System, USA, 2016, p. 166;
- 5h. Martin L. J., Koegl M., Bader G., et al., J. Med. Chem. 2016, 59, 4462–4475; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5i. Crawford T. D., Tsui V., Flynn E. M., et al., J. Med. Chem. 2016, 59, 5391–5402; [DOI] [PubMed] [Google Scholar]
- 5j. Cox O. B., Krojer T., Collins P., et al., Chem. Sci. 2016, 7, 2322–2330; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5k. Chen P., Chaikuad A., Bamborough P., et al., J. Med. Chem. 2016, 59, 1410–1424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5l. Chaikuad A., Lang S., Brennan P. E., et al., J. Med. Chem. 2016, 59, 1648–1653; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5m. Bamborough P., Chung C. w., Demont E. H., et al., Angew. Chem. Int. Ed. 2016, 55, 11382–11386; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11554–11558. [Google Scholar]
- 6.
- 6a. Bannister A. J., Kouzarides T., Nature 1996, 384, 641–643; [DOI] [PubMed] [Google Scholar]
- 6b. Ogryzko V. V., Schiltz R. L., Russanova V., et al., Cell 1996, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.L. Kruidenier, K. Lee, D. F. Tough, et al., Glaxo Group Limited, UK, 2014, p. 38;
- 7b.B. K. Albrecht, A. Cote, T. Crawford, et al., Genentech, Inc., USA, Constellation Pharmaceuticals, Inc., USA, 2016, p. 179;
- 7c.B. K. Albrecht, A. Cote, T. Crawford, et al., Genentech, Inc., USA, Constellation Pharmaceuticals, Inc. USA, 2016, p. 95.
- 8.
- 8a. Mujtaba S., He Y., Zeng L., et al., Mol. Cell 2002, 9, 575–586; [DOI] [PubMed] [Google Scholar]
- 8b. Quy V. C., Pantano S., Rossetti G., et al., Biology 2012, 1, 277–296; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Wang Q., Wang R., Zhang B., et al., MedChemComm 2013, 4, 737–740; [Google Scholar]
- 8d. Dorr A., Kiermer V., Pedal A., et al., EMBO J. 2002, 21, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.M.-M. Zhou, G. Gerona-Navarro, Y. Rodriguez-Fernandez, et al., Icahn School of Medicine at Mount Sinai, USA, 2015, p. 87.
- 10. Vidler L. R., Brown N., Knapp S., et al., J. Med. Chem. 2012, 55, 7346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Bastiaansen A. J. N. M., Ewing M. M., de Boer H. C., et al., Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1902–1910; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Deng W.-G., Zhu Y., Wu K. K., Blood 2004, 103, 2135–2142. [DOI] [PubMed] [Google Scholar]
- 12. Hu P., Wang X., Zhang B., et al., ChemMedChem 2014, 9, 928–931. [DOI] [PubMed] [Google Scholar]
- 13.B. K. Albrecht, D. J. Burdick, A. Cote, et al., Genentech, Inc., USA, Constellation Pharmaceuticals, Inc. USA, 2016, p. 117.
- 14.
- 14a. http://www.thesgc.org/chemical-probes/Bromosporine;
- 14b. Picaud S., Leonards K., Lambert J.-P., et al., Sci. Adv. 2016, 2, e1600760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fedorov O., Lingard H., Wells C., et al., J. Med. Chem. 2014, 57, 462–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neves M. A. C., Totrov M., Abagyan R., J. Comput.-Aided Mol. Des. 2012, 26, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Southwick P. L., Anderson J. E., J. Am. Chem. Soc. 1957, 79, 6222–6229. [Google Scholar]
- 18. https://www.chemaxon.com/library/pka-and-logp-property-prediction-and-training/.
- 19. Hopkins A. L., Groom C. R., Alex A., Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- 20. Machleidt T., Woodroofe C. C., Schwinn M. K., et al., ACS Chem. Biol. 2015, 10, 1797–1804. [DOI] [PubMed] [Google Scholar]
- 21. Meléndez R. E., Lubell W. D., Tetrahedron 2003, 59, 2581–2616. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary