

Abstract
Obesity and its associated complications such as insulin resistance and non-alcoholic fatty liver disease are reaching epidemic proportions. In mice, the TGF-β superfamily is implicated in the regulation of white and brown adipose tissue differentiation. The kielin/chordin-like protein (KCP) is a secreted regulator of the TGF-β superfamily pathways that can inhibit both TGF-β and activin signals while enhancing bone morphogenetic protein (BMP) signaling. However, KCP's effects on metabolism and obesity have not been studied in animal models. Therefore, we examined the effects of KCP loss or gain of function in mice that were maintained on either a regular or a high-fat diet. KCP loss sensitized the mice to obesity and associated complications such as glucose intolerance and adipose tissue inflammation and fibrosis. In contrast, transgenic mice that expressed KCP in the kidney, liver, and adipose tissues were resistant to developing high-fat diet-induced obesity and had significantly reduced white adipose tissue. Moreover, KCP overexpression shifted the pattern of SMAD signaling in vivo, increasing the levels of phospho (P)-SMAD1 and decreasing P-SMAD3. Adipocytes in culture showed a cell-autonomous effect in response to added TGF-β1 or BMP7. Metabolic profiling indicated increased energy expenditure in KCP-overexpressing mice and reduced expenditure in the KCP mutants with no effect on food intake or activity. These findings demonstrate that shifting the TGF-β superfamily signaling with a secreted protein can alter the physiology and thermogenic properties of adipose tissue to reduce obesity even when mice are fed a high-fat diet.
Keywords: adipocyte, bone morphogenetic protein (BMP), metabolic syndrome, obesity, transforming growth factor β (TGF-β)
Introduction
Energy balance is critical for maintaining normal body weight and homeostasis. When caloric intake chronically exceeds energy expenditure, white adipose tissue (WAT)2 stores excess energy in the form of triglycerides, leading to obesity and complications related to insulin resistance such as type 2 diabetes and metabolic syndrome (1). Metabolic disease is reaching epidemic proportions in the developed world, primarily due to the increased availability of high caloric foods and the decrease in daily physical activity (2, 3). WAT functions not only as a nutrient storage center but also as an endocrine organ by secreting proteins such as leptin and adiponectin that provide signals to regulate appetite and nutrient metabolism. In contrast to nutrient storage, brown adipose tissue (BAT) functions as a thermogenic organ by converting triglycerides into heat to maintain body temperature in homotherms (4). In humans, physiologically active BAT was identified in adults and inversely correlated with age and body mass index (5–7). Recent investigations into the origin and regulation of brown adipocytes have revealed pathways for brown adipocyte differentiation and molecular and physiological distinctions between developmentally programmed BAT, postnatally generated BAT, and inducible or beige/brite BAT (8, 9).
Among the most debilitating effects of obesity are inflammation and fibrosis in adipose tissue, liver, and other organs (10–13). In adipose tissue, increased numbers of macrophages are associated with the secretion of proinflammatory cytokines and increased extracellular matrix deposition (14, 15). A more recent study suggests that inflammation in adipose tissue does have an adaptive advantage for tissue remodeling in the absence of which more ectopic lipid accumulation is seen in other tissues (16). Obesity and insulin resistance are also closely linked to non-alcoholic fatty liver disease, which covers a broad spectrum of liver pathology that includes inflammation, fibrosis, hepatic steatosis, and cirrhosis and may ultimately progress to hepatocellular carcinoma (17).
TGF-β is a proinflammatory cytokine whose role in kidney, liver, lung, and cardiac fibrosis is well documented. More recently, the importance of TGF-β superfamily signaling in determining the types of adipose tissue and the effects of a high-fat diet (HFD) on obesity and associated complications have been studied in several different experimental model systems (18, 19). A deletion of SMAD3 in mice prevented HFD-induced obesity, hepatic steatosis, and insulin resistance with phenotypic changes in WAT tissue that resembled BAT (20). The BMP proteins are also implicated in the transition of WAT to a more brown or beige phenotype. Elevated expression of BMP4 in adipose tissue of transgenic mice reduced the mass of gonadal WAT and promoted a beige phenotype (21). Strikingly, a deletion of the BMP type 1A receptor in brown fat depletes these tissues in mice but results in a compensatory change in white fat to mimic a brownlike, or beige, fat phenotype (22). Induction of BMP8 through a HFD can increase thermogenesis by enhanced signaling through the p38MAPK/cAMP-response element-binding protein pathway in BAT and regulation of neuropeptides in response to norepinephrine in the hypothalamus (23).
The regulation of TGF-β superfamily signaling is complex and encompasses mechanisms that function both within and outside of the cell. The TGF-β family ligands are disulfide-linked homo- or heterodimers that form heterotetramers with type I and type II transmembrane receptor kinases. Activated type II receptor phosphorylates the type I receptor, a serine/threonine kinase that phosphorylates the receptor-activated SMAD proteins, which transduce the signal to the nucleus. The accessibility of either TGF-β or BMPs to their corresponding receptors can be inhibited or enhanced by the actions of secreted proteins. Extracellular inhibitors of BMP signaling include vertebrate chordin, which binds directly to BMPs through the cysteine-rich (CR) domains containing CXXCXC and CCXXC motifs (24–26). Although chordin blocks BMP/receptor interactions, the multi-CR domain protein KCP enhances BMP/receptor interactions to increase the efficacy of signaling (27). However, KCP is also able to inhibit TGF-β and activin signaling (28). Conversely, the CR domain protein connective tissue growth factor enhances TGF-β-mediated signaling while suppressing the BMP-dependent pathway (29). Mice homozygous for a mutant kcp allele show no gross developmental abnormalities, although kcp mutations can enhance the renal developmental phenotype in mutants of Cv2 (30), a gene that encodes a related multi-CR domain activator of BMPs. In animal models of renal disease, kcp−/− mice are sensitized to developing renal interstitial fibrosis (27), a process regulated by both BMPs and TGF-β. The kcp−/− mice exhibit lower BMP signaling and higher TGF-β signaling as measured by specific P-SMAD readouts after renal injury. Conversely, in mice that express a KCP transgene, a reciprocal shift in the balance of TGF-β and BMP signaling made animals more resistant to developing interstitial fibrosis in the kidney (31). In the livers of aged mice or in mice fed a high-fat diet to induce non-alcoholic fatty liver disease, loss of KCP promoted hepatic steatosis and liver fibrosis, whereas expression of a KCP transgene was protective (32).
In this report, we describe how loss and gain of KCP function impact body mass, body composition, and the effects of diet-induced obesity on nutrient metabolism. Initial observations indicated that kcp−/− mice gained more weight over time, whereas KCP transgenic mice (kcpTg) remained lean. The KCP protein altered the amount and type of body fat to greatly impact metabolism. Thus, transgenic KCP expression protected mice from HFD-induced obesity, whereas the KCP-deficient mutant mice are more susceptible to metabolic dysfunction even on a normal diet (ND). By modulating KCP protein to alter the signaling balance between BMPs and TGF-β and associated P-SMADs, fat composition, thermoregulation, and metabolism were all impacted. These data suggest that altering TGF-β superfamily signaling by secreted regulatory proteins can attenuate the negative effects of obesity-induced metabolic syndrome.
Results
KCP transgenic mice are protected from diet-induced obesity
We had created a kcp−/− allele by conventional gene _targeting (27) and a kcpTg allele using the mouse PEPCK promoter driving the KCP cDNA coding region with an amino-terminal human Igκ secretory signal and a carboxyl-terminal myc epitope tag (31). Both kcp−/− and kcpTg mice were viable and fertile with no gross morphological defects. As reported previously, aged kcp−/− mice exhibit liver pathology that was accentuated when the mice were put on a high-fat diet to induce obesity (32). To more precisely quantitate the changes in body mass, growth curves were determined for kcp−/−, kcp+/+, and kcpTg mice when fed a ND or HFD (Fig. 1, A and B). On normal diet, both kcp+/+ and kcp−/− mice were on average about 25% heavier than kcpTg. After 12 weeks on a high-fat diet, both kcp+/+ and kcp−/− were more than 80% heavier than the kcpTg mice. In fact, the kcpTg mice did not gain significantly more weight on the high-fat diet compared with the normal diet. We did not observe any gross skeletal abnormalities or evidence of dwarfism as body lengths were not significantly different between the genotypes. Body composition analyses indicated that the difference in body weights on both diets between WT or kcp−/− and the kcpTg mice was due in large part to less white adipose tissue in the kcpTg mice with no significant difference in fluid composition (Fig. 1, C and D).
Figure 1.
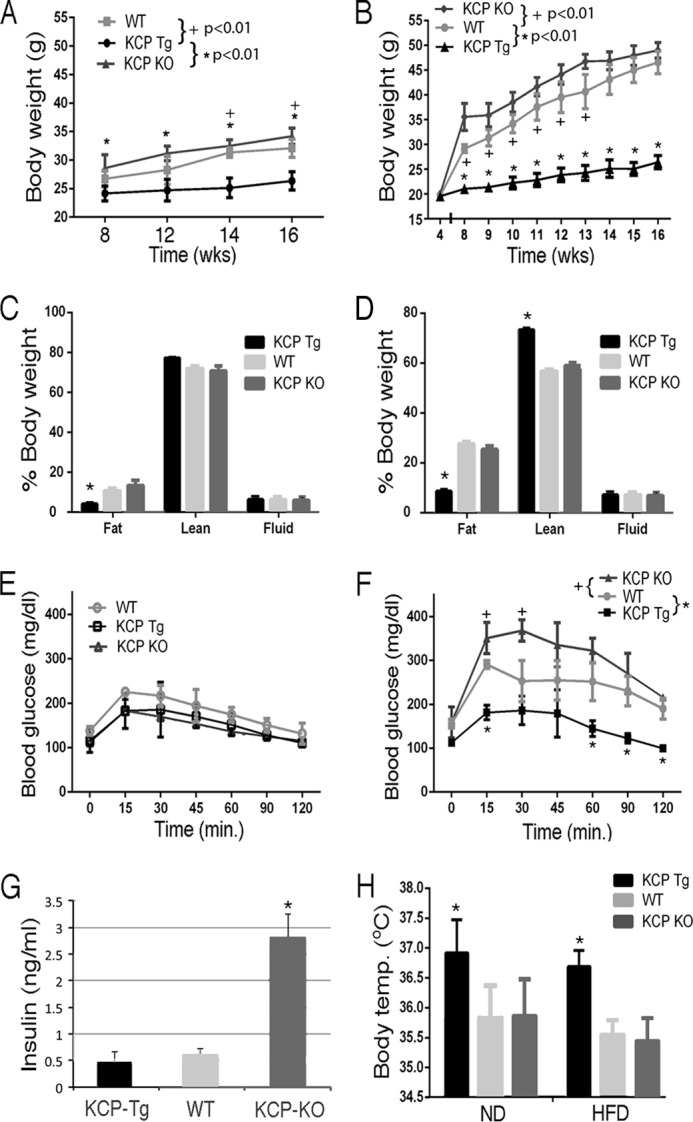
Growth curves, body composition, glucose tolerance, and body temperatures. A, body mass of kcp+/+, kcpTg, and kcp−/− mice from 8 to 16 weeks of age while maintained on a ND. Statistically significant p values are as indicated. B, body mass of kcp+/+, kcpTg, and kcp−/− mice from 8 to 16 weeks while maintained on a HFD. Statistically significant p values are as indicated. C, percent body composition of kcp+/+, kcpTg, and kcp−/− mice after 8 weeks on a ND as in A. Significant differences between kcpTg and kcp+/+ and kcp−/− are indicated (*, p < 0.05). D, body composition after 8 weeks on a HFD as in B. Significant differences between kcpTg and kcp+/+ and kcp−/− are indicated (*, p < 0.05). E, glucose tolerance test of kcp+/+, kcpTg, and kcp−/− mice after 12 weeks on a ND as in A. No significant differences are indicated. F, glucose tolerance test of kcp+/+, kcpTg, and kcp−/− mice after 12 weeks on a HFD as in B. Significant differences are observed at the indicated time points between cohorts as indicated (+ and *, p < 0.05). G, average fasting insulin levels as measured in kcp+/+, kcpTg, and kcp−/− mice (n = 3). Significant differences were observed for kcp−/− mice (*, p < 0.05, ANOVA). H, mean body temperatures (Temp.) of kcp+/+, kcpTg, and kcp−/− mice after 12 weeks of a ND or 8 weeks of a HFD. Significant differences are indicated for kcpTg (*, p < 0.05, ANOVA). Error bars in all panels are one standard deviation from the mean.
Obesity-induced metabolic disorder in mice can lead to glucose intolerance and insulin resistance. To assess the effects of KCP loss and overexpression on glucose metabolism, we performed glucose tolerance testing (Fig. 1, E and F). kcp+/+, kcp−/−, and kcpTg mice maintained on normal diets had similar glucose tolerance. However on the high-fat diets, the kcp+/+ and kcp−/− mice had impaired glucose tolerance compared with their lean counterparts, whereas the kcpTg mice showed glucose uptake similar to the mice on a normal diet (180 ± 25 mg/dl after 15 min). In those mice, fasting insulin levels were significantly higher in the kcp−/− animals, consistent with the development of glucose intolerance (Fig. 1G).
Metabolic profiles of KCP transgenic and mutant mice
To evaluate the mechanism of impaired weight gain in the kcpTg mice, we first measured average body temperatures as an indicator of metabolic function and thermoregulation. The kcpTg mice had significantly higher average body temperatures, almost a full degree more, than kcp+/+ or kcp−/− mice regardless of the diet conditions (Fig. 1H). Subsequently, mice of all three genotypes were fed either normal or high-fat diets for 8 weeks and then adapted to single metabolic cages for 4 weeks. Multiple metabolic parameters were then measured over a 3-day time period (Fig. 2A). Among the mice fed a normal diet, the only significant difference among the genotypes was that the kcpTg mice oxidized more glucose compared with kcp+/+ and kcp−/−. However, the mice kept on a HFD exhibited multiple differences among the three genotypes. The kcpTg mice had higher total energy expenditure compared with kcp+/+, whereas the kcp−/− mice expended less energy than kcp+/+. The differences are best illustrated in average oxygen consumption, as adjusted for lean body mass, over the light and dark cycles of a 3-day test period (Fig. 2B). The kcpTg mice used ∼14% more oxygen compared with kcp+/+, whereas the kcp−/− mice used 25% less oxygen over the 3-day period. Similar statistically significant data can be seen when energy expenditure is averaged over time for the three genotypes (Fig. 2C). The increased energy expenditure in kcpTg mice was not reflected by any more activity or any more food consumption as there were no significant differences among the three genotypes. The kcpTg mice were able to oxidize more glucose and fat compared with kcp+/+, whereas kcp−/− mice oxidized less glucose but similar amounts of fat compared with kcp+/+.
Figure 2.

Metabolic analyses. A, mice kept for 8 weeks on either a ND or a HFD were then singly housed in metabolic cages to measure energy expenditure, food intake, activity, VO2, glucose oxidation, and fat oxidation. Chamber sampling frequencies were every 20 min over a 3-day period, whereas activity measurements were taken every second. Graphs display the 3-day sampling period for kcp+/+, kcpTg, and kcp−/− mice. Mice kept on a ND showed no significant differences except for increased glucose oxidation in kcpTg mice. On a HFD, kcpTg mice had higher energy expenditure and oxygen consumption compared with kcp+/+, whereas kcp−/− mice exhibited values lower than those for kcp+/+ for both parameters. kcpTg also showed higher rates of fat oxidation, consistent with their leaner body mass. B, average oxygen consumption over a 3-day period as adjusted for lean body mass (LBM). Data are shown for light and dark cycles and for the total 3-day periods. Statistically significant differences are indicated by red and blue bars. C, total energy expenditure (Exp.) averaged over 3 days for light and dark cycles from five mice for each genotype. Statistically significant comparisons are indicated. D, NE challenge experiments in which mice were kept at 30 °C, anesthetized for baseline (BL) readings, and given a single dose of NE (1 mg/kg) subcutaneously and metabolic parameters were measured for 30 h. E, temperature challenge experiments in which mice were kept at 22, 30, and 4 °C as indicated. All three genotypes showed significant increases in energy expenditures at 4 °C, but differences among genotypes were not deemed significant. F, subcutaneous body temperature (Subcut. Temp.) readings during the temperature challenge experiments indicated significantly increased body temperature for kcpTg mice at 22 °C, no significant differences at 30 °C, but a lower average temperature at 4 °C.
The increased energy and oxygen consumption, despite no change in food intake or activity, are consistent with the significantly increased body temperature in kcpTg mice and suggest altered thermoregulation. To determine whether this was due to a neuroendocrine effect or responsiveness to norepinephrine (NE), we challenged the mice with a single dose of NE and measured the metabolic rates (Fig. 2D). Mice were kept at a thermoneutral temperature of 30 °C and anesthetized for baseline readings. Subsequently, a single injection of NE significantly increased respiration and energy expenditure in all three genotypes with no significant differences. The three genotypes were also subjected to a series of temperature challenges by first housing mice at 22 °C for 1 day, 30 °C for another day, and then 4 °C for 2 days (Fig. 2E). All three genotypes exhibited a decrease in energy expenditure and oxygen consumption at 30 °C followed by a nearly 3-fold increase in energy expenditure at 4 °C. As previous results indicated, average body temperatures at 22 °C were significantly higher in kcpTg mice, but this effect was reduced at 30 °C (Fig. 2F). Strikingly, at 4 °C, average subcutaneous body temperatures appeared lower in the kcpTg mice, which may reflect the reduced amount of insulating subcutaneous body fat in these animals.
KCP alters white and brown adipose tissues
To determine the mechanisms underlying the reduced amount of white adipose tissue in kcpTg mice, we characterized both white and brown adipose tissues in mice kept on a HFD. Although kcpTg mice had less visceral fat, we were able to isolate enough epididymal WAT (eWAT) for histology and protein expression analyses. By Western blot analyses, kcpTg eWAT had significantly lower levels of P-SMAD3 but higher levels of P-SMAD1 compared with either kcp+/+ or kcp−/− eWAT (Fig. 3, A and B). These data are entirely consistent with prior results showing inhibition of TGF-β signaling and enhancement of BMP signaling by KCP protein. Furthermore, the kcpTg eWAT had higher levels of UCP1, which increases heat production and is usually associated with brown fat (Fig. 3, A and C). The kcpTg eWAT tissues also had higher levels of PPARγ, PGC1, P-AKT, and cytochrome c oxidase subunit IV, all proteins associated with a more beige fat phenotype (Fig. 3, A and C). Also of note were the increased levels of P-ERK in the kcp−/− eWAT; P-ERK is known to phosphorylate PPARγ and increase expression of diabetes-associated genes (33). We also used quantitative RT-PCR to assess gene expression for adiponectin, kcp, and pgc1 (Fig. 3D), all of which were higher in the kcpTg mice and lower in the kcp−/− mice compared with the kcp+/+ animals.
Figure 3.
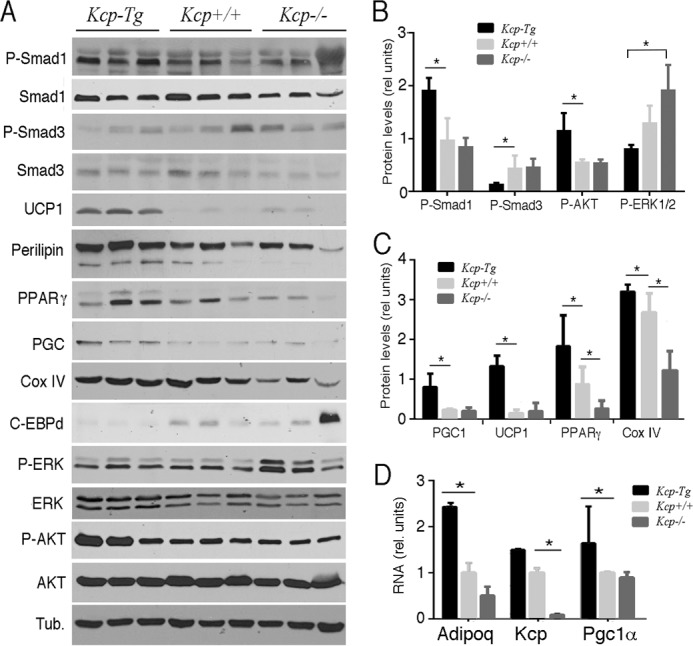
Western analyses of proteins from epididymal WAT on a HFD. A, total protein lysates were isolated from kcp+/+, kcpTg, and kcp−/− male mice and immunoblotted with the indicated antibodies. Three independent isolates were run for each of the indicated genotypes. Tub, tubulin; Cox, cytochrome c oxidase; C-EBPd, CCAAT/enhancer-binding protein δ. B, Western blot results for phosphoproteins were quantified by densitometry scanning, and the results are shown graphically with error bars representing one standard deviation. Statistical significance was determined using Student's t test (*, p < 0.05) between the indicated pair of data sets. C, quantitation of adipocyte-specific marker proteins was done as in B (*, p < 0.05) with error bars representing one standard deviation. D, quantitative RT-PCR for select genes is shown. Note the increase in adiponectin and PGC1 in kcpTg mice (*, p < 0.05) with error bars representing one standard deviation. rel., relative; Adipoq, Adiponectin, C1Q and Collagen domain-containing protein.
