

Abstract
The putative glycine dehydrogenase of Mycobacterium tuberculosis catalyzes the reductive amination of glyoxylate to glycine but not the reverse reaction. The enzyme was purified and identified as the previously characterized alanine dehydrogenase. The Ald enzyme was expressed in Escherichia coli and had both pyruvate and glyoxylate aminating activities. The gene, ald, was inactivated in M. tuberculosis, which resulted in the loss of all activities. Both enzyme activities were found associated with the cell and were not detected in the extracellular filtrate. By using an anti-Ald antibody, the protein was localized to the cell membrane, with a smaller fraction in the cytosol. None was detected in the extracellular medium. The ald knockout strain grew without alanine or glycine and was able to utilize glycine but not alanine as a nitrogen source. Transcription of ald was induced when alanine was the sole nitrogen source, and higher levels of Ald enzyme were measured. Ald is proposed to have several functions, including ammonium incorporation and alanine breakdown.
INTRODUCTION
Mycobacterium tuberculosis is the causative agent of tuberculosis, and one of the most successful human pathogens. It was responsible for approximately 2 million deaths in 2008, while currently almost one-third of the world's population is infected with this organism. Research with M. tuberculosis has described a pathogen uniquely adapted to the wide range of harsh environments presented by the host. Much of this work has focused on the microbe's metabolism, with the idea of identification of novel enzymes or pathways to _target for drug development.
One of these environmental factors is nitrogen availability. Very little is known about the nitrogen sources used by M. tuberculosis in vivo. M. tuberculosis can utilize many amino acids for nitrogen, including alanine and glycine (29). Mutants of M. tuberculosis unable to synthesize proline retained the ability to replicate in the human macrophage cell line THP-1 (35), while other amino acid auxotroph mutants were attenuated (3, 17, 22, 52). A Mycobacterium bovis BCG mutant unable to make methionine showed survival in mice similar to the wild-type strain (32). This suggests some amino acids are available in vivo and serve as nutrients for M. tuberculosis.
The enzyme glycine dehydrogenase was first described in M. tuberculosis in 1962 (16). This enzyme was detected by the reductive amination of glyoxylate to glycine concurrent with the oxidation of NADH to NAD+ (Fig. 1). This reaction represents glyoxylate reductive aminase (GxRA) activity. The activity corresponding to the reverse reaction, catalyzed by glycine dehydrogenase (GDH), was not detected. The expression of glyoxylate reductive amination by a putative glycine dehydrogenase in M. tuberculosis has been characterized in nonreplicating persistent (NRP) cultures (58). In the Wayne model of dormancy, sealed cultures of M. tuberculosis create a microaerobic environment (NRP-1), which subsequently develops into the anaerobic stage (NRP-2) (58). GxRA activity was induced during microaerobic NRP-1, with the strongest activity in anaerobic NRP-2 cultures. It was proposed that the role of this enzyme was to maintain redox balance by recycling NADH/NAD+ during interruption of aerobic respiration (59, 60).
Fig 1.

Possible reactions of alanine dehydrogenase. GDH activity was not detected.
The naming of the glycine dehydrogenase was based on the similarity of the glyoxylate reductive aminase reaction to that catalyzed by l-alanine dehydrogenase (Ald; l-alanine:NAD+ oxidoreductase; EC 1.4.1.1) (15). Alanine dehydrogenase catalyzed the reductive amination of pyruvate to l-alanine (PvRA), but the reverse reaction, the oxidative dehydrogenation of l-alanine (ALD), was also detected. Alanine dehydrogenases are well-studied enzymes found in a wide range of bacterial species. In mycobacteria, it was first identified as an enzyme absent from the vaccine strains of M. bovis BCG but present in virulent M. tuberculosis (2). It was suggested that impairment of M. bovis BCG replication in humans due to the lack of a functional alanine dehydrogenase inhibited the development of protective immunity (44).
There have been several attempts to identify the gene encoding the putative glycine dehydrogenase. gcvB (Rv1832), annotated in the M. tuberculosis genome as a glycine dehydrogenase gene, most likely encodes the P protein of the glycine cleavage system (60). In Mycobacterium smegmatis pyruvate and glyoxylate aminase activities comigrated on a native polyacrylamide gel (53), suggesting one enzyme for both activities. However, a knockout mutant of the alanine dehydrogenase gene ald in M. smegmatis lost alanine dehydrogenase activity but retained glycine dehydrogenase activity (14). Moreover, M. bovis does not produce alanine dehydrogenase, but glycine dehydrogenase activity has been reported (4, 27). Thus, the identity of this unique enzyme is unknown.
In this study ald was shown to encode both alanine dehydrogenase and glyoxylate reductive aminase (glycine dehydrogenase) activities. This was determined by both biochemical and genetic methods. This dual function enzyme was localized to the cell membrane and cytosol. It plays an essential role in the utilization of alanine, but not glycine, as a nitrogen source.
MATERIALS AND METHODS
Strains and media.
M. tuberculosis H37Rv and Erdman strains were from the culture collection of this laboratory. Mycobacterial cultures were grown at 37°C in Dubos Tween-albumin broth (DTA; Difco, Detroit, MI). For studies with nitrogen or carbon sources, minimal lysis-inducing medium (LIM) was used with amino acids at 10 mM (45). Aerobic cultures were incubated on a model G24 rotary shaker-incubator (New Brunswick Scientific, Edison, NJ). For hypoxic cultures (Wayne model), slow magnetic stirring in sealed tubes with a headspace ratio of 0.5 were prepared as previously described (58).
Hygromycin was used at a concentration of 50 μg/ml, and gentamicin was used at 10 μg/ml. All antibiotics and chemicals were from Sigma (St. Louis, MO).
Purification of glycine dehydrogenase.
M. tuberculosis was grown in flasks of 400 ml of DTA in the Wayne model (58). Cells were harvested by centrifugation, washed twice with 100 mM phosphate buffer (pH 7.0), and disrupted by sonication (48). DEAE-Sephacel was used to remove excessive lipids. The gel was first equilibrated with 10 mM phosphate buffer (pH 7.0), then the cell extracts were loaded, and finally protein was eluted with 200 mM phosphate buffer (pH 7.0) (16). Glycine dehydrogenase was further purified at Athena Environmental Sciences, Inc. (Baltimore, MD) by using a two-step chromatographic fractionation procedure. In the first step, hydrophobic interaction chromatography was used. Cell extract was fractionated on a phenyl-Sepharose HiTrap column (Amersham Pharmacia Biotech, Piscataway, NJ) at pH 7.0. Next, anion-exchange chromatography was used. Samples with glyoxylate reductive aminase activity were fractionated on a 1-ml Mono Q HR5/5 column (Amersham Pharmacia Biotech). The fraction with the greatest activity was concentrated by ultrafiltration with a Microcon YM-3 column (Millipore, Billerica, MA) and washed three times with 10 mM ammonium acetate (pH 6.8). The sample was subjected to tryptic digestion, fractionated by microcapillary reverse-phase high-performance liquid chromatography, and then run through a nano-electrospray ionization source of an ion trap mass spectrometer. The tandem mass spectrometry data were correlated with known sequences by using the Sequest and HMF computer algorithms (13).
Preparation of cell extracts for enzyme studies.
M. tuberculosis was grown aerobically to either mid-log phase with an approximate optical density at 580 nm (OD580) of 0.4 or to NRP-2 after 250 h in the Wayne model. To harvest, cell suspensions were placed on ice for 2 h and then washed twice by centrifugation with cold 100 mM phosphate buffer (pH 7.0) containing 0.05% Tween 80. Cells were disrupted using a minibeadbeater (BioSpec Products, Bartlesville, OK) with 6 repetitions of 30-s bursts and with cooling on ice between each burst. Extracts were clarified by centrifugation and then sterilized by filtration through a 0.22-μm-pore-size SpinX filter (Corning, Corning, NY). The protein concentration was determined with the Bio-Rad (Hercules, CA) protein assay reagents.
Assay of enzyme activity.
The assay for GxRA activity for the identification of the putative glycine dehydrogenase was based on the original protocol for measuring the rate of oxidation of NADH during the reductive amination of glyoxylate (16). Approximately 50 μg of cell extract protein or 1 μg of His-tagged Ald was added to a 1-ml UV-transparent cuvette containing 100 mM phosphate buffer. Ammonium sulfate to 400 mM and sodium glyoxylate (pH 6.4) to 50 mM were added. The reactions were started with the addition of NADH to 80 μM and measured by the rate of decrease of the A340 on a Libra S32PC spectrophotometer (Biochrome, Cambridge, United Kingdom). A control reaction without ammonium sulfate was included to measure the background, which was subtracted from the experimental values. Glutamine, when employed, was used at 40 mM.
Reductive amination of pyruvate to alanine (PvRA) was measured in an identical manner to that of GxRA activity, except that sodium pyruvate was used in place of glyoxylate. In some experiments pyruvate was replaced with α-ketoglutaric acid, lithium hydroxypyruvate, sodium glycolate, sodium acetate, sodium oxaloacetate, or methylglyoxal at a final concentration of 20 mM.
For the oxidative deamination reaction (ALD) in which alanine was converted to pyruvate, l-alanine at 100 mM and NAD+ at 1 mM were used (15). Glycine, in place of alanine, was used to detect GDH activity. In some experiments alanine was replaced with aspartate, glutamate, glycerate, proline, or serine at 40 mM.
To determine the pH profile, 4 different buffers were used with overlapping pH ranges: 100 mM phosphate buffer (pH 4.5 to 9.0), 100 mM citrate buffer (pH 3.0 to 6.0), 100 mM Tris-HCl buffer (pH 7.0 to 9.0), and 100 mM carbonate buffer (pH 8.0 to 11.0). Other components were at the same concentrations listed above. All assays were performed in triplicate at room temperature.
To determine the Km values, the reductive amination reaction or oxidative deamination reaction was performed in 100 mM phosphate buffer at pH 8.5. The concentrations of pyruvate, glyoxylate, ammonium, alanine, NADH, and NAD+ were varied at least 10-fold above and below the apparent Km. Other substrates were at the standard concentrations previously mentioned. Km and Vmax values were determined from a Lineweaver-Burk plot.
For all reactions, activities were determined from the linear sections of curves. An extinction coefficient of 6,300 M−1 cm−1 was used. Specific activity was expressed as μmoles of NADH oxidized per min per mg of cell extract protein. The pH was measured after each reaction was complete. All reactions were performed at room temperature.
Three-dimensional crystal structures were analyzed for substrate specificity by using the program COOT (12).
Cloning and expression of histidine-tagged alanine dehydrogenase.
The ald gene was amplified from M. tuberculosis H37Rv by PCR using the primers AscI and NotI (Table 1). The fragment produced was digested with NotI and AscI, ligated into pET DUET (New England BioLabs, Ipswich, MA), and electroporated into Escherichia coli ER2566. The resulting plasmid was named pAld-His1.
Table 1.
Oligonucleotide primers used in this study
| Name | Sequence (5′→3′)a | Purpose in this studyb |
|---|---|---|
| p58 | ACTCGAGTGGCGAACGGGTGAGTAACACGT | Q-PCR of 16S |
| p59 | AGGCCGTCACCCCACCAACAAGCTGATAGG | Q-PCR of 16S |
| p169 | GCCGCGTCACGTGