

Abstract
Objective
Autoimmune encephalitis (AE) is an important and treatable cause of acute encephalitis. Diagnosis of AE in a developing child is challenging because of overlap in clinical presentations with other diseases and complexity of normal behavior changes. Existing diagnostic criteria for adult AE require modification to be applied to children, who differ from adults in their clinical presentations, paraclinical findings, autoantibody profiles, treatment response, and long-term outcomes.
Methods
A subcommittee of the Autoimmune Encephalitis International Working Group collaborated through conference calls and email correspondence to consider the pediatric-specific approach to AE. The subcommittee reviewed the literature of relevant AE studies and sought additional input from other expert clinicians and researchers.
Results
Existing consensus criteria for adult AE were refined for use in children. Provisional pediatric AE classification criteria and an algorithm to facilitate early diagnosis are proposed. There is also discussion about how to distinguish pediatric AE from conditions within the differential diagnosis.
Conclusions
Diagnosing AE is based on the combination of a clinical history consistent with pediatric AE and supportive diagnostic testing, which includes but is not dependent on antibody testing. The proposed criteria and algorithm require validation in prospective pediatric cohorts.
Autoimmune encephalitis (AE) refers to an increasingly recognized group of inflammatory brain diseases. Children with AE present with acute or subacute onset of neuropsychiatric symptoms due to an underlying abnormal immune response to the CNS.1,2 Many AE associate with antibodies directed toward extracellular antigens, such as synaptic receptors and ion channels.2,3 Autoantibodies that bind to extracellular antigens are generally pathogenic, whereas antibodies that bind intracellular antigens are not considered pathogenic, instead general markers of autoimmunity.
A number of different antibodies have been described in children with AE.4–21 Currently, the most common autoantibodies in children _target the N-methyl-D-aspartate receptor (NMDAR), myelin oligodendrocyte glycoprotein (MOG), and glutamic acid decarboxylase 65 (GAD65).5–12 It is also recognized that not all children with a clinical phenotype of AE have a known autoantibody.1,4
Diagnosing AE is challenging because of overlap in clinical presentations between the types of AE, other inflammatory brain diseases, infections, metabolic diseases, and psychiatric disorders.1 It is especially difficult in children because of the complexity of normal behavioral changes during childhood and the limited capacity of younger children to describe their symptoms.1 Compared to adults with AE, children may manifest important differences in symptoms, paraclinical findings, comorbidities, treatment response, and prognosis.4–7,22–24 There is an urgent need to recognize pediatric AE because treatment delays worsen prognosis and increase the risk of permanent neurocognitive deficits.6,25,26
In this article, we build on existing consensus criteria for adult AE by refining them for use in children.27 We propose provisional pediatric AE classification criteria and an algorithm to facilitate early diagnosis. Diagnosing AE is based on the combination of a clinical history consistent with the disease and supportive diagnostic testing, which includes but is not dependent on antibody testing. We also discuss the differential diagnosis in children with suspected AE.
Methodology
At the 2014 Autoimmune Encephalitis Alliance (AEalliance.org) conference in North Carolina, the Autoimmune Encephalitis International Working Group was formed and initiated discussions around developing diagnostic criteria for AE. A subcommittee of pediatric neurologists and rheumatologists identified that adult-focused criteria may not apply well to children. As a result, this subcommittee collaborated through conference calls and email correspondence to consider the pediatric-specific approach to AE. The subcommittee reviewed the literature on relevant AE studies and sought additional input from other experts. The first author (T.C.) developed a draft based on the preceding discussions that was subsequently reviewed and modified by all authors.
Existing diagnostic criteria for AE
The International Encephalitis Consortium 2013 diagnostic criteria for encephalitis of presumed infectious or autoimmune etiology require patients to have altered mental status lasting more than 24 hours with no alternative cause identified.28 Confirmation of this diagnosis requires at least 3 minor criteria, including fever within 72 hours of presentation; new onset focal neurologic findings; CSF leukocytosis; acute new neuroimaging abnormality suggestive of encephalitis; or EEG abnormalities consistent with encephalitis.28 These criteria do not differentiate autoimmune from infectious encephalitis.
More recently, an international group developed diagnostic criteria for early diagnosis of AE in adults, which require (1) subacute onset over less than 3 months of working memory deficits, altered mental status, or psychiatric symptoms; (2) at least one of the following: new focal CNS findings, seizures not explained by a preexisting disorder, CSF pleocytosis, and/or MRI features suggestive of encephalitis; and (3) reasonable exclusion of alternative causes.27 Specific neurologic syndromes were given criteria, including limbic encephalitis, anti-NMDAR encephalitis, and autoantibody-negative AE.27
These AE criteria required modification to be applied to children. For example, deficits in working memory are challenging to identify in younger children. Also, children are less likely to present with a well-defined neurologic syndrome and, even in anti-NMDAR encephalitis, the sequence of symptom development may differ from adults.5–7 Furthermore, the differential diagnosis for a child presenting with temporal lobe seizures and cognitive slowing is broad, whereas this presentation in adults suggests limbic encephalitis or acquired temporal pathology.
Clinical features distinguishing adults and children with AE
Typically, children with AE are previously healthy and present with rapid onset of neuropsychiatric symptoms. Prodromal symptoms including fever occur in over 50% of patients.2,4–6 Between disease onset and initiation of therapy, symptoms typically persist over time. This distinguishes AE from pediatric acute-onset neuropsychiatric syndrome (PANS), where patients often experience a relapsing-remitting course with rapid progression to maximum symptom severity and rapid return to previous function over hours or days, sometimes without therapy.
Neurologic manifestations of AE include altered level of consciousness, confusion, disturbed sleep, movement disorders and seizures. Seizures are the most common feature in AE and may be the predominant manifestation.4–7,10–21 Seizures may be focal or generalized and are often multifocal.4–7,10–21 Over one third of patients with AE have abnormal movements, such as ataxia, chorea, dystonia, myoclonus, or tremor.4–7,13,15 Both seizures and movement disorders can be highly refractory to standard treatments in children with AE.10,14,16,24 Some degree of cognitive impairment is seen in the overwhelming majority of AE patients and is considered a cardinal symptom.4,5,13,14,16,19,21 As such, a diagnosis of AE would be highly questionable in patients with documented normal cognition, again differentiating AE from PANS where cognition is often preserved. Assessing memory deficits in young children may be challenging; however, developmental regression, language loss or speech impairments may be presenting features of pediatric AE.5–7,29
Behavioral changes, such as repetitive or stereotypical behaviors, irritability, hyperactivity, hypersexuality, insomnia and anger outbursts, are common in pediatric AE.4–7 Psychiatric symptoms may range from mood swings and mild personality changes to fulminant psychosis and occur in over 50% of AE patients.4–7 New-onset psychosis in children younger than 13 years is uncommon and considered a red flag for an underlying medical, rather than primary psychiatric, condition. It is critical to assess for cognitive changes, seizures, movement abnormalities, or other neurologic symptoms in children with acute psychiatric symptoms, as these symptoms are suggestive of AE.
Children with AE likely differ from adults in their clinical presentations due to evolution of neuronal circuits, neuroreceptor densities and myelination during normal development. Children with AE are more likely to present with multifocal neuropsychiatric symptoms, rather than isolated clinical syndromes. For example, children with GAD65 antibodies may not present with the classic stiff-person syndrome or cerebellar degeneration seen in adults.11,12,22 Children with anti–NMDAR-associated encephalitis are more likely to present with movement abnormalities, agitation, insomnia, seizures, speech deficits, ataxia, and/or hemiparesis, whereas memory deficits, psychiatric manifestations, and central hypoventilation are more common in adults with the same antibody.5–7 Pediatric AE is less associated with tumors compared with adults.4–7
Diagnostic evaluation of children and teenagers with suspected AE
Although no single investigation is diagnostic of pediatric AE, the presence of a suggestive clinical phenotype and supportive paraclinical testing is essential to diagnose an underlying inflammatory process and to exclude alternative diagnoses. Initial investigations to be considered for any child with suspected AE are listed in table 1, although diagnostic workup should be tailored to the individual.
Table 1.
Recommended investigations for children with suspected AE

Blood tests are helpful to assess for systemic inflammatory changes, autoantibodies associated with systemic autoimmune diseases, vitamin B12 deficiency, markers of infection, elevated lactate due to metabolic conditions, and recreational drug use. Erythrocyte sedimentation rate, C-reactive protein, leukocyte counts, and platelet counts may be normal in children with AE.1,4–21
CSF pleocytosis and/or elevated protein levels may be seen at diagnosis or during disease course, but are not uniformly present.1,4–21 Recommended tests to assess for infectious encephalitis were based on population-based studies in California and England (table 1).30,31 However, workup for infectious etiologies varies depending on the season and region where the patient lives or has traveled. A recent report suggests that anti-NMDAR encephalitis may be more common in children than any specific infectious encephalitis, further highlighting the importance of considering AE when evaluating for infectious encephalitis.32 CSF neopterin is a useful but not rapidly accessible biomarker that is frequently elevated in anti-NMDAR encephalitis and other encephalitides, but normal in PANS.33 There is evidence that AE (particularly anti-NMDAR encephalitis) may be triggered by herpes simplex virus encephalitis and Japanese encephalitis.34
All patients should have a brain MRI with and without gadolinium. Over half of patients with AE will have a normal brain and spine MRI at diagnosis.4–7,16,21,22 Inflammatory lesions (high signal on T2 and fluid-attenuated inversion recovery sequences) may develop over time, and cerebral atrophy may occur months later.4,6,7,15 MRI lesions are most likely to be present in those with antibodies to MOG or the gamma-aminobutyric acid-A receptor (GABAAR).9,14,15 Neuroimaging findings are not limited to the temporal lobe or cortex.1,5–21 A normal MRI lessens suspicion for CNS vasculitis, demyelinating diseases, infections, and malignancies.1 In contrast, restriction on diffusion-weighted imaging reduces the likelihood of pediatric AE and should prompt consideration of other etiologies, such as infection-associated encephalopathies and vasculitis.1 Small retrospective adult AE studies have proposed that functional PET and SPECT studies may demonstrate brain dysfunction, but experience is limited in pediatric AE.35,36
A normal EEG is unusual in children with AE during active disease, although prolonged EEG may be needed for improved sensitivity. Therefore, focal or generalized seizures, epileptiform discharges, and encephalopathic changes, such as diffuse or focal slowing, may help to distinguish AE from primary psychiatric disorders or PANS. Adults with AE are more likely to have EEG changes predominantly involving the temporal lobes, whereas EEG findings in children may be more generalized.4–7,14–21 Specific EEG features, such as the “delta brush” pattern and extreme spindles, have been linked to anti-NMDAR encephalitis, but sensitivity is low.6,22,23
Neurocognitive testing may identify deficits in memory, attention, problem solving, language, and processing speed, particularly in younger children. A change in neurocognitive function supports a diagnosis of pediatric AE and may differentiate these patients from those with primary psychiatric disorders. However, interpretation of neurocognitive testing at diagnosis should be undertaken with caution, as there is often no premorbid testing for comparison.
Other diagnostic tests may be considered. Most children with AE do not require brain biopsy. However, a _targeted brain biopsy of MRI abnormalities may be needed when the diagnosis remains uncertain after initial workup. The diagnostic yield of brain biopsy is higher in pediatric patients than in adults.37
Antibody testing and interpretation in children and teenagers with suspected AE
Antibodies associated with pediatric AE are listed in tables 2 and 3. Each antibody is associated with characteristic symptoms, seizure types, and other clinical findings. However, there is significant overlap between the different disorders and so testing a panel of neural autoantibodies is recommended for any child with suspected AE. The most common autoantibodies identified in children _target NMDAR, MOG, GAD65, and GABAAR. Given the rarity of other autoantibodies, further testing should be considered only if antibodies to these _targets are negative and suspicion of AE persists (table 3).
Table 2.
Antibodies that are commonly identified in pediatric AE

Table 3.
Antibodies that are identified less frequently in pediatric autoimmune encephalitis

Antibody testing should be performed in both CSF and serum to avoid false-negative and false-positive results. For example, testing for NMDAR antibodies typically has higher sensitivity in CSF compared with serum, with up to 15% of patients having negative serum results.5–7 In contrast, MOG autoantibodies have higher sensitivity in serum.9
Interpretation of antibody test results should carefully consider the child's clinical presentation, especially when more than 1 antibody is identified. For example, GAD65 antibodies tend to be associated with personal or familial autoimmunity and low titers, such as those seen in type 1 diabetes mellitus, are not neurologically relevant.22 The presence of more than 1 antibody in some patients with AE has been recognized and may be associated with overlapping syndromes. Antibody specificity is also important when interpreting antibody test results. For instance, only IgG isotype antibodies to the GluN1 subunit of the NMDAR on a cell-based assay are specifically associated with AE.5,38
In adults with AE, most antibodies to the voltage-gated potassium channel complex (VGKCC) do not bind to the channel, but to proteins in the complex, particularly leucine-rich glioma-inactivated protein 1 (LGI1) and contactin-associated protein-like 2 (Caspr2).39 In children, VGKCC antibodies rarely _target LGI1 or Caspr2.40,41 It has been argued that VGKCC antibodies without specific binding to LGl1 or Caspr2 have limited clinical significance.40
Proposed classification criteria and algorithm for diagnosis of pediatric AE
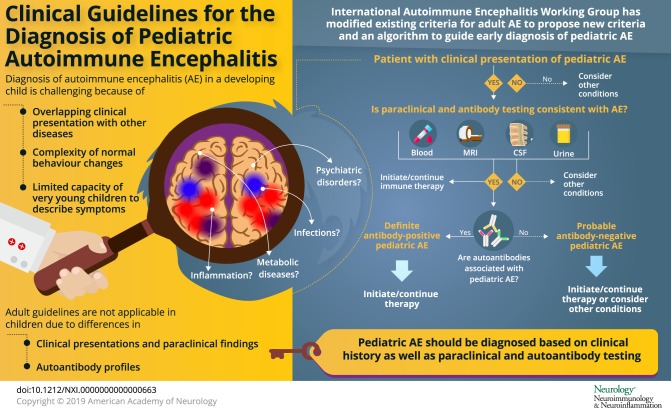
We modified the criteria for adult AE and propose provisional classification criteria for possible pediatric AE, probable antibody-negative pediatric AE, and definite antibody-positive pediatric AE in table 4.27 A diagnostic algorithm is also provided in figure. The provisional criteria and algorithm should be assessed prospectively in future cohorts.
Table 4.
Proposed classification criteria for possible, definite antibody-positive and probable antibody-negative pediatric AE

Figure. Algorithm for diagnostic workup of children with suspected AE using provisional criteria.

AE = autoimmune encephalitis.
A diagnosis of pediatric AE should be considered in previously healthy children who present with acute or subacute (less than 3 months) onset of new focal or diffuse neurologic deficits, cognitive difficulties, developmental regression, movement abnormalities, psychiatric symptoms, and/or seizures. Although children with preexisting developmental delay or chronic behavior/psychiatric abnormalities may develop AE, alternative diagnoses, such as genetic, metabolic, or neurodegenerative etiologies, should be considered in these patients.
Children with a clinical presentation suggestive of AE should have serum and CSF examined for neuronal antibodies, undergo paraclinical testing for neuroinflammation, and have disease mimics excluded (tables 1 and 4). EEG is not included as paraclinical evidence of neuroinflammation because EEG cannot differentiate AE from other encephalopathies. However, EEG encephalopathic features are allowable as an alternative for clinical features of encephalopathy. If a patient fulfills criteria for possible pediatric AE (table 4) and is functionally impaired, therapy may be started while awaiting the results of antibody and other testing, given the importance of early treatment to improve outcomes.4,25,26 If a patient with possible AE subsequently does not have positive antibodies or paraclinical testing for neuroinflammation, a diagnosis of AE is not supported. For these children, careful further consideration of the differential diagnosis is warranted, and additional immune therapy should only be undertaken with caution (table 5, figure).
Table 5.
Differential diagnosis of AE in children and adolescents

Children may have AE caused by antibodies that have not yet been identified and may meet criteria for probable antibody-negative pediatric AE (table 4). These patients will have 1 or more positive paraclinical tests for neuroinflammation, but negative antibody testing. Children who meet the criteria for definite antibody-positive pediatric AE will have positive antibody testing. If CSF antibodies are present (e.g., NMDAR and GAD65), no other paraclinical evidence of neuroinflammation is required for a diagnosis of definite AE (table 4). If only serum antibodies are present, 1 or more paraclinical tests of neuroinflammation must be abnormal. There should be caution in diagnosing AE when only serum antibodies (particularly NMDAR, GABAAR, and glycine receptor) are found in the absence of paraclinical evidence of neuroinflammation.
The proposed pediatric AE criteria differ from the adult criteria in several ways (table 4, table e-5, links.lww.com/nxi/A184).27 First, the pediatric criteria include both acute and subacute time frames for symptom onset, reflecting the range in disease course observed in children. Adult AE criteria were developed for several well-defined syndromes (i.e., limbic encephalitis, acute disseminated encephalomyelitis [ADEM], and anti-NMDAR encephalitis) and the associated algorithm focuses on whether patients meet criteria for these syndromes.27 In contrast, many pediatric patients with AE do not present with a well-defined syndrome and so the pediatric criteria were devised to capture the breadth of clinical and paraclinical findings reported in children. Similarly, the pediatric AE algorithm (figure) does not focus on syndrome identification, but is intended to guide a clinician in assessing clinical features and in paraclinical and antibody testing, so as to determine whether an AE diagnosis is appropriate. The adult AE criteria group clinical and paraclinical markers together, whereas the pediatric criteria distinguish clinical evidence of neurologic dysfunction from paraclinical evidence of neuroinflammation.