In mice fed a HFD, the eWAT tissue also exhibited differences in histology and in markers for inflammation among the three genotypes (Fig. 4). Trichrome staining showed extracellular collagen deposition in kcp−/− eWAT with many large adipocytes and evidence of enhanced inflammation. The average cross-sectional area of eWAT cells was approximately half in kcpTg adipocytes, whereas the kcp−/− adipocytes were even larger than kcp+/+ cells (Fig. 4, A–C and J). Whole-mount immunofluorescence examined macrophage infiltration and crownlike structures in eWAT as a measure of the inflammatory response that correlates with high-fat diet-induced obesity and glucose intolerance (Fig. 4, D–F and K). The kcpTg mice had few if any crownlike structures and MAC2-positive macrophages in eWAT. However, the obese kcp+/+ and especially the kcp−/− animals had large numbers of macrophages present in eWAT. We also looked for evidence of fibrosis by Picrosirius Red staining for collagen deposition. The kcp−/− animals showed increased collagen matrix deposition in eWAT compared with kcp+/+ mice, whereas the kcpTg mice had significantly less collagen deposition when fed a high-fat diet (Fig. 4, G–I and L).
Figure 4.

Morphology of white adipose tissue. Epididymal WAT was isolated from kcp+/+, kcpTg, and kcp−/− male mice maintained on a HFD and embedded in paraffin blocks. Sections were stained with trichrome (A–C), immunostained with anti-caveolin (Cav1) and anti-MAC2 (D–F), or stained with Picrosirius Red (G–I). Genotypes are as indicated. Note the increased adipocyte size, enhanced inflammatory response, and increased evidence of fibrosis in the kcp−/− mice. The kcpTg mice showed no MAC2-positive macrophages in WAT. Scale bars, 50 μm. J, quantitative morphometry of average cell surface area from histological sections. More than 100 cells were counted with p < 0.05 (*) for all pairs. sq. mic., square μm. K, quantitative analyses of MAC2-positive cell staining showing the average number of crownlike structures (CLS) per field. For each genotype, at least 10 sections were counted from three different mice with error bars representing one standard deviation; p < 0.01 (*; ANOVA) for all pairs tested. L, quantitative morphometry of Sirius Red-stained surface area in micrographs of the indicated genotypes maintained on a HFD. Error bars are one standard deviation with p < 0.05 for the indicated pairs (*; ANOVA).
Given the increased body temperature and metabolism in kcpTg mice, we also examined interscapular brown adipose tissue (iBAT) for protein expression and histology. Protein expression levels in iBAT of mice fed a ND showed that kcpTg mice had elevated levels of P-SMAD1 and PPARγ but no significant differences in UCP1 (Fig. 5, A–C). The kcp−/− mice showed slightly more P-SMAD3 and less PPARγ but no significant differences in P-SMAD1 or UCP1 compared with kcp+/+ animals (Fig. 5, A–C). The kcpTg mice also demonstrated myc-KCP expression in iBAT, indicating that the PEPCK promoter used for the transgene is actively expressed in this tissue. When mice were kept on a HFD, differences in protein expression levels in iBAT from kcpTg and kcp−/− mice were more pronounced (Fig. 5, D–F). kcp−/− mice had less P-SMAD1 and higher levels of P-SMAD3 compared with kcpTg mice (Fig. 5F). In addition, levels of P-ERK were higher in kcp−/− mice. Levels of UCP1 were significantly higher in kcpTg mice as were PGC1 and PPARγ (Fig. 5E) compared with kcp−/− mice.
Figure 5.

Western analyses of interscapular BAT. A, protein lysates from three independent samples of BAT isolated from kcpTg, kcp+/+, and kcp−/− mice fed a ND. Genotypes are as indicated, and blots were probed with antibodies for proteins as marked. Tub., tubulin. B, quantitation of protein levels by image analyses shows increased levels of PPARγ and PGC1 in kcpTg mice and a decrease in kcp−/− mice (*, p < 0.05, with error bars representing one standard deviation). Rel., relative. C, quantitation of phosphoprotein levels shows an increased level of P-SMAD1 in kcpTg BAT (*, p < 0.05, with error bars representing one standard deviation). D, protein lysates from three independent samples of BAT isolated from kcpTg, kcp+/+, and kcp−/− mice fed a HFD. E, quantitation of protein levels indicates an increase in PPARγ and PGC1in kcpTg mice and a decrease in UCP1 in kcp−/− mice (*, p < 0.05, with error bars representing one standard deviation). F, phosphoprotein quantitation shows an increase in P-SMAD1 levels in kcpTg mice and an increase in P-SMAD3 levels for kcp−/− mice. P-ERK levels are also significantly increased in kcp−/− mice (*, p < 0.05, with error bars representing one standard deviation).
Histology of iBAT indicated that the kcp−/− mice fed a normal diet exhibited increased lipid storage in iBAT, compared with WT, that became even more pronounced on the HFD (Fig. 6). Histology of BAT generally shows densely staining cytoplasm because of the density of mitochondria that fuel thermoregulation. Consistent with our metabolic data, kcpTg mice had very densely staining iBAT compared with WT on both normal diet and HFD (Fig. 6) with only minimal lipid accumulation.
Figure 6.
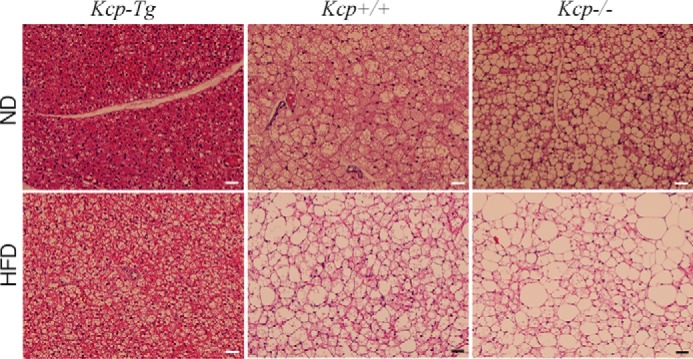
Morphology of interscapular BAT. Representative hematoxylin and eosin stained histological sections of BAT from kcpTg, kcp+/+, and kcp−/− mice as indicated are shown. Mice were maintained on a ND or a HFD for 8 weeks prior to tissue isolation. Scale bars, 100 μm.
Finally, to test whether the differences in brown and white adipocytes were cell-autonomous and could reflect the response to TGF-β superfamily ligands, we cultured differentiated adipocytes isolated from inguinal WAT and interscapular BAT tissues of all three genotypes kept on a ND (Fig. 7). Differentiated adipocytes isolated from inguinal white adipose tissue (iWAT) showed that the kcpTg cells had a stronger response to BMP4 with higher levels of P-SMAD1 (Fig. 7A). Cells isolated from kcp−/− mice showed a higher basal level of P-SMAD3 and a stronger response to TGF-β despite having less total SMAD3. Also of note, the kcpTg cells expressed more UCP1, suggesting a more beige phenotype. The differentiated adipocytes isolated from iBAT showed a weaker BMP4 response in kcp−/− cells but no difference in the kcpTg cells. P-SMAD3 was not significantly affected in iBAT cells. However, kcp−/− cells had significantly lower UCP1 and PGC1 levels. The protein lysates were collected 2 h after addition of ligands, which did not affect expression of the non-phosphorylated proteins. Thus, the adipocytes that express myc-KCP have altered cell-autonomous responses to TGF superfamily ligands, consistent with the known role for KCP in modifying the receptor/ligand interactions. That these effects were observed on a ND indicates that they are not due to obesity or associated metabolic syndrome. The cultured cells are also removed from any infiltrating immune cells and associated cytokines, suggesting a direct effect of KCP on TGF-β superfamily signaling in adipocytes.
Figure 7.

Gene expression and signaling responses in cultured adipocytes. A, adipocytes were differentiated from the stromal vascular fraction isolated from the iWAT from kcpTg, kcp+/+, and kcp−/− mice kept on a ND and cultured with increasing amounts of BMP4 or TGF-β1 as indicated. Protein lysates were probed with the indicated antibodies. Note the increased P-SMAD1 response in kcpTg mice and increased levels of UCP1, whereas kcp−/− mice show more P-SMAD3 despite less total SMAD3. B, adipocytes were differentiated from the stromal vascular fraction isolated from the iBAT from kcpTg, kcp+/+, and kcp−/− mice kept on a ND and cultured with increasing amounts of BMP4 or TGF-β1 as indicated. Protein lysates were probed with the indicated antibodies. Note the decreased P-SMAD1 response in kcp−/− mice and the lower levels of UCP1 and PGC1. Representative blots from duplicate experiments are shown.
Discussion
We have utilized both gain and loss of KCP function in genetically engineered mice to demonstrate that KCP can regulate the TGF-β superfamily signaling profiles in adult tissues. This dramatically alters the metabolic profile in mice and their responses to a high-fat diet. As reported previously, KCP loss of function makes mice more susceptible to fibrotic disease in the kidney (27) and liver (32), whereas gain of function increases resistance to fibrotic disease (31). Here, we show that KCP expression in liver and iBAT can lessen the severity of HFD-induced obesity and associated metabolic disease. Conversely, a loss of KCP function already predisposes mice to obesity and increases the severity of HFD-induced obesity and its associated complications. Consistent with our biochemical models for KCP function, P-SMAD1 is generally increased in tissues of kcpTg mice, and P-SMAD3 is decreased, whereas the kcp−/− mice show the reciprocal effects. Despite these alterations, young kcp−/− and kcpTg mice were viable and fertile and showed few other phenotypic changes in growth, skeletal patterning, or physiology when maintained on a normal diet. However, as reported previously, aging kcp−/− mice had significant liver pathology (32).
The secreted cysteine-rich domain protein KCP, whose gene was identified based on sequence homology to the potent BMP inhibitor chordin (34, 35), can bind to both BMPs and TGF-β to regulate the activation of downstream receptor-activated SMAD proteins. Unlike chordin, KCP is a potent activator of BMP and an inhibitor of TGF-β- and activin-dependent signaling in cell culture (27, 28). How KCP is able to distinguish among these ligands is unclear. However, in vitro pulldown experiments show that KCP promotes BMP/BMPr1a interactions, an effect likely to underlie the mechanisms of enhanced SMAD1 phosphorylation. The expression of KCP is seen in embryonic development of the kidney, limb buds, and parts of the central nervous system, although prior studies did not address expression in adipocytes or their progenitors (27). Initial characterization of the kcp−/− mice focused on kidney development, which appeared normal. This lack of developmental phenotype in kcp−/− kidneys can be explained in part by the redundant function of the cysteine-rich domain protein Cv2 as compound kcp/Cv2 mutants do exhibit severe renal developmental defects (30). However, endogenous KCP expression in adult tissues is low but was shown to increase upon acute and chronic kidney injury (27, 28).
The endogenous KCP protein has a signal peptide and is secreted in cell culture, suggesting that there may be systemic effects of circulating KCP in vivo regardless of the tissue of origin. In the kcpTg mice maintained on a normal diet, myc-KCP is detected in iBAT, liver, and kidney and in primary inguinal adipocyte cultures from the kcpTg mice. By quantitative RT-PCR, we can detect KCP in eWAT of WT mice and increased levels in the kcpTg mice, whereas kcp−/− mice have undetectable levels. Whether the shift in P-SMAD levels observed in eWAT of the kcpTg reflects this low level of KCP expression in eWAT or is due to systemic circulating KCP from the liver is difficult to discern. Antibodies generated against KCP do recognize the higher protein levels in embryos but not in the disease models where KCP mRNA is only detected by more sensitive methods (27). Nevertheless, the increase in P-SMAD3 signaling and the decrease in P-SMAD1 detected in kcp−/− mice are consistent with a role for KCP in mediating the responses to endogenous BMPs and TGF-β in disease regardless of the source. Alternatively, KCP might affect the level of TGF receptors, although activation and recycling can also affect type I and type II receptor levels, thus making it difficult to rule out this possibility (36). Given the profibrotic effects of TGF-β in a variety of tissues and the recently established link of the TGF-β/SMAD3 axis to eWAT physiology and HFD-induced obesity (18, 19, 21, 22, 37), our results confirm that regulating TGF-β/BMP signaling through the extracellular proteins that inhibit or enhance receptor/ligand interactions is a viable strategy to attenuate both fibrotic and metabolic disease.
The origin of white and brown fat tissues has been examined with molecular and genetic techniques in detail (38). Mesenchymal cells of lateral plate mesodermal origin are thought to generate the white preadipocytes, which express PPARγ (39, 40), PDGF receptor β (41), and Zfp423 (42). There is also evidence that white adipocyte progenitors are closely associated with the developing vasculature (43). In contrast, brown adipocytes share a common ancestry with the muscle lineage (44) and depend on the actions of the transcriptional cofactor PRDM16 (45). More recently, beige or brite fat is a term used to describe brownlike adipocytes within the adult WAT that may be derived from transdifferentiation of white adipocyte progenitors (46, 47). A number of TGF-β superfamily ligands and effectors have been implicated in the transition of white to beige adipocytes, specifically loss of SMAD3 (20) or growth and differentiation factor 3 (37) or expression of BMP4 (21).
Transgenic expression of KCP alters the ability of adipocytes to respond to TGF superfamily ligands in vivo and in vitro. We believe this is cell-autonomous, at least in part, because differentiated adipocytes isolated from the three different genotypes respond differently to BMP4 or TGF-β in vitro. Protein analyses from tissue of kcpTg mice show increased levels of UCP1 and PPARγ in WAT of mice fed a HFD, suggesting a more beige fat phenotype. This is consistent with the metabolic data that indicate an effect on thermal regulation. kcpTg mice burn more energy and have a higher resting body temperature, which could explain the lack of weight gain on a HFD. Conversely, the kcp−/− mice show less UCP1 in BAT in vivo and in vitro, consistent with the histology of BAT, which shows more lipid accumulation and less densely staining mitochondria. Thus, the protective effects of KCP expression on HFD-induced obesity and metabolic disease may be due solely to the increased thermogenesis due to browning of WAT and enhanced energy consumption by iBAT. In the liver, the protective effects of KCP may be due to a suppression of fibrosis as TGF-β superfamily signaling has been linked to fibrotic disease in a variety of tissues (48, 49). Alternatively, the suppression of hepatic steatosis in kcpTg mice may also be secondary to the increased energy expenditure and the resistance to obesity.
In summary, the expression of a secreted TGF-β superfamily regulatory protein for shifting the signaling paradigm from P-SMAD3 to P-SMAD1 in HFD-induced obesity has not been reported previously. The ability to shift TGF-β superfamily signaling by extracellular secreted regulatory proteins may prove a valuable tool to study adipocyte differentiation, maintenance, and obesity-related complications in animal models. Given the plasticity of adipocyte progenitors and the renewal of adipose tissue in adults, the significance of our findings may translate into alternative therapies for obesity and related disorders and clearly merits further development.
Experimental procedures
Animals
All animal use was in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the University Committee on Use and Care of Animals at the University of Michigan. The kcpTg (31) and kcp−/− (27) alleles were described previously and were bred onto a C57/Bl6 genetic background. Mice were housed in a specific pathogen-free facility with a 12-h light, 12-h dark cycle and given free access to food and water. The KCP-Tg, KCP-KO, and wild-type males were maintained on a regular diet (5008, Purina LabChow; 16.7% calories from fat) or on a high-fat diet (D12451, Research Diets Inc.; 45% calories from fat) starting at 8 weeks of age for 12–14 weeks.
Glucose tolerance test
Mice were fasted for 6 h then injected i.p. with 0.7g/kg body weight d-glucose. Just before injection, fasting glucose was taken at time 0. With the minimum amount of stress possible, tails were snipped with scissors and gently milked for blood droplets. The first drop was wiped off, and the second drop of blood was collected and read with a glucometer, and the measurement was recorded. Fresh blood droplets were collected every 15 min for 60 min and then every 30 min for another hour.
Metabolic testing
Oxygen consumption (VO2), carbon dioxide production (VCO2), spontaneous motor activity, and food intake were measured using the Comprehensive Laboratory Monitoring System (CLAMS, Columbus Instruments), an integrated open-circuit calorimeter equipped with an optical beam activity monitoring device. Mice were weighed each time before the measurements and individually placed into the sealed chambers (7.9 × 4 × 5 inches) with free access to food and water. The study was carried out in a temperature-controlled (5–40 °C) enclosure (CLAMS-ENC, Columbus Instruments) with an operating temperature selected at 22, 30, or 5 °C and lighting set at 12-12 h (6:00 p.m.–6:00 a.m.) dark-light cycles. The measurements were carried out continuously for 72 h. During this time, animals were provided with food and water through the equipped feeding and drinking devices located inside the chamber. The amount of food of each animal was monitored through a precision balance attached below the chamber. The system was routinely calibrated each time before the experiment using a standard gas (20.5% O2 and 0.5% CO2 in N2). VO2 and VCO2 in each chamber were sampled sequentially for 5 s in a 20-min interval, and the motor activity was recorded every second in x and z dimensions. The airflow rate through the chambers was adjusted at the level to keep the oxygen differential around 0.3% at resting conditions. Respiratory quotient, also known as respiratory exchange ratio, was calculated as VCO2/VO2. Total energy expenditure, carbohydrate oxidation, and fatty acid oxidation were calculated, respectively, using equations based on the values of VO2 and VCO2 assuming that the contribution of protein breakdown to energy expenditure under the study condition is negligible and with little variation among the groups (50, 51). For the measurement of non-shivering thermogenesis, mice were kept at a thermoneutral temperature of 30 °C and anesthetized via i.p. injection of 1:2 pentobarbital-saline dilution at 70 mg/kg. After the baseline recording of VO2 and VCO2, a single injection of NE (0.5 mg/1 ml) solution was given subcutaneously at a dose of 1 mg/kg. Subsequently, the increases of respiration and energy expenditure were recorded.
Body temperature was measured using a temperature transponder (IPTT-300, Bio Medic Data Systems) that was surgically implanted subcutaneously on the back of the neck in the vicinity of BAT. Mice were briefly anesthetized for less than a minute using an isoflurane vaporizer, and the transponder loaded in the delivering syringe was injected into the subcutaneous space about 1 cm from the injecting site. Mice were replaced back in their home cages and recovered within 5 min. The implantation was done 5 days prior to the CLAMS experiment. During the measurement of VO2 and VCO2, temperature was manually recorded using the DAS-7007 (Bio Medic Data Systems) reader scanning from the outside of the CLAMS chamber.
Body fat, lean mass, and free fluid were measured using an NMR analyzer (Minispec LF90II, Bruker Optics). The measurement takes less than 2 min while conscious mice were placed individually into the measuring tube with minimum restraint. The machine was calibrated daily using a reference sample as recommended by the manufacturer.
Antibodies and Western blotting
The following antibodies were purchased from Cell Signaling Technologies: SMAD1 (9743), P-SMAD1 (9511), SMAD3 (9523S), AKT (9272), P-AKT (Ser-473; 4060), cytochrome c oxidase subunit IV (4844), CCAAT/enhancer-binding protein δ (2318), perilipin (9349), PPARγ (2443), and P-ERK1/2 (9101S). Additional antibodies were PGC1α (AB3242, Millipore), UCP1 (U6382, Sigma), myc epitope 9E10 (Sigma), β-tubulin (T4026, Sigma), and P-SMAD3 (ab52903, Abcam).
Tissues were isolated from iBAT and eWAT and homogenized in radioimmune precipitation assay buffer (R0278, Sigma) supplemented with freshly added protease inhibitor mixtures (Roche Diagnostics). Crude lysates were centrifuged at 14,000 × g for 10 min twice, and the protein concentration was measured using the BCA Protein Assay kit (23227, Pierce). Samples were diluted in 6× SDS-PAGE sample buffer, separated on polyacrylamide gels, and transferred to PVDF membranes (IPFL00010, Millipore). Specific proteins were detected with primary antibodies and horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) using Western Lightning enhanced chemiluminescence substrate (Pierce). Blots were scanned and analyzed for band intensities using UN-SCAN-IT version 5.1 software (Silk Scientific, Orem, UT) and confirmed with ImageJ in a blinded fashion. Alternatively, infrared fluorescent secondary antibodies were used for detection and quantification of specific protein on the Odyssey CLx imager (LI-COR Biosciences).
Histology and immunostaining
eWAT fixed in 1% PFA for 1 h at room temperature or overnight in Bouin's fixative. Liver and iBAT were fixed overnight in 4% PFA at 4 °C. After the specific fixation time period for each tissue, samples were processed for paraffin embedding. Picrosirius Red staining was done according to the manufacturer's recommended instructions (24901, Polysciences Inc.).
For immunostaining, fat pads were excised and fixed for 1 h in 1% PFA at room temperature with rocking. Tissues were washed two times in PBS and blocked for 30 min at room temperature with 5% BSA (A7030, Sigma) in PBS with 0.1% saponin (47036, Sigma). Primary and secondary antibodies were diluted in 5% BSA, 0.1% saponin. Samples were incubated for 1 h with primary antibodies at room temperature followed by three washes and incubation with fluorescent conjugated secondary antibodies. After an additional three washes, samples were stored at 4 °C protected from light until imaging. Antibodies used were rabbit anti-caveolin1(610060, BD Transduction Laboratories) and rat anti-galectin-3 (14-5301-85, eBioscience).
RNA analyses
RNA from tissues was extracted using RNeasy Midi kits (Qiagen), and cDNA was generated from 0.5–1.0 μg of total RNA using high-capacity cDNA reverse transcription kits (Applied Biosystems). Power SYBR Green PCR Master Mix (Applied Biosystems) and the StepOnePlus System (Applied Biosystems) were used for real-time quantitative PCR. Acidic ribosomal phosphoprotein expression was used as an internal control for data normalization. Samples were assayed in triplicate, and relative expression was determined using the 2−ΔΔCT method. Primers pairs were as follows: PGC1, TATGGAGTGACATAGAGTGTGCT and CCACTTCAATCCACCCAGAAAG; KCP, CCACAATGGGCAGTCTTATGG and TGGCACTTCACAGGCACACATC; adiponectin, GCAGGCATCCCAGGACATC and GCGATACATATAAGCGGCTTCT.
Primary preadipocyte isolation and differentiation
Adipocytes were cultured as described previously (52). Mice were sacrificed, and tissue was isolated from iWAT and iBAT depots. Tissue was minced and digested in a collagenase D (1108888201, Roche Applied Science)/Dispase II (4942078, Roche Applied Science) solution. After the tissue was fully digested, cells were resuspended in wash medium (DMEM/F-12 + GlutaMAX; 10565042, Invitrogen) + 10% newborn calf serum (N4762, Sigma)), filtered through a 100-μm cell strainer, and centrifuged at 300 × g for 5 min. The supernatant was removed, and the pellet was disrupted, resuspended in wash media, filtered through a 40-μm cell strainer, and again centrifuged. The resulting pellet was disrupted, resuspended in growth media (DMEM/F-12 + GlutaMAX + 15% FBS (F2442, Sigma), and plated onto 10-cm dishes that had previously been coated with rat tail collagen type 1 (CB-40236, Fisher) following the protocol contained in the product information. Cells were washed after 1 day of growth to remove floating cells and then underwent at least one round of subculture before being seeded onto collagen-coated 12-well plates for differentiation. The day after seeding the cells were stimulated in DMEM/F-12 + GlutaMAX supplemented with 10% FBS, 5 μm dexamethasone (D4902, Sigma), 0.5 μg/ml insulin (15500, Sigma), 0.5 mm isobutylmethylxanthine (17018, Sigma), and 1 μm rosiglitazone (71740, Cayman). After 2 days, cells were maintained in DMEM/F-12 + GlutaMAX with 10% FBS and 0.5 μg/ml insulin. Mature adipocytes were cultured with increasing doses of BMP4 or TGF-β1 (R&D Systems, Inc.) 6–7 days after beginning stimulation, and protein lysates were made 2 h post-stimulation.
Statistical analyses
For quantitative readouts, including body mass, glucose, insulin, body temperature, and quantitative RT-PCR, three to five mice of each genotype were used. For micrographs, specific areas were calculated by ImageJ over eight to 10 images from three animals. For Western blotting, three independent samples were run, and protein levels were measured by densitometry scanning of autoradiographs at appropriate exposures in the linear range. Averages and standard deviations were calculated, and significance was assessed by ANOVA for multiple comparisons or Student's two-tailed t test for independent variables when doing pairwise comparisons with three data points each.
Author contributions
A. S. coordinated the study and performed experiments for Figs. 1 and 3–6. K. I. W. performed experiments and analyzed data for Figs. 2, 3, and 6. M. P. E. performed experiments and analyzed data in Fig. 7. N. Q. analyzed data and coordinated experiments in Fig. 2. G. M.-S. performed experiments for Figs. 1 and 3. E. G. performed experiments for Figs. 1 and 3. W. O. performed experiments for Figs. 3 and 4. G. R. D. organized the study, analyzed the data, prepared figures, and wrote the manuscript.
Acknowledgments
We thank E. Limback and the University of Michigan Animal Phenotyping Core for metabolic analyses, J. Poore and J. Harrison from the University of Michigan Microscopy and Imaging Analysis core for histology and imaging, S. Reilly for discussion and insights, T. Mergian and J. DelProposto for help with the glucose tolerance test, and E. Ranghini for advice and assistance. The core services were supported by National Institutes of Health Grants DK020572 and DK089503 (to the University of Michigan).
This work was supported by National Institutes of Health Grants DK062914 and DK073722 (to G. R. D.) and 1F31DK112625 (to M. P. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- WAT
- white adipose tissue
- BAT
- brown adipose tissue
- HFD
- high-fat diet
- Tg
- transgenic
- BMP
- bone morphogenetic protein
- CR
- cysteine-rich
- KCP
- kielin/chordin-like protein
- ND
- normal diet
- PEPCK
- phosphoenolpyruvate carboxykinase
- NE
- norepinephrine
- eWAT
- epididymal WAT
- PPARγ
- peroxisome proliferator-activated receptor γ
- iBAT
- interscapular brown adipose tissue
- P-
- phospho-
- VO2
- oxygen consumption
- VCO2
- carbon dioxide production
- PFA
- paraformaldehyde
- iWAT
- inguinal white adipose tissue
- ANOVA
- analysis of variance
- PGC
- peroxisome proliferator-activated receptor-γ coactivator.
References
- 1. Rosen E. D., and Spiegelman B. M. (2014) What we talk about when we talk about fat. Cell 156, 20–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doria A., Patti M. E., and Kahn C. R. (2008) The emerging genetic architecture of type 2 diabetes. Cell Metab. 8, 186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saltiel A. R. (2012) Insulin resistance in the defense against obesity. Cell Metab. 15, 798–804 [DOI] [PubMed] [Google Scholar]
- 4. Cannon B., and Nedergaard J. (2004) Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 [DOI] [PubMed] [Google Scholar]
- 5. Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., Kolodny G. M., and Kahn C. R. (2009) Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., and Teule G. J. (2009) Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508 [DOI] [PubMed] [Google Scholar]
- 7. Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T., Taittonen M., Laine J., Savisto N. J., Enerbäck S., and Nuutila P. (2009) Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 8. Schulz T. J., and Tseng Y. H. (2013) Brown adipose tissue: development, metabolism and beyond. Biochem. J. 453, 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu J., Cohen P., and Spiegelman B. M. (2013) Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 27, 234–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donath M. Y., and Shoelson S. E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107 [DOI] [PubMed] [Google Scholar]
- 11. Lumeng C. N., and Saltiel A. R. (2011) Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 121, 2111–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun K., Tordjman J., Clément K., and Scherer P. E. (2013) Fibrosis and adipose tissue dysfunction. Cell Metab. 18, 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Divoux A., and Clément K. (2011) Architecture and the extracellular matrix: the still unappreciated components of the adipose tissue. Obes. Rev. 12, e494–e503 [DOI] [PubMed] [Google Scholar]
- 14. Keophiphath M., Achard V., Henegar C., Rouault C., Clément K., and Lacasa D. (2009) Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol. Endocrinol. 23, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spencer M., Yao-Borengasser A., Unal R., Rasouli N., Gurley C. M., Zhu B., Peterson C. A., and Kern P. A. (2010) Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am. J. Physiol.. Endocrinol. Metab. 299, E1016–E1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wernstedt Asterholm I., Tao C., Morley T. S., Wang Q. A., Delgado-Lopez F., Wang Z. V., and Scherer P. E. (2014) Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 20, 103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Asrih M., and Jornayvaz F. R. (2015) Metabolic syndrome and nonalcoholic fatty liver disease: is insulin resistance the link? Mol. Cell. Endocrinol. 418, 55–65 [DOI] [PubMed] [Google Scholar]
- 18. Yadav H., and Rane S. G. (2012) TGF-β/Smad3 signaling regulates brown adipocyte induction in white adipose tissue. Front. Endocrinol. 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zamani N., and Brown C. W. (2011) Emerging roles for the transforming growth factor-β superfamily in regulating adiposity and energy expenditure. Endocr. Rev. 32, 387–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yadav H., Quijano C., Kamaraju A. K., Gavrilova O., Malek R., Chen W., Zerfas P., Zhigang D., Wright E. C., Stuelten C., Sun P., Lonning S., Skarulis M., Sumner A. E., Finkel T., et al. (2011) Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 14, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qian S. W., Tang Y., Li X., Liu Y., Zhang Y. Y., Huang H. Y., Xue R. D., Yu H. Y., Guo L., Gao H. D., Liu Y., Sun X., Li Y. M., Jia W. P., and Tang Q. Q. (2013) BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 110, E798–E807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schulz T. J., Huang P., Huang T. L., Xue R., McDougall L. E., Townsend K. L., Cypess A. M., Mishina Y., Gussoni E., and Tseng Y. H. (2013) Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 495, 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whittle A. J., Carobbio S., Martins L., Slawik M., Hondares E., Vázquez M. J., Morgan D., Csikasz R. I., Gallego R., Rodriguez-Cuenca S., Dale M., Virtue S., Villarroya F., Cannon B., Rahmouni K., et al. (2012) BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 149, 871–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Larraín J., Bachiller D., Lu B., Agius E., Piccolo S., and De Robertis E. M. (2000) BMP-binding modules in chordin: a model for signalling regulation in the extracellular space. Development 127, 821–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia Abreu J., Coffinier C., Larraín J., Oelgeschläger M., and De Robertis E. M. (2002) Chordin-like CR domains and the regulation of evolutionarily conserved extracellular signaling systems. Gene 287, 39–47 [DOI] [PubMed] [Google Scholar]
- 26. Shi Y., and Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 27. Lin J., Patel S. R., Cheng X., Cho E. A., Levitan I., Ullenbruch M., Phan S. H., Park J. M., and Dressler G. R. (2005) Kielin/chordin-like protein, a novel enhancer of BMP signaling, attenuates renal fibrotic disease. Nat. Med. 11, 387–393 [DOI] [PubMed] [Google Scholar]
- 28. Lin J., Patel S. R., Wang M., and Dressler G. R. (2006) The cysteine-rich domain protein KCP is a suppressor of transforming growth factor β/activin signaling in renal epithelia. Mol. Cell. Biol. 26, 4577–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abreu J. G., Ketpura N. I., Reversade B., and De Robertis E. M. (2002) Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat. Cell Biol. 4, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikeya M., Kawada M., Kiyonari H., Sasai N., Nakao K., Furuta Y., and Sasai Y. (2006) Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development 133, 4463–4473 [DOI] [PubMed] [Google Scholar]
- 31. Soofi A., Zhang P., and Dressler G. R. (2013) Kielin/chordin-like protein attenuates both acute and chronic renal injury. J. Am. Soc. Nephrol. 24, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soofi A., Wolf K. I., Ranghini E. J., Amin M. A., and Dressler G. R. (2016) The kielin/chordin-like protein KCP attenuates nonalcoholic fatty liver disease in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G587–G598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Banks A. S., McAllister F. E., Camporez J. P., Zushin P. J., Jurczak M. J., Laznik-Bogoslavski D., Shulman G. I., Gygi S. P., and Spiegelman B. M. (2015) An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature 517, 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sasai Y., Lu B., Steinbeisser H., and De Robertis E. M. (1995) Regulation of neural induction by the Chd and Bmp-4 antagonistic patterning signals in Xenopus. Nature 376, 333–336; Corrections (1995) 377, 757 and (1995) 378, 419 [DOI] [PubMed] [Google Scholar]
- 35. Sasai Y., Lu B., Steinbeisser H., Geissert D., Gont L. K., and De Robertis E. M. (1994) Xenopus chordin: a novel dorsalizing factor activated by organizer-specific homeobox genes. Cell 79, 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Budi E. H., Xu J., and Derynck R. (2016) Regulation of TGF-β receptors. Methods Mol. Biol. 1344, 1–33 [DOI] [PubMed] [Google Scholar]
- 37. Shen J. J., Huang L., Li L., Jorgez C., Matzuk M. M., and Brown C. W. (2009) Deficiency of growth differentiation factor 3 protects against diet-induced obesity by selectively acting on white adipose. Mol. Endocrinol. 23, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berry R., Jeffery E., and Rodeheffer M. S. (2014) Weighing in on adipocyte precursors. Cell Metab. 19, 8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosen E. D., Sarraf P., Troy A. E., Bradwin G., Moore K., Milstone D. S., Spiegelman B. M., and Mortensen R. M. (1999) PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617 [DOI] [PubMed] [Google Scholar]
- 40. Tontonoz P., Hu E., and Spiegelman B. M. (1994) Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 41. Berry R., and Rodeheffer M. S. (2013) Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 15, 302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta R. K., Arany Z., Seale P., Mepani R. J., Ye L., Conroe H. M., Roby Y. A., Kulaga H., Reed R. R., and Spiegelman B. M. (2010) Transcriptional control of preadipocyte determination by Zfp423. Nature 464, 619–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang W., Zeve D., Suh J. M., Bosnakovski D., Kyba M., Hammer R. E., Tallquist M. D., and Graff J. M. (2008) White fat progenitor cells reside in the adipose vasculature. Science 322, 583–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S., Scimè A., Devarakonda S., Conroe H. M., Erdjument-Bromage H., Tempst P., Rudnicki M. A., Beier D. R., and Spiegelman B. M. (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seale P., Kajimura S., Yang W., Chin S., Rohas L. M., Uldry M., Tavernier G., Langin D., and Spiegelman B. M. (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab. 6, 38–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Q. A., Tao C., Gupta R. K., and Scherer P. E. (2013) Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 19, 1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenwald M., Perdikari A., Rülicke T., and Wolfrum C. (2013) Bi-directional interconversion of brite and white adipocytes. Nat. Cell Biol. 15, 659–667 [DOI] [PubMed] [Google Scholar]
- 48. Dooley S., and ten Dijke P. (2012) TGF-β in progression of liver disease. Cell Tissue Res. 347, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gewin L., and Zent R. (2012) How Does TGF-β mediate tubulointerstitial fibrosis? Semin. Nephrol. 32, 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferrannini E. (1988) The theoretical bases of indirect calorimetry: a review. Metabolism 37, 287–301 [DOI] [PubMed] [Google Scholar]
- 51. Simonson D. C., and DeFronzo R. A. (1990) Indirect calorimetry: methodological and interpretative problems. Am. J. Physiol. Endocrinol. Metab. 258, E399–E412 [DOI] [PubMed] [Google Scholar]
- 52. Emont M. P., Yu H., Jun H., Hong X., Maganti N., Stegemann J. P., and Wu J. (2015) Using a 3D culture system to differentiate visceral adipocytes in vitro. Endocrinology 156, 4761–4768 [DOI] [PMC free article] [PubMed] [Google Scholar]