

ABSTRACT
Cerebral ischemia induces massive mitochondrial damage. These damaged mitochondria are cleared, thus attenuating brain injury, by mitophagy. Here, we identified the involvement of BNIP3L/NIX in cerebral ischemia-reperfusion (I-R)-induced mitophagy. Bnip3l knockout (bnip3l−/−) impaired mitophagy and aggravated cerebral I-R injury in mice, which can be rescued by BNIP3L overexpression. The rescuing effects of BNIP3L overexpression can be observed in park2−/− mice, which showed mitophagy deficiency after I-R. Interestingly, bnip3l and park2 double-knockout mice showed a synergistic mitophagy deficiency with I-R treatment, which further highlighted the roles of BNIP3L-mediated mitophagy as being independent from PARK2. Further experiments indicated that phosphorylation of BNIP3L serine 81 is critical for BNIP3L-mediated mitophagy. Nonphosphorylatable mutant BNIP3LS81A failed to counteract both mitophagy impairment and neuroprotective effects in bnip3l−/− mice. Our findings offer insights into mitochondrial quality control in ischemic stroke and bring forth the concept that BNIP3L could be a potential therapeutic _target for ischemic stroke, beyond its accepted role in reticulocyte maturation.
KEYWORDS: BNIP3L/NIX, cerebral ischemia, mitophagy, PARK2/PARKIN, phosphorylation
Introduction
Cerebral ischemia is a leading cause of mortality and disability, yet only limited therapies are currently available. Mitochondrial homeostasis is critical for neuronal survival in ischemic brains. Macroautophagy/autophagy machinery surveys mitochondrial quality by recognizing and delivering defective mitochondria to lysosomes for destruction. Selective mitochondria autophagy (herein referred to as mitophagy) is activated in ischemic brains.1-4 In the scenario known as ischemia-reperfusion (I-R), mitophagy inhibition exacerbates ischemic brain injury.1 Conversely, reinforced mitophagy confers benefits for neuronal survival.5,6 These findings indicate that mitophagy activation may hold promise as a potential therapeutic strategy against ischemic brain injury.7,8 Nevertheless, the mechanisms underlying mitophagy in ischemic neurons are not fully understood. Recent investigations highlighted the roles of PARK2, an E3 ubiquitin ligase, in mitophagy.9-11 Our previous findings demonstrated the involvement of PARK2 in I-R-induced mitophagy.1,5 However, PARK2 levels were found to decrease rapidly with reperfusion after cerebral ischemia,12 and persistent mitophagy was observed during this process.1 Furthermore, increasing numbers of investigations have documented mitophagy without the involvement of PARK2 in mammalian cells,13-16 which suggest that some PARK2-independent mechanisms may be responsible for the mitophagy observed in cerebral ischemia.
BNIP3L/NIX is a BH3-only proapoptotic protein.17 It has recently become clear that BNIP3L mediates mitophagy in erythroblast maturation, indicating its critical roles in development.18,19 BNIP3L locates on the mitochondrial outer membrane, where it recruits phagophores, the precursors to autophagosomes, to mitochondria by directly binding with Atg8 family proteins.20 BNIP3L may coordinate with PARK2 in mitophagy induction. BNIP3L primes mitochondrial PARK2 in mouse embryonic fibroblasts with CCCP treatment,21 and a more recent study identified BNIP3L as a PARK2 substrate in the promotion of mitophagy.22 BNIP3L transcription is induced under hypoxia in a variety of cells,23-26 implying the involvement of BNIP3L in hypoxia-induced mitophagy. However, it seems that the contributions of BNIP3L, as a mitophagy-related protein, have been underestimated under pathological conditions such as cerebral ischemia. Therefore, further identification of the molecular mechanisms of BNIP3L in the induction of mitophagy promise to establish a biological rationale for the development of cerebral ischemia therapies to modulate the mitophagy process.
Results
BNIP3L is required in ischemia-reperfusion-induced mitophagy
To investigate whether BNIP3L participates in brain ischemia-reperfusion-induced mitophagy, we performed transient middle cerebral artery occlusion (tMCAO) on Bnip3l+/+, Bnip3l+/− and bnip3l−/− mice. BNIP3L protein loss confirmed the gene deletion. Unlike PARK2, BNIP3L was not degraded by tMCAO (Fig. 1A). The protein levels of SQSTM1, TOMM20, and COX4I1 were examined to assess mitophagy activation. The reductions in the levels of the mitochondrial marker proteins COX4I1 and TOMM20, as well as the reduction of the autophagy substrate SQSTM1 by tMCAO, were significantly reversed in bnip3l−/− mice, suggesting the requirement of BNIP3L in tMCAO-induced mitophagy (Fig. 1 A-D). We found the MAP1LC3B-II/LC3B-II upregulation by tMCAO was not disturbed by Bnip3l deletion (Fig. 1E), confirming the previous notion that BNIP3L does not activate the autophagy machinery.18
Figure 1.

BNIP3L is required in ischemia-reperfusion-induced mitophagy. (A) Bnip3l+/+, Bnip3l+/−, and bnip3l−/− mice were subjected to transient middle cerebral artery occlusion (tMCAO) for 1 h, and expression of SQSTM1, TOMM20, COX4I1, and LC3B in ischemic penumbra was assessed by western blot after 6 h of reperfusion. (B–E) Semi-quantitative analysis of SQSTM1, TOMM20, COX4I1, and LC3B bands are shown, respectively. (F and G) Primary cultured mice cortical neurons were subjected to 2 h of oxygen and glucose deprivation followed by 6 h of reperfusion (OGD-Rep). (F) The SQSTM1, TOMM20, and COX4I1 protein levels in both Bnip3l+/+ and bnip3l−/− neurons treated with OGD-Rep were determined. (G) The relative mitochondrial DNA (mtDNA) levels, which as indicated by the mt-Atp6 (mitochondria-encoded DNA):Rpl13 (nucleus-encoded DNA) ratio, were assessed by real-time PCR in both Bnip3l+/+ and bnip3l−/− neurons. (H) Adeno-associated viruses expressing GFP-BNIP3L or GFP were intracerebrally injected into Bnip3l+/+ and bnip3l−/− mice 2 wk before tMCAO. After 6 h of reperfusion, the TOMM20, COX4I1, and cleaved-CASP3 expression in ischemic penumbra were determined by western blot. The data are expressed as means ± SD. Statistical comparisons were performed with one-way ANOVA followed by Dunnett's t test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the indicated group.
The primary cultured cortical neurons from Bnip3l+/+ and bnip3l−/− mice were subjected to oxygen and glucose deprivation reperfusion (OGD-Rep.) treatment, an in vitro model of cerebral ischemia. The loss of both TOMM20 and COX4I1 was compromised in bnip3l−/− neurons (Fig. 1F). To evaluate mitophagy defects, the abundance of the mitochondrial-DNA encoded gene ATPase6 was normalized to the level of the genomic gene Rpl13 by RT-qPCR; this method reflects the relative amount of mitochondria. We found the reduction in the ATPase6:Rpl13 ratio that occurred with OGD-Rep was significantly reversed by Bnip3l deletion (Fig. 1G). These results implicate the involvement of BNIP3L in ischemia-reperfusion-induced mitophagy. To further confirm this assertion, the striatum of Bnip3l+/+ and bnip3l−/− mice were infected, respectively, with AAV-GFP-BNIP3L and AAV-GFP. The infection efficiency was confirmed (Fig. S1A). Exogenous expression of BNIP3L rescued the mitophagy impairment of bnip3l−/− mice. We previously reported that mitophagy inhibition reinforced ischemia-reperfusion-induced neuronal apoptosis.1 In accordance with this finding, we here found activated CASP3 in bnip3l−/− mice with tMCAO, and this could be rescued by compensatory expression of BNIP3L (Fig. 1H). Taken together, these data suggest that BNIP3L is required in ischemia-reperfusion-induced mitophagy.
BNIP3L deficiency aggravates ischemic brain injury
Mitophagy attenuates ischemia-reperfusion-induced brain injury.1,5 To verify the contributions of BNIP3L-mediated mitophagy to ischemic brain injury, Bnip3l+/+, Bnip3l+/− and bnip3l−/− mice were subjected to transient middle cerebral artery occlusion (tMCAO). BNIP3L deficiency significantly exacerbated ischemic brain injury as revealed by increased infarct volumes and neurological deficit scores, pointing to a neuroprotective effect of BNIP3L. To further confirm the apparent benefits of BNIP3L in ischemic brains, AAV-GFP-BNIP3L was injected to the striatum of bnip3l−/− mice to force BNIP3L expression (Fig. S1). The deleterious effects of Bnip3l deletion were completely rescued (Fig. 2A-C). Further, the contributions of BNIP3L to ischemic neuronal injury were evaluated in vitro. Compared with wild-type primary cultured neurons, bnip3l−/− neurons showed weaker tolerance to OGD-Rep insults, and this could be rescued by AAV-GFP-BNIP3L infection. The benefits of BNIP3L are closely related to mitophagy as the rescue effects of AAV-GFP-BNIP3L were reversed by the autophagy inhibitor 3-methyladenine (3-MA; Fig. 2D).
Figure 2.
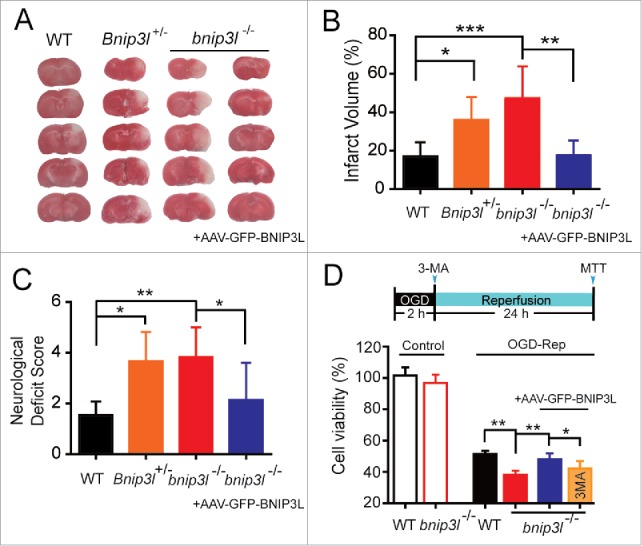
BNIP3L deficiency aggravates ischemic brain injury. (A) Mice were subjected to 1 h of tMCAO followed by 24 h reperfusion. The cerebral infarct volumes were determined by TTC staining; representative TTC-stained brain slices from each group are shown. (B and C) The infarct volumes and neurological deficit scores of each group were determined (n = 5–8). (D) A timeline scheme showed the procedures of this experiment. Primary cultured bnip3l−/− neurons were infected with adeno-associated viruses expressing GFP-BNIP3L or GFP 48 h before 2 h of oxygen and glucose deprivation treatment. 3-MA (10 μg) was injected intracerebroventricularly at the onset of reperfusion. After 24 h of reperfusion, the cell viability in both Bnip3l+/+ and bnip3l−/− neurons was measured by MTT assay. The data are expressed as means ± SD. Statistical comparisons were performed with one-way ANOVA followed by Dunnett's t test. *p < 0.05, **p< 0.01, ***p < 0.001 vs. the indicated group.
PARK2 is dispensable in BNIP3L-mediated mitophagy evoked by ischemia-reperfusion
We have previously reported the involvement of PARK2 in ischemia-reperfusion-evoked mitophagy in brains.1,5 To establish whether BNIP3L-mediated mitophagy requires PARK2, we conducted experiments with park2−/− mice. The colocalization of BNIP3L and LC3B was observed in both Park2+/+ and park2−/− neurons subjected to OGD-Rep (Fig. 3A and B), indicating that the ability of BNIP3L to bind with LC3B is not influenced by PARK2. Additionally, mitophagy was still observed in park2−/− mice subjected to tMCAO, suggesting the presence of alternative mitophagy mechanisms. Moreover exogenous expression of GFP-BNIP3L in the striatum reinforced ischemia-reperfusion-induced mitophagy in park2−/− mice (Fig. 3C). Consistently, forced BNIP3L expression also increased mitophagy in primary cultures of park2−/− neurons (Fig. 3D), indicating the dispensability of PARK2 in BNIP3L-mediated mitophagy. We tested the neuroprotection effects of BNIP3L-mediated mitophagy in park2−/− mice and found that the extent of brain injury was significantly reduced by GFP-BNIP3L overexpression (Fig. 3E). Similar results were obtained in cultured park2−/− neurons (Fig. 3F). These results further support the assertion that PARK2 is dispensable for BNIP3L-mediated mitophagy in ischemic brains.
Figure 3.

PARK2 is dispensable in BNIP3L-mediated mitophagy evoked by ischemia-reperfusion. (A) Park2+/+ and park2−/− neurons were treated with 2 h of OGD. LC3B (green) and BNIP3L (red) were stained by immunocytochemistry at the indicated time after reperfusion, and the images were taken by confocal microscopy. (B) Columns represent the Manders overlap coefficient of BNIP3L and LC3B in Park2+/+and park2−/− neurons. At least 30 cells from 3 independent experiments for each group were included. (C and D) park2−/− mice and primary cultured neurons were infected with AAVs expressing GFP-BNIP3L or GFP. The mice were subjected to one h of tMCAO with 6 h of reperfusion. The cultured neurons were subjected to 2 h of OGD and 6 h of reperfusion. The SQSTM1, TOMM20 and COX4I1 levels in (C) brain tissue and (D) cultured neurons were determined by western blot. (E) Another set of mice were killed 24 h after MCAO and infarct volumes were determined by TTC staining. (F) Primary cultured wild-type and park2−/− neurons were infected with GFP-BNIP3L or GFP-expressing AAVs 48 h in advance. After 2 h of OGD and 24 h of reperfusion, the cell viability was measured by MTT assay. The data are expressed as means ± SD. Statistical comparisons were performed with unpaired Student t tests. *p < 0.05 vs. the indicated group.
Given the involvement of PARK2 in ischemia-reperfusion-induced mitophagy, we were curious about whether PARK2-mediated mitophagy, conversely, depends on BNIP3L. We thus examined the recruitment of PARK2 to mitochondria, which has been demonstrated as a prerequisite for its subsequent biological functions.27 By immunostaining PARK2 and TOMM20 in primary cultured neurons, we found the translocation of PARK2 to mitochondria increased along with reperfusion within 3 h, and decreased afterward, suggesting mitophagic degradation. Interestingly, Bnip3l deletion did not disturb the observed trends of PARK2 recruitment to mitochondria (Fig. 4A and B). These results suggest that BNIP3L is indeed dispensable for PARK2-dependent mitophagy in ischemic brains. This supposition was further confirmed by the fact that forced expression of PARK2 significantly attenuated infarct volumes in bnip3l−/− mice (Fig. 4C). Taken together, these results strongly suggest that BNIP3L and PARK2 function in mutually independent pathways to mediate mitophagy in ischemic brains.
Figure 4.

BNIP3L is dispensable in PARK2-dependent mitophagy evoked by ischemia-reperfusion. (A) Bnip3l+/+ and bnip3l−/− neurons were treated with 2 h of OGD. DAPI (blue), PARK2 (green), and the mitochondrial marker TOMM20 (red) were stained by immunocytochemistry at the indicated times after reperfusion. The images were taken by confocal microscopy. (B) Columns represent the Manders overlap coefficient of PARK2 and TOMM20 in Bnip3l+/+ and bnip3l−/− neurons. At least 44 cells from 3 independent experiments for each group were included. (C) bnip3l−/− mice were infected with GFP- or GFP-PARK2-expressing AAVs, and were then subjected to tMCAO for one h. The mice were killed 24 h after MCAO, and infarct volumes were determined by TTC staining. *p < 0.05 vs. the indicated group. The data are expressed as means ± SD. Statistical comparisons were performed with one-way ANOVA followed by Dunnett's t test. *p < 0.05 vs. the indicated group.
Mitophagy in ischemic brains is synergistically impaired in bnip3l and park2 double-knockout mice
To further study the association of BNIP3L with PARK2 in mitophagy induction in ischemic brains, we crossed bnip3l−/− and park2−/− mice to generate bnip3l and park2 double-knockout (DKO, Fig. S1B) mice. Primary cultured neurons from mice of various germlines were previously transfected with plasmids encoding GFP-LC3B and Mito-DsRed, and the extent of mitophagy was examined by the ratio of LC3 puncta on mitochondria:total LC3 puncta. After OGD-Rep treatment, the ratio was significantly decreased in bnip3l−/− and park2−/− neurons as compared with wild-type neurons. Of note, the ratio in DKO neurons was reduced to a further extent relative to that observed in either bnip3l or park2 single deletion cells (Fig. 5A and B), indicating a synergistic reduction in mitophagy. We next subjected brain corticostriatal slices from the bnip3l−/−, park2−/− and DKO mice to OGD-Rep., and assessed the SQSTM1, TOMM20, and COX4I1 levels. Although bnip3l and park2 deletion alone partly compromised OGD-Rep-induced mitophagy, mitophagy was further diminished in DKO slices (Fig. 5C). However, as revealed by monitoring the number of GFP-LC3B puncta, autophagic flux was not blocked in these mutant neurons (Fig. 5B), indicating specific impairment of mitophagy. Consistently, DKO corticostriatal slices showed a greater extent of ischemic injury as compared with the injury in slices of the single-gene deletion genotypes, as determined by 2, 3, 5-triphenyltetrazolium hydrochloride (TTC) conversion assay (Fig. 5D), a redox indicator that measures metabolic activity. Importantly, exogenous expression of either BNIP3L or PARK2 was sufficient to rescue the ischemic brain injury in DKO mice (Fig. 5E). Taken together, these data further confirm the hypothesis that BNIP3L and PARK2 function in mutually-independent pathways to mediate mitophagy in ischemic tissues, and establish that the neuroprotective effects of BNIP3L-mediated mitophagy are functionally independent of PARK2 in the protection of ischemic brains.
Figure 5.
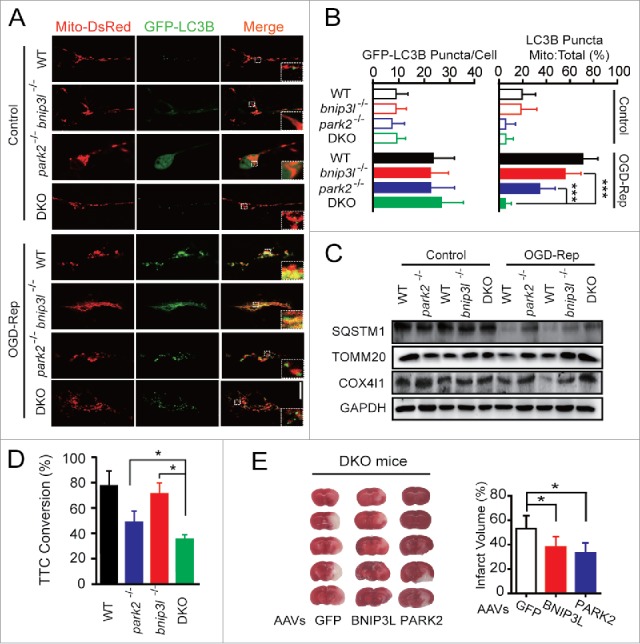
Mitophagy is further impaired in bnip3l and park2 double-knockout ischemic brain. park2 and bnip3l double-knockout mice (DKO) were generated by hybridizing park2−/− with bnip3l−/− mice. (A) Primary cultured cortical neurons from the indicated germline mice were previously transfected with GFP-LC3B and Mito-DsRed by viral vector infection. After 3 h of reperfusion, fluorescent images were captured by confocal microscopy. Images show representative examples from 3 independent experiments. (B) Columns represent the number of GFP-LC3B-positive puncta per cell (left panel) and the proportion of LC3B puncta on mitochondria of the total LC3B puncta (right panel). At least 5 random fields from one section and 3 to 6 sections were averaged in each independent experiment. The bar chart shows mean ± SD values of puncta number from at least 3 independent experiments; at least 50 cells were analyzed in each group. (C and D) Brain corticostriatal slices from the indicated mice were subjected to 15 min of OGD plus 1 h reperfusion. (C) The protein levels of SQSTM1, TOMM20, and COX4I1 in the indicated mice were assessed by western blot. (D) After one h of reperfusion, tissue viability was quantified by TTC conversion assay. (E) DKO mice were injected with AAVs expressing GFP, GFP-BNIP3L or GFP-PARK2. The mice were then subjected to one h of tMCAO with 24 h of reperfusion. The representative images of ischemic brains are shown and the infarct volumes were determined by TTC staining. The data are expressed as means ± SD. Statistical comparisons were performed with one-way ANOVA followed by Dunnett's t test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the indicated group.
Phosphorylation of BNIP3L Ser81 is required for BNIP3L-mediated mitophagy
The mechanisms through which BNIP3L induces mitophagy are not fully understood. It was reported that phosphorylation of key mitophagy-related proteins, including AMBRA1, FUNDC1, and BNIP3, are critical for the regulation of mitophagy.28-30 We therefore hypothesized that BNIP3L phosphorylation may also play a role in ischemia-induced mitophagy. To test this, we assessed BNIP3L phosphorylation via Phos-tag assays. In line with our hypothesis, we detected the appearance of a shifted phosphorylated BNIP3L band following tMCAO treatment in mice brains. Interestingly, the level of this phosphorylated BNIP3L increased along with reperfusion, and increased following the inhibition of autophagy with 3-MA (Fig. 6A), suggesting that BNIP3L phosphorylation appears to be related to mitophagy. To explore the phosphorylation sites of BNIP3L, we screened the phosphorylated serine residues of BNIP3L revealed by phosphoproteome studies.31-34 We constructed plasmids expressing nonphosphorylated BNIP3L mutations by replacing the candidate serine residues with alanine. A screen of these mutants revealed that serine 81 was required for BNIP3L-mediated mitophagy (Fig. S3), and the alignment analysis showed it was conserved among species (Fig. 6B). We transfected COS7 cells with a plasmid encoding BNIP3LS81A and found that this mutation reversed BNIP3L-induced mitophagy. Similar results were reproduced in HeLa cells lacking PARK2, again supporting the independence of PARK2 in BNIP3L-mediated mitophagy (Fig. 6C). To specifically detect the phosphorylation of BNIP3L (Ser81), we developed an antibody against phosphorylated Ser81 of BNIP3L. The specificity of this antibody was confirmed because only wild-type but not the mutant BNIP3LS81A in HeLa cells could be detected (Fig. 6D, IB: Phos-BNIP3L [S81]). The phosphorylation of BNIP3L (Ser81) decreased with CCCP treatment in a time-dependent manner, while the total BNIP3L level remained intact (Fig. 6D, IB: Flag). These data indicated the selective degradation of phosphorylated BNIP3L (Ser81), suggesting its association with mitophagy. Phosphorylation of BNIP3L (Ser81) was also observed in both Park2+/+ and park2−/− brains. Importantly, phospho-BNIP3L (Ser81), rather than total BNIP3L was reduced after tMCAO, which further confirmed the close link of phospho-BNIP3L (Ser81) with mitophagy in ischemic brains (Fig. 6E). Previous studies have demonstrated that the binding of BNIP3L with LC3A and LC3B mediate the recruitment of phagophores to mitochondria.20 Here, we found that the BNIP3LS81A mutant protein could hardly bind with MYC-LC3A and MYC-LC3B whereas the phosphomimic mutant BNIP3LS81E showed stronger interaction with both GFP-LC3A and GFP-LC3B (Fig. 6F). In addition, in HeLa cells, recruitment of phagophores (marked by GFP-LC3B) to BNIP3L-labeled mitochondria (marked by Flag-BNIP3L and TOMM20) was evident, but this could not be detected in cells transfected with the BNIP3LS81A mutant protein (Fig. 6G). Together, these results support the characterization of Ser81 as a novel phosphorylation site of BNIP3L and confirm that phosphorylation at this site is required for BNIP3L-mediated mitophagy.
Figure 6.

Phosphorylation of BNIP3L (Ser81) is responsible for BNIP3L-mediated mitophagy. (A) Wild-type mice were subjected to one h tMCAO, 10 µg 3-MA was injected at the onset of reperfusion, and phosphorylation of BNIP3L was determined by Phos-tag analysis at the indicated reperfusion time. (B) Alignment of BNIP3L sequences from the indicated species. Identical amino acids residues are highlighted in black. Highly conserved residues are labeled by “*” while less conserved ones are labeled by “•." Serine 81 is marked in red. Indicated domains of BNIP3L are underlined. (C) Plasmids expressing Flag (vector, VEC), Flag-BNIP3L, Flag-BNIP3LS81A and Flag-BNIP3LS81D were transfected into COS7 and HeLa cells for 36 h, and 12 h after 20 µM CCCP treatment the TOMM20 level was assessed by western blot. (D) Anti-phosphorylated BNIP3L (Ser81) antibody was confirmed in HeLa cells expressing Flag-BNIP3L or Flag-BNIP3LS81A after 20 µM CCCP treatment of the indicated hours. (E) The phosphorylation of BNIP3L on Ser81 was detected in both Park2+/+ and park2−/− mice after tMCAO, sample from bnip3l−/− mice were taken as a negative control. (F) Protein-protein interactions of both LC3A and LC3B with BNIP3L, BNIP3LS81A and BNIP3LS81E were confirmed by co-immunoprecipitation in HeLa cells expressing the indicated plasmids. (G) Plasmids expressing Flag, Flag-BNIP3L and Flag-BNIP3LS81A were co-transfected with GFP-LC3B plasmid to HeLa cells for 36 h. Six hours after 20 µM CCCP treatment, Flag (blue) and the mitochondrial marker TOMM20 (red) were stained by immunocytochemistry and the images were taken by confocal microscopy.
Phosphorylation of BNIP3L on Ser81 is responsible for ischemia-reperfusion-induced mitophagy and its neuroprotective effects
We next investigated the involvement of the phosphorylation of BNIP3L (Ser81) in BNIP3L-mediated mitophagy in ischemic scenarios. Cultured bnip3l−/− neurons transfected with wild-type BNIP3L enhanced OGD-Rep-induced mitophagy, as revealed by the reductions in the levels of TOMM20 and COX4I1. However, these effects were not observed following transfection with the BNIP3LS81A mutant protein (Fig. 7A). These data clearly indicated the requirement of BNIP3L (Ser81) phosphorylation for BNIP3L-mediated mitophagy in ischemic neurons. We next explored this in ischemic mice brains and found that the results of experiments with GFP-BNIP3L, rather than the GFP-BNIP3LS81A, expression in bnip3l−/− mice reinforced the idea of tMCAO-induced mitophagy (Fig. 7C). Finally, the forced expression of GFP-BNIP3L, but not the forced expression GFP-BNIP3LS81A, reversed both neuronal cell viability and brain infarct volumes (Fig. 7B and D). These results indicate that phosphorylation of BNIP3L at Ser81 is required for its mitophagy activity and neuroprotective effects in ischemic brains.
Figure 7.

Phosphorylation of BNIP3L on Ser81 is required for the protection of mitophagy in ischemia-reperfusion. (A and B) bnip3l−/− neurons were transfected with adeno-associated viruses (AAVs) expressing GFP, GFP-BNIP3L, or GFP-BNIP3LS81A. (A) After 2 h of OGD followed with 6 h of reperfusion, the TOMM20 and COX4I1 protein levels were assessed by western blot. (B) After 24 h of reperfusion, the neuronal viability was measured by MTT assay. (C and D) bnip3l−/− mice brains were infected with AAVs expressing GFP, GFP-BNIP3L, or GFP-BNIP3LS81A and were then subjected to one h of tMCAO. (C) After 6 h of reperfusion, TOMM20 and COX4I1 levels were assessed by western blot. (D) After 24 h of reperfusion, the infarct volumes were determined by TTC staining. (E and F) Wild-type neurons were electroporated with plasmids expressing Flag (vector, VEC), Flag-BNIP3L, Flag-BNIP3LS81A or Flag-BNIP3LS81E. (E) After 2 h of OGD followed with 6 h of reperfusion, the TOMM20 and Flag-BNIP3L proteins were assessed by western blot. (F) After 24 h of reperfusion, the neuronal viability was measured by MTT assay. The data are expressed as means ± SD. Statistical comparisons were performed with one-way ANOVA followed by Dunnett's t test. *p < 0.05, **p < 0.01 vs. the indicated group.
We next asked whether the phosphomimetic BNIP3L mutant showed additional neuroprotective effects. We transfected the wild-type neurons with Flag, Flag-BNIP3L, Flag-BNIP3LS81A and Flag-BNIP3LS81E plasmids. Compared to vector, BNIP3L and Flag-BNIP3LS81E reinforced the OGD-Rep-induced mitophagy to a similar extent, but failed to show further neuroprotective effects, while Flag-BNIP3LS81A reversed BNIP3L-mediated mitophagy and aggravated injury (Fig. 7E and F). These results indicated that BNIP3L-mediated mitophagy is required for but not sufficient to protect ischemic neurons.
Discussion
BNIP3L is a mitophagy receptor that removes mitochondria during the development of reticulocytes.18,35 However, the significance of BNIP3L-mediated mitophagy in diseases is largely unclear. Here we found that bnip3l deletion compromised ischemia-reperfusion-induced mitophagy, both in vivo and in vitro (Fig. 1). Conversely, exogenous BNIP3L expression in brains rescued mitophagy impairment in bnip3l−/− mice (Fig. 1H). These observations provide a previously undiscovered implication of BNIP3L-mediated mitophagy in cerebral ischemia. Furthermore, BNIP3L overexpression rescued ischemic injury in bnip3l−/− mice (Fig. 2). These data clearly point to a neuroprotective role of BNIP3L in ischemic stroke. Consistently, we found that exogenous BNIP3L expression attenuated CASP3 activation in ischemic tissues after MCAO, suggesting suppression of apoptosis. We further found that the neuroprotective effect of BNIP3L-mediated mitophagy was abolished by 3-MA, a broad autophagy inhibitor, in OGD-treated primary cultured neurons. These findings challenge the widely held notion of the pro-apoptotic roles of BNIP3L in ischemic brains36 and, for the first time, indicate that BNIP3L-mediated mitophagy protects against ischemic brain injury. Hence, the present findings provide further evidence that confirmed our previous notion that mitophagy protects ischemic brains1 and shed light on the opportunities of considering BNIP3L as a potential _target for stroke therapy.
Current knowledge indicates that PARK2 is closely associated with mitophagy induction. However, it is not clear whether BNIP3L-mediated mitophagy associates with PARK2 in cerebral ischemia. We found an overlap of endogenous LC3B and BNIP3L by OGD-reperfusion in park2−/− neurons. Because most BNIP3L localizes in mitochondria,17 this result suggested PARK2 has little impact on phagophore _targeting to BNIP3L-labeled mitochondria. Furthermore, we interestingly find that exogenous BNIP3L expression still promoted mitophagy in park2−/− brains and neurons with ischemia-reperfusion (Fig. 3C and D), which confirmed that BNIP3L-mediated mitophagy remained competent in the absence of PARK2. In contrast, previous studies indicate BNIP3L-mediated mitophagy largely counts on PARK2 in non-neuronal cells,21,37 in which intracellular PARK2 levels are intact or overexpressed. Together with the present data, it seems that BNIP3L may play a compensatory role to mediate mitophagy with a low PARK2 level. This compensation could be critical because remarkable PARK2 loss is observed in ischemic brain tissue12 and this was confirmed by the present results (Fig. 1A and Fig. 4A). Interestingly, there was no significant loss of BNIP3L in ischemic brains (Fig. 1A and Fig. 6E). Even after 6 h of reperfusion, when remarkable PARK2 was lost, BNIP3L was colocalized with LC3B on mitochondria in OGD-Rep-treated neurons (Fig. 3A). These observations strongly indicated that BNIP3L could be involved in a successive mechanism of mitophagy induction after PARK2 is exhausted during ischemia-reperfusion. The significance of BNIP3L-mediated mitophagy was confirmed by the results that the overexpression of BNIP3L attenuated ischemic injury of park2−/− mice (Fig. 3E and F). Conversely, by further deletion of Bnip3l from park2−/− mice, we found synergistic mitophagy impairment in OGD-Rep-treated DKO neurons (Fig. 5A-C), as well as increased vulnerability to ischemia reperfusion of DKO mice as compared with either single gene deletion. These data strongly suggested that BNIP3L-mediated mitophagy is PARK2-independent and could be an important compensation for PARK2-mediated mitophagy in ischemic brains. Some other proteins, including FUNDC1 and BNIP3, have also been documented to induce mitophagy.29,30,38,39 We confirmed the presence of both FUNDC1 and BNIP3 in tMCAO-treated mice brains. BNIP3 was found downregulated along with reperfusion after ischemia and was accumulated with 3-MA treatment, suggesting autophagy-dependent degradation of BNIP3. However, the expression of FUNDC1 was not regulated with these manipulations (Fig. S2). We found that mitophagy was almost completely dysfunctional in DKO neurons subjected to OGD-Rep (Fig. 5B, C). Although the contributions of other mitophagy receptors cannot be fully excluded, we indicated that BNIP3L and PARK2 are prominent proteins responsible for mitophagy in cerebral ischemia.
Mitochondrial transmembrane potential (ΔΨm) loss could be a critical trigger for PARK2 recruitment to damaged mitochondria. In ischemic neurons, the dramatically decreased ΔΨm is transiently hyperpolarized upon reperfusion.40,41 BNIP3L was reported to depolarize mitochondria42 and mitochondrial PARK2 accumulation is reversible with ΔΨm regulation.43 However, we found bnip3l deletion showed no impact on PARK2 recruitment to mitochondria in OGD-reperfusion-treated neurons (Fig. 4A and B). More importantly, PARK2 overexpression still rescued ischemic brain injury in bnip3l−/− mice (Fig. 4C). These data exclude the possibility that BNIP3L may facilitate PARK2 recruitment to mitochondria and thus reinforced the idea that BNIP3L and PARK2 are mutually independent pathways for mitophagy induction in ischemic brains. In addition, hypoxia has previously been proposed to induce BNIP3L expression, while we did not find significant BNIP3L upregulations with ischemia-reperfusion insults (Fig. 1A). Several studies support this idea; for instance, BNIP3L shows reduced (or no) sensitivity to hypoxia as compared with its homolog protein BNIP3 in either tumor cells or cardiomyocytes.24,44 It is worth noting that park2−/− mice showed a slightly more extensive ischemic injury than bnip3l−/− mice (Fig. 2B vs. Fig. 3E). In the corticostriatal slices, park2−/− mice also showed more severe injury than bnip3l−/− ones (Fig. 5D). Also, exogenous expression of BNIP3L showed comparable levels of neuroprotection to that observed with PARK2 overexpression (Fig. 5E). Similarly, park2−/− neurons seemed to show a more extensive mitophagy impairment compared with bnip3l−/− neurons (Fig. 5B). Although we found 2 mutually independent and compensatory pathways in ischemic brains, these observations might also highlight the importance of PARK2-mediated mitophagy in brain stroke.
The molecular basis for the BNIP3L-mediated mitophagy in ischemic neurons is unidentified. Here, we provided evidence to indicate that phosphorylation of BNIP3L (Ser81) was required for its mitophagy activity (Fig. 6C, Fig. 7A and C), and subsequent neuroprotective effects (Fig. 7B and D). The phosphorylation of BNIP3L (Ser81) in park2−/− brains (Fig. 6E) further confirmed the dispensability of PARK2 in BNIP3L-mediated mitophagy. Moreover, the Ser81-phosphorylated BNIP3L was dramatically decreased upon ischemia-reperfusion but the total BNIP3L level was intact (Fig. 6E). Therefore, compared with PARK2, it seems that BNIP3L has an apparent redundancy in mitophagy execution in cerebral ischemia. This redundancy may also explain why forced BNIP3L expression conferred neuroprotective effects in bnip3l−/− mice (Fig. 7D), but neither BNIP3L nor BNIP3LS81E protected the wild-type mice and neurons (Fig. S4 and Fig. 7F). In addition, Ser81 is an evolutionarily conserved residue within the minimal-essential region (MER) of BNIP3L, which was previously demonstrated to be essential in reticulocyte mitophagy.19 The MER in BNIP3L has been predicted to form an α-helix and may fold with the LC3-interacting region motif when binding with other _targets, given that MER per se does not bind with LC3 directly.19 It is likely the phosphorylation of Ser81 in MER regulates the binding affinity of the LC3-interacting region motif with Atg8 family members; further studies regarding the BNIP3L protein structure may help to address this issue. In contrast, the S81A mutation seems to have no impact on mitophagy in erythroid development; this discrepancy may be attributable to different cell types and/or, more likely, the ischemic environment. Although we did not here identify the kinases responsible for phosphorylation of BNIP3L (Ser81), our results establish a foundation for future investigation of this topic.
The present study demonstrated the previously unidentified neuroprotective effects of BNIP3L-mediated mitophagy in cerebral ischemia-reperfusion injury. BNIP3L and PARK2 were found to function in mutually independent pathways for the induction of mitophagy in ischemic brains. The phosphorylation of Ser81 of BNIP3L was found to be required for its mitophagy activity. These findings underscore the complex nature of mitophagy induction in ischemic neurons and highlight the potential value of BNIP3L as a _target for therapies for ischemic stroke.
Materials and methods
Animals
Male C57BL/6 mice and male BALB/C mice weighing 22–25 g were used. The park2−/− mice were kindly provided by Prof Zhuohua Zhang from Central South China University; the bnip3l−/- mice were kindly provided by Prof. Ney Paul from St. Jude Children's Research Hospital. We then crossed park2−/− and bnip3l−/− mice to generate the park2 bnip3l double-knockout mice. For the culture of primary cortical neurons, pregnant mice with embryonic (E17) fetuses were used. All experiments were approved by and conducted in accordance with the ethical guidelines of the Zhejiang University Animal Experimentation Committee.
Transient MCAO mouse models
Mice were anesthetized by inhalation of isoflurane for surgery. Cerebral blood flow (CBF) was evaluated in the area of the middle cerebral artery (MCA). Transient focal cerebral ischemia was induced by MCAO, as described previously with minor modifications.45 Briefly, a 6–0 nylon monofilament suture was inserted 10 mm into the internal carotid to occlude the origin of the MCA. Animals with less than 80% reduction in CBF were excluded from the study. Body temperature was maintained at 37°C during surgery and for 2 h after the start of reperfusion. To measure infarct volume, coronal mice brain slices, at 2-mm intervals, were stained with 0.25% TTC (Sigma, T8877). Infarcted areas were analyzed using Image Pro Plus 7.0 and measured by the indirect method, which corrects for edema. Neurological deficit scores were evaluated after 24 h of reperfusion.
Viral delivery
Under sodium pentobarbital anesthesia (50 mg/kg, intraperitoneal), mice were mounted in a stereotaxic apparatus and adeno-associated virus (AAV, designed, produced and identified by Obio Technology Corp., Ltd., Shanghai, China) including AAV-GFP, AAV-GFP-PARK2, AAV-GFP-BNIP3L, and AAV-GFP-BNIP3L_S81A, were injected into the corpus striatum (AP +0.5 mm; L -2.0 mm; V -3.0 mm). The viruses were infected for a minimum of 2 wk before further experiments to allow time for sufficient protein accumulation.
Preparation of brain slices and oxygen-glucose deprivation
Acute brain slices were prepared from adult male mice. The oxygen–glucose deprivation (OGD) procedures were performed as we described previously.46 Briefly, corticostriatal slices (400-mm thick) were immersed in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 5 mM KCl, 1.25 mM KH2PO4, 2 mM MgSO4, 26 mM NaHCO3, 2 mM CaCl2, 10 mM glucose (pH 7.4, all chemicals were purchased from Sigma) for one h and then incubated at 37°C for 15 min before further experiments. For OGD, slices were transferred into glucose-free ACSF bubbled with 5% CO2 and 95% N2 for 15 min and then returned to oxygenated ACSF for one h. Slice viability was determined by 0.25% TTC (Sigma, T8877) staining as described previously.47 Formazan extracted was measured at 490 nm and normalized to the dry weight of the slice.
Cell culture, OGD procedures, and cell viability
The primary cortical neuronal culture experiments were performed as described previously.45 Briefly, the dissected cortex from E17 fetal mice was digested with 0.25% trypsin (Invitrogen, 25200–056). Approximate 2 × 105 cells/cm2 were seeded onto poly-L-lysine (10 µg/ml)-coated plates (Sigma, P1399) and dishes. The neurons were grown in Neurobasal medium (Invitrogen, 21103–049) supplemented with 2% B27 (Invitrogen, 17504–044) and 0.5 mmol/L glutamine (Invitrogen, 21051–024). Cultures were maintained for 8 d before treatment. Human HeLa cells and COS7 cells were routinely cultured in DMEM containing 10% fetal bovine serum (Invitrogen, 10099141).
For OGD treatment, primary neurons were refreshed with O2- and glucose-free DMEM. Cells were then immediately placed in a sealed chamber (Billups-Rothenburg, MIC-101) loaded with mixed gas containing 5% CO2 and 95% N2. For reperfusion, neurons were refreshed with normal culture medium. 3-MA (5 mM; dissolved in normal culture medium) was added to the cells at the onset of reperfusion.
Transfection
Primary cultured neurons were transfected with AAVs containing GFP-LC3B (Hanbio, China). For Mito-DsRed delivery, the Mito-DsRed DNA sequence was cloned from pDsRed2-Mito (Clontech, 632421) and inserted into the pLVX-puro plasmid (Clontech, 632164) and then transfected into HEK293T cells with a set of plasmids for lenti-virus packaging (Clontech, 631247). The lentivirus and/or the AAVs were added to neurons at 6 d in vitro.
BNIP3L, BNIP3L_S81A, and BNIP3L_S81D or S81E were ligated into the p3 × FLAG-CMV-7.1 vector (Sigma, E7533) and transfected into primary cultured neurons using Amaxa electroporation (VPG-1001, Lonza, Switzerland) according to the manufacturer's protocol. Briefly, after digestion of the cortex, 5 × 106 cells were collected and resuspended in 100 μl room temperature Nucleofector Solution (Lonza, VPG-1001) containing 3 μg DNA per sample. The cell/DNA suspension was then transferred into a cuvette. The cuvette with cell/DNA suspension was inserted into the Nucleofector Cuvette Holder. After being electroporated using program O-005, the cells were resuspended in the medium mentioned above and then transferred into the prepared culture dishes. The plasmids were transfected into COS7 or HeLa cells according to the jetPRIME (n114–15, Polyplus, France) protocol.
Immunoblotting and co-immunoprecipitation
The brain tissues and cells were homogenized in RIPA buffer (Sangon Biotech, C500005). A 40-µg aliquot of protein from each sample was separated using SDS-PAGE. The following primary antibodies were used: phos-BNIP3L (S81, 1:2000; Abcam, ab208190), LC3 (1:1,000; Sigma, L7543), SQSTM1 (1:1,000; Cell Signaling Technology, 5114), cleaved CASP3 (1:1,000; Cell Signaling Technology, 9661), COX4I1 (1:1,000; Cell Signaling Technology, 4850), TOMM20 (1:1,000; Anbo Biotechnology, c16678), PARK2 (1:1,000; Sigma, P5748), or GAPDH (1:3,000; KangChen, KC-5G4). Secondary antibodies conjugated with HRP against either rabbit or mouse IgG (1:5,000; Cell Signaling Technology, 7071 and 7072) were applied. Digital images were quantified using densitometric measurement with Quantity-One software (Bio-Rad).
For co-immunoprecipitation, HeLa cells were transiently transfected. At 24 h post transfection, the cells were lysed and immunoprecipitated using a FLAG Immunoprecipitation Kit (Sigma, FLAGIPT1). Thereafter, the precipitants were washed 3 times with lysis buffer and the immune complexes were eluted with sample buffer containing 1% SDS (AMRESCO, 0227) for 5 min at 100°C and analyzed by SDS-PAGE.
Detection of phosphorylated BNIP3L by using Phos-tag SDS-PAGE was performed according to the manufacturer's protocol (Wako Pure Chemical Industries, Japan). Briefly, a 50-µg aliquot of protein from each sample was separated using 8.5% polyacrylamide gels containing 100 µM Phos-tag acrylamide (Wako Pure Chemical Industries, AAL-107) and 200 µM MnCl2 (Sigma, 244589). Gels were washed with transfer buffer containing 1 mM EDTA (Sigma, E6758) for 10 min at room temperature and then with transfer buffer without EDTA for another 10 min. Proteins were transferred to PVDF membranes (Millipore, IPVH00010) and immunoblotting performed using a chemiluminescent digital imaging system (6100, Tanon, China).
Immunocytochemistry and confocal microscopy
For immunostaining, cells were incubated with antibodies against Flag (1:200; Cell Signaling Technology, 2368S), PARK2 (1:200; Sigma, P5748), or TOMM20 (1:100; Abcam, ab56783). Secondary antibodies labbled with Alex Fluor 488, 596 or 647 (Invitrogen, A32731, A21203 and A32733) were subsequently added to the cells. Coverslips were observed on a confocal microscope (Fluoview FV1000, Olympus, Japan). Mander's overlap efficiency was measured and analyzed with Image Pro-Plus 7.0 software. Five randomly selected fields from one coverslip were included to calculate an average, and experiments were repeated independently at least 3 times.
Real-time PCR
After 6 h of OGD-reperfusion, total cellular DNA was extracted with a DNeasy Blood & Tissue kit (Qiagen, DP304–03). Aliquots of 20 ng total DNA were analyzed via real-time PCR to evaluate the expression of the mitochondrial gene mt-Atp6 and the genomic gene Rpl13, as described previously.1 The copy numbers of Atp6 and Rpl13 were calculated and normalized by the standard curve. Relative expression was presented as mt-Atp6:Rpl13.
Statistical analysis
All data were collected and analyzed in a blind manner. Data are presented as mean ± SD. One-way ANOVA (analysis of variance) with Dunnett's T3 post-hoc test was applied for multiple comparisons. p < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- AAV
adeno-associated virus
- BNIP3L/NIX
BCL2/adenovirus E1B interacting protein 3-like
- CASP3
caspase 3
- COX4I1
cytochrome c oxidase subunit 4I1
- CQ
chloroquine
- DKO
Park2 and Bnip3l double knockout
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- I-R
ischemia-reperfusion
- MAP1LC3A/LC3A
microtubule-associated protein 1 light chain 3 α
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- MER
minimal essential region
- mt-Atp6
mitochondrially encoded ATP synthase 6
- mtDNA
mitochondrial DNA
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- OGD-Rep.
oxygen and glucose deprivation-reperfusion
- PARK2
Parkinson disease (autosomal recessive, juvenile) 2, parkin
- SQSTM1
sequestosome 1
- tMCAO
transient middle cerebral artery occlusion
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- TTC
2, 3, 5-triphenyltetrazolium hydrochloride
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Prof. Paul Ney from St. Jude Children's Research Hospital for kindly offering the bnip3l−/− mice. The authors are grateful to Dr. Zhuohua Zhang for offering the Park2 gene knockout mice. Thanks for Dr. Hanmin Shen from National University of Singapore for reading the paper. We thank Abcam for generating rabbit monoclonal antibody against the phosphorylated BNIP3L (Ser81). We are grateful to the Core Facilities of Zhejiang University Institute of Neuroscience and Imaging Facilities, Zhejiang University School of Medicine for the help in confocal microscopy.
Funding
This work was funded by the National Natural Science Foundation of China (81273506, 81521062, 81373393 and 81573406), the Zhejiang Provincial Natural Science Foundation (LR15H310001).
References
- [1].Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al.. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321-33. doi: 10.4161/auto.25132. PMID:23800795 [DOI] [PubMed] [Google Scholar]
- [2].Yu S, Zheng S, Leng J, Wang S, Zhao T, Liu J. Inhibition of mitochondrial calcium uniporter protects neurocytes from ischemia/reperfusion injury via the inhibition of excessive mitophagy. Neurosci Letters. 2016;628:24-9. doi: 10.1016/j.neulet.2016.06.012 [DOI] [PubMed] [Google Scholar]
- [3].Zuo W, Zhang S, Xia CY, Guo XF, He WB, Chen NH. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology. 2014;86:103-15. doi: 10.1016/j.neuropharm.2014.07.002. PMID:25018043 [DOI] [PubMed] [Google Scholar]
- [4].Li Q, Zhang T, Wang J, Zhang Z, Zhai Y, Yang GY, Sun X. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun. 2014;444:182-8. doi: 10.1016/j.bbrc.2014.01.032. PMID:24440703 [DOI] [PubMed] [Google Scholar]
- [5].Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, Zheng Y, Deng T, Yan H, Li W, et al.. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014;10:1801-13. doi: 10.4161/auto.32136. PMID:25126734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li J, Lu J, Mi Y, Shi Z, Chen C, Riley J, Zhou C. Voltage-dependent anion channels (VDACs) promote mitophagy to protect neuron from death in an early brain injury following a subarachnoid hemorrhage in rats. Brain Res. 2014;1573:74-83. doi: 10.1016/j.brainres.2014.05.021. PMID:24880016 [DOI] [PubMed] [Google Scholar]
- [7].Yuan Y, Zhang X, Zheng Y, Chen Z. Regulation of mitophagy in ischemic brain injury. Neurosci Bull. 2015;31:395-406. doi: 10.1007/s12264-015-1544-6. PMID:26219224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467-84. doi: 10.1038/nrn.2016.51. PMID:27256553 [DOI] [PubMed] [Google Scholar]
- [9].Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5:706-8. doi: 10.4161/auto.5.5.8505. PMID:19377297 [DOI] [PubMed] [Google Scholar]
- [10].Tanaka A. Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Letters. 2010;584:1386-92. doi: 10.1016/j.febslet.2010.02.060. PMID:20188730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson's disease: the PINK1-parkin link. Biochim Biophys Acta. 2011;1813:623-33. doi: 10.1016/j.bbamcr.2010.08.007. PMID:20736035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mengesdorf T, Jensen PH, Mies G, Aufenberg C, Paschen W. Down-regulation of parkin protein in transient focal cerebral ischemia: A link between stroke and degenerative disease? Proc Natl Acad Sci U S A. 2002;99:15042-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A. 2011;108:12937-42. doi: 10.1073/pnas.1103295108. PMID:21768369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14:1127-35. doi: 10.1038/embor.2013.168. PMID:24176932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, et al.. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798-813. doi: 10.15252/embj.201488658. PMID:25349190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309-14. doi: 10.1038/nature14893. PMID:26266977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem 1999;274:7-10. doi: 10.1074/jbc.274.1.7. PMID:9867803 [DOI] [PubMed] [Google Scholar]
- [18].Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al.. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500-5. doi: 10.1073/pnas.0708818104. PMID:18048346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy. 2012;8:1325-32. doi: 10.4161/auto.20764. PMID:22906961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al.. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45-51. doi: 10.1038/embor.2009.256. PMID:20010802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879-90. doi: 10.1074/jbc.M110.119537. PMID:20573959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, Wang G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genetics. 2015;24:2528-38. doi: 10.1093/hmg/ddv017 [DOI] [PubMed] [Google Scholar]
- [23].Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082-7. doi: 10.1073/pnas.97.16.9082. PMID:10922063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669-73. doi: 10.1016/j.ccr.2004.10.012. PMID:11559532 [DOI] [PubMed] [Google Scholar]
- [25].Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, Buzzai M, Dicker DT, McKenna WG, Bernhard EJ, et al.. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004;6:597-609. PMID:15607964 [DOI] [PubMed] [Google Scholar]
- [26].Papandreou I, Krishna C, Kaper F, Cai D, Giaccia AJ, Denko NC. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res. 2005; 65:3171-8. doi: 10.1158/0008-5472.CAN-04-3395. PMID:15833847 [DOI] [PubMed] [Google Scholar]
- [27].Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795-803. doi: 10.1083/jcb.200809125. PMID:19029340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, et al.. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22:517. doi: 10.1038/cdd.2014.190. PMID:25661525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, et al.. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Reports. 2014;15:566-75. doi: 10.1002/embr.201438501. PMID:24671035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Thomas RL, Kubli DA, Gustafsson AB. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy. 2011;7:775-7. doi: 10.4161/auto.7.7.15536. PMID:21460627 [DOI] [PubMed] [Google Scholar]
- [31].Dai J, Jin WH, Sheng QH, Shieh CH, Wu JR, Zeng R. Protein phosphorylation and expression profiling by Yin-yang multidimensional liquid chromatography (Yin-yang MDLC) mass spectrometry. J Proteome Res. 2007;6:250-62. doi: 10.1021/pr0604155. PMID:17203969 [DOI] [PubMed] [Google Scholar]
- [32].Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007;104:1488-93. doi: 10.1073/pnas.0609836104. PMID:17242355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wisniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9:3280-9. doi: 10.1021/pr1002214. PMID:20415495 [DOI] [PubMed] [Google Scholar]
- [34].Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012;11:215-29. doi: 10.1074/mcp.O112.018366. PMID:22645316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232-5. doi: 10.1038/nature07006. PMID:18454133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Birse-Archbold JL, Kerr LE, Jones PA, McCulloch J, Sharkey J. Differential profile of Nix upregulation and translocation during hypoxia/ischaemia in vivo versus in vitro. J Cereb Blood Flow Metab. 2005;25:1356-65. doi: 10.1038/sj.jcbfm.9600133. PMID:15902200 [DOI] [PubMed] [Google Scholar]
- [37].Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, Wang G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. 2015;24(9):2528-38. doi: 10.1093/hmg/ddv017. PMID:25612572 [DOI] [PubMed] [Google Scholar]
- [38].Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al.. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177-85. doi: 10.1038/ncb2422. PMID:22267086 [DOI] [PubMed] [Google Scholar]
- [39].O'Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity. 2015;43:331-42. doi: 10.1016/j.immuni.2015.07.012. PMID:26253785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Huttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47:9-23. doi: 10.1007/s12035-012-8344-z. PMID:23011809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al.. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431-5. doi: 10.1038/nature13909. PMID:25383517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang J, Ney PA. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008;4:354-6. doi: 10.4161/auto.5552. PMID:18623629 [DOI] [PubMed] [Google Scholar]
- [43].Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012;22:320-33. doi: 10.1016/j.devcel.2011.12.014. PMID:22280891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW 2nd. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem. 2006;281:1442-8. doi: 10.1074/jbc.M509056200. PMID:16291751 [DOI] [PubMed] [Google Scholar]
- [45].Fan YY, Hu WW, Dai HB, Zhang JX, Zhang LY, He P, Shen Y, Ohtsu H, Wei EQ, Chen Z. Activation of the central histaminergic system is involved in hypoxia-induced stroke tolerance in adult mice. J Cereb Blood Flow Metab. 2011;31:305-14. doi: 10.1038/jcbfm.2010.94. PMID:20588322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fan YY, Shen Z, He P, Jiang L, Hou WW, Shen Y, Zhang XN, Hu WW, Chen Z. A novel neuroprotective strategy for ischemic stroke: transient mild acidosis treatment by CO2 inhalation at reperfusion. J Cereb Blood Flow Metab. 2014;34:275-83. doi: 10.1038/jcbfm.2013.193. PMID:24192637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055-62. doi: 10.1097/ALN.0b013e3181730257. PMID:18497606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.