Patients with definite AE may benefit from continued or advanced immunosuppressive therapy, although specific protocols are not yet validated. Identification of an antibody associated with AE may facilitate counseling regarding expected course and outcomes. Timing of clinical responses to immunotherapy in children with AE may vary from immediate to months after starting.5–7,24,42 Therefore, using response to therapy as confirmatory support for a diagnosis of AE may be misleading.
Approach to clinically recognizable syndromes
Anti-NMDAR encephalitis
Anti-NMDAR encephalitis is the most common pediatric AE. The current adult diagnostic criteria for anti–NMDAR-associated encephalitis have been tested and apply well in children.43 However, children are more likely to present with neurologic symptoms, instead of psychiatric symptoms, and may not present with the classic sequence of symptoms described in adults—for example, movement disorders and autonomic dysfunction occur earlier in children.5–7
AE associated with antibodies to MOG, including acute disseminated encephalomyelitis
The most common autoantibody associated with autoimmune demyelination _targets MOG.8,9,42 Patients who have ADEM associated with MOG autoantibodies are more likely to exhibit large globular lesions and long segment myelitis compared with those without these antibodies.44 Children with MOG antibodies are also less likely to have oligoclonal bands than those with MS.42,44 However, the spectrum of brain disease associated with MOG antibodies in adults and children has broadened to include ADEM, meningoencephalitis, cortical encephalitis with seizures, brainstem encephalitis, and mimics of vasculitis.45–47 Some of these patients will evolve into more typical demyelinating phenotypes, such as ADEM; therefore, MOG antibodies should be considered in pediatric AE presentations beyond ADEM.45–47 MOG autoantibodies are typically transient in monophasic ADEM, but remain positive in relapsing phenotypes.8,9,42
Limbic encephalitis
The clinical, EEG, and radiologic features of limbic encephalitis are uncommon in children.48 Autoantibodies associated with adult limbic encephalitis include those that _target LGI1, GAD65, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, gamma-aminobutyric acid-B receptor, Caspr2, Hu, and Ma2.3 With the exception of GAD65, these specific antibodies are rare in children.48
Hashimoto encephalopathy
Hashimoto encephalopathy (HE) presents with nonspecific neuropsychiatric symptoms accompanied by antithyroid antibodies, which are considered markers of autoimmunity, rather than pathogenic. Patients may develop seizures, altered mental status, cognitive decline, psychosis, paranoia, focal neurologic defects, and movement disorders.49,50 Over 70% of children with HE have a normal brain MRI, CSF rarely shows pleocytosis, and EEG often shows generalized or focal slowing without seizures.49,50 Most children have normal thyroid function despite having antithyroid antibodies.49,50 Thoughtful interpretation is required because serum thyroid autoantibodies have been identified in healthy children.49,50
Approach to probable antibody-negative pediatric AE
Children with a clinical phenotype of AE and paraclinical findings of neuroinflammation, but negative testing for neural antibodies, may meet criteria for probable antibody-negative pediatric AE (table 4). It is well recognized that not all neural autoantibodies have been identified. Having CSF and serum testing in a research laboratory may identify patients who have antibodies against neural cell surface antigens of yet unknown identity and who may respond to immunotherapy.
Probable antibody-negative AE is one of the most challenging clinical scenarios. It is appropriate that a child presenting with new onset encephalopathy, neuropsychiatric features, and changes in function be investigated for possible AE. However, the differential diagnosis in children is arguably broader than in adults, and so it is important to ensure that other diagnoses have been excluded before giving an AE diagnosis. Pathologic entities that often cause diagnostic difficulty are cortical dysplasias and genetic epilepsies presenting with fever-provoked symptomatic focal seizures, infection-provoked encephalopathy and PANS. In these syndromes, CSF pleocytosis or oligoclonal bands are usually absent, and MRI is either normal or demonstrates alternative pathology. Therefore, critical examination of paraclinical tests for evidence of CNS inflammation is mandatory to avoid unnecessary immune suppression. A diagnosis of probable antibody-negative pediatric AE should also be reassessed in children with atypical features.
Differential diagnosis of AE
The spectrum of inflammatory brain diseases in children has rapidly expanded as new diseases and new etiologies for existing conditions have been described. The underlying pathogenic mechanisms that lead to CNS inflammation may involve vessel wall inflammation, demyelination, or an immune response directed against neurons and supporting structures.1,3 Inflammation may also occur secondary to infection, malignancy, or a systemic inflammatory disease. Diagnosing pediatric AE is especially challenging because of the clinical overlap between conditions in the differential diagnosis (table 5) and the clinical heterogeneity within patients having the same disease.
Specific conditions within the differential diagnosis of AE
Comprehensive evaluation is required to distinguish children with AE from those who have other inflammatory brain diseases. For example, children with large-vessel CNS vasculitis typically demonstrate a stroke phenotype, including paresis and speech deficits, and may be distinguished by the presence of ischemic changes on MRI and angiographic abnormalities, such as aneurysm and beading.51 In contrast, children with small-vessel CNS vasculitis present with cognitive dysfunction, seizures, vision abnormalities, and bilateral nonischemic lesions on MRI and have inflammatory vessel wall changes identified on brain biopsy.51
Infection-associated encephalopathy disorders include febrile infection-related epilepsy syndrome (FIRES), acute necrotizing encephalopathy, mild encephalopathy with reversible splenium lesion, and acute encephalopathy with biphasic seizures and diffusion restriction.52 These syndromes have typical clinical and radiologic features, often with diffusion restriction on imaging, which may infer cytotoxicity and distinguish these patients from those with AE. For example, children with FIRES develop a nonspecific febrile illness followed by sustained refractory status and then progress to chronic, drug-resistant epilepsy with neuropsychological impairment.52 Neuroimaging and brain biopsy in FIRES are usually normal.52 The pathogenesis of these diseases is unresolved, but may include genetic vulnerability leading to an infection-triggered “cytokine storm.”52
Other diagnoses within the differential are PANS and pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS). These conditions describe an idiopathic or postinfectious onset of obsessive-compulsive disorder, eating restriction, other emotional syndromes, tics, loss of skills, or personality change.53 Both clinical phenotypes lack robust biomarkers, and pathogenesis remains disputed; however, there is some evidence of immune mediation and immunotherapy responsiveness.53,54 Although patients may appear to have an acquired brain syndrome, most children with PANDAS or PANS would not fulfill the proposed pediatric AE criteria.
Also, monogenic autoinflammatory syndromes may involve the brain, such as the genetic interferonopathies, vasculopathies, and hemophagocytic lymphohistiocytosis.55 These disorders typically present in early childhood, result in chronic progressive disease, often involving increasing spasticity, intracranial calcifications and microcephaly, and are associated with persistent CSF immune activation.55 These syndromes are distinguished from AE by the presence of non-neurologic features, such as skin lesions, cytopenias, hepatosplenomegaly, and lung disease.55
Finally, neuropsychiatric symptoms are common in pediatric AE and are also the hallmark of primary psychiatric disorders. Delusions, hallucinations, reduced speech, sleep disturbance, and cognitive difficulties may be seen in both disease groups. Features that distinguish patients with AE from those with psychiatric disease include autonomic instability, hyperkinesia, dyskinesia, rapid progression of psychosis despite therapy, seizures, slowing or epileptic activity on EEG, CSF pleocytosis, CSF oligoclonal bands, and MRI abnormalities.56
Discussion
Proposed pediatric AE criteria are intended to address differences in clinical presentations, paraclinical findings, and autoantibody profiles between children and adults. The accompanying algorithm aims to guide diagnostic workup and facilitate earlier initiation of therapy.
Glossary
- AE
autoimmune encephalitis
- Caspr2
contactin-associated protein-like 2
- FIRES
febrile infection-related epilepsy syndrome
- GABAAR
gamma-aminobutyric acid A receptor
- GAD65
glutamic acid decarboxylase 65
- HE
Hashimoto encephalopathy
- LGI1
leucine-rich glioma-inactivated protein 1
- MOG
myelin oligodendrocyte glycoprotein
- NMDAR
N-methyl-D-aspartate receptor
- PANDAS
pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections
- PANS
pediatric acute-onset neuropsychiatric syndrome
- VGKCC
voltage-gated potassium channel complex
Appendix. Authors
Study funding
There was no external funding for this manuscript.
Disclosure
The authors have no conflicts of interest relevant to this article to disclose, with the exception of Dr. Dalmau being the editor of Neurology: Neuroimmunology & Neuroinflammation. Go to Neurology.org/NN for full disclosures.
References
- 1.Van Mater H. Pediatric inflammatory brain disease: a diagnostic approach. Curr Opin Rheumatol 2014;26:553–561. [DOI] [PubMed] [Google Scholar]
- 2.Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018;378:840–851. [DOI] [PubMed] [Google Scholar]
- 3.Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell-surface proteins in autoimmune diseases of the central nervous system. Physiol Rev 2017;97:839–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hacohen Y, Wright S, Waters P, et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry 2013;84:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armangue T, Titulaer MJ, Malaga I, et al. Pediatric anti-N-methyl-D-aspartate receptor encephalitis – clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Florance NR, Davis RL, Lam C, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 2009;66:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pröbstel AK, Dornmair K, Bittner R, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 2011;77:580–588. [DOI] [PubMed] [Google Scholar]
- 9.Brilot F, Dale RC, Selter RC, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol 2009;66:833–842. [DOI] [PubMed] [Google Scholar]
- 10.Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 2010;67:470–478. [DOI] [PubMed] [Google Scholar]
- 11.Mishra N, Rodan LH, Nita DA, et al. Anti-glutamic acid decarboxylase antibody associated limbic encephalitis in a child: expanding the spectrum of pediatric inflammatory brain diseases. J Child Neurol 2014;29:677–683. [DOI] [PubMed] [Google Scholar]
- 12.Gresa-Arribas N, Ariňos H, Martinez-Hernández E, et al. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One 2015;10:e0121364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dale RC, Merheb V, Pillai S, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain 2012;135:3453–3468. [DOI] [PubMed] [Google Scholar]
- 14.Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterization of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spatola M, Petit-Pedrol M, Simabakuro MM, et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology 2017;88:1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancaster E, Lai M, Peng X, et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: case series and characterization of the antigen. Lancet Neurol 2010;9:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013;81:1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damásio J, Leite MI, Coutinho E, et al. Progressive encephalomyelitis with rigidity and myoclonus: the first pediatric case with glycine receptor antibodies. JAMA Neurol 2013;70:498–501. [DOI] [PubMed] [Google Scholar]
- 19.Carvajal-González A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 2014;137:2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lancaster E, Martinez-Hernandez E, Titulaer MJ, et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology 2011;77:1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spatola M, Sabater L, Planagumà J, et al. Encephalitis with mGluR5 antibodies: symptoms and antibody effects. Neurology 2018;90:e1964–e1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suleiman J, Dale RC. The recognition and treatment of autoimmune epilepsy in children. Dev Med Child Neurol 2015;57:431–440. [DOI] [PubMed] [Google Scholar]
- 23.Armangue T, Petit-Pedrol M, Dalmau J. Autoimmune encephalitis in children. J Child Neurol 2012;27:1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nosadini M, Mohammad SS, Ramanathan S, Brilot F, Dale RC. Immune therapy in autoimmune encephalitis: a systematic review. Expert Rev Neurother 2015;15:1391–1419. [DOI] [PubMed] [Google Scholar]
- 25.Breese EH, Dalmau J, Lennon VA, Apiwattanakul M, Sokol DK. Anti-N-methyl-D-aspartate receptor encephalitis: early treatment is beneficial. Pediatr Neurol 2010;42:213–214. [DOI] [PubMed] [Google Scholar]
- 26.Byrne S, Walsh C, Hacohen Y, et al. Earlier treatment of NMDAR antibody encephalitis in children results in a better outcome. Neurol Neuroimmunol Neuroinflamm 2015;2:e130 doi: 10.1212/NXI.0000000000000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatesan A, Tunkel AR, Bloch KC, et al. Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the International Encephalitis Consortium. Clin Infect Dis 2013;57:1114–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldberg EM, Titulaer M, de Blank PM, Sievert A, Ryan N. Anti-N-methyl-D-aspartate receptor-mediated encephalitis in infants and toddlers: case report and review of the literature. Pediatr Neurol 2014;50:181–184. [DOI] [PubMed] [Google Scholar]
- 30.Glaser CA, Gilliam S, Schnurr D, et al. In search of encephalitis etiologies: diagnostic challenges in the California Encephalitis Project, 1998-2000. Clin Infect Dis 2003;36:731–742. [DOI] [PubMed] [Google Scholar]
- 31.Granerod J, Ambrose HE, Davies NW, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10:835–844. [DOI] [PubMed] [Google Scholar]
- 32.Gable MS, Sheriff H, Dalmau J, et al. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis 2012; 54:899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kothur K, Wienholt L, Mohammad SS, et al. Utility of CSF cytokine/chemokines as markers of active intrathecal inflammation: comparison of demyelinating, anti-NMDAR and enterviral encephalitis. PLoS One 2016; 11:e0161656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armangue T, Spatola M, Vlagea A, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol 2018;17:760–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Probasco JC, Solnes L, Nalluri A, et al. Abnormal brain metabolism on FDG-PET/CT is a common early finding in autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm 2017;4:e352 doi: 10.1212/NXI.0000000000000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solnes LB, Jones KM, Rowe SP, et al. Diagnostic value of 18F-FDG PET/CT versus MRI in the setting of antibody-specific autoimmune encephalitis. J Nucl Med 2017;58:1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkateswaran S, Hawkins C, Wassmer E. Diagnostic yield of brain biopsies in children presenting to neurology. J Child Neurol 2008;23:253–258. [DOI] [PubMed] [Google Scholar]
- 38.Hara M, Martinez-Hernandez E, Ariño H, et al. Clinical and pathogenic significance of IgG, IgA, and IgM antibodies against the NMDA receptor. Neurology 2018;90:e1386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired myotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hacohen Y, Singh R, Rossi M, et al. Clinical relevance of voltage-gated potassium channel–complex antibodies in children. Neurology 2015;5:967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López-Chiriboga AS, Klein C, Zekeridou A, et al. LGl1 and CASPR2 neurological autoimmunity in children. Ann Neurol 2018;84:473–480. [DOI] [PubMed] [Google Scholar]
- 42.Hacohen Y, Wong YY, Lechner C, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated diseases. JAMA Neurol 2018;75:478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho ACC, Mohammad SS, Pillai SC, et al. High sensitivity and specificity in proposed clinical diagnostic criteria for anti-N-methyl-D-aspartate receptor encephalitis. Dev Med Child Neurol 2017;59:1256–1260. [DOI] [PubMed] [Google Scholar]
- 44.Baumann M, Sahin K, Lechner C, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry 2015;86:265–272. [DOI] [PubMed] [Google Scholar]
- 45.Matesanz S, Kotch C, Perrone C, et al. Expanding the MOG phenotype: brainstem encephalitis with punctate and curvilinear enhancement. Neurol Neuroimmunol Neuroinflamm 2019;6:e619 doi: 10.1212/NXI.0000000000000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Budhram A, Mirian A, Le C, et al. Unilateral cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol 2019;266:2481–2487. [DOI] [PubMed] [Google Scholar]
- 47.Patterson K, Iglesias E, Nasrallah M, et al. Anti-MOG encephalitis mimicking small vessel CNS vasculitis. Neurol Neuroimmunol Neuroinflamm 2019;6:e538 doi: 10.1212/NXI.0000000000000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haberlandt E, Bast T, Ebner A, et al. Limbic encephalitis in children and adolescents. Arch Dis Child 2011;96:186–191. [DOI] [PubMed] [Google Scholar]
- 49.Mamoudjy N, Korff C, Maurey H, et al. Hashimoto's encephalopathy: identification and long-term outcome in children. Eur J Paediatr Neurol 2013;17:280–287. [DOI] [PubMed] [Google Scholar]
- 50.Laurent C, Capron J, Quillerou B, et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT): characteristics, treatment and outcome in 251 cases from the literature. Autoimmun Rev 2016;15:1129–1133. [DOI] [PubMed] [Google Scholar]
- 51.Cellucci T, Tyrrell PN, Twilt M, Sheikh S, Benseler SM. Distinct phenotype clusters in childhood inflammatory brain diseases: implications for diagnostic evaluation. Arthritis Rheumatol 2014;66:750–756. [DOI] [PubMed] [Google Scholar]
- 52.Saitoh M, Kobayashi K, Ohmori I, et al. Cytokine-related and sodium channel polymorphism as candidate predisposing factors for childhood encephalopathy FIRES/AERRPS. J Neurol Sci 2016;368:272–276. [DOI] [PubMed] [Google Scholar]
- 53.Chang K, Frankovich J, Cooperstock M, et al. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol 2015;25:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swedo SE, Frankovich J, Murphy TK. Overview of treatment of pediatric acute-onset neuropsychiatric syndrome. J Child Adolesc Psychopharmacol 2017;27:562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crow YJ, Chase DS, Lowenstein Schmidt J, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 2015;167A:296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herken J, Prüss H. Red flags: clinical signs for identifying autoimmune encephalitis in psychiatric patients. Front Psychiatry 2017;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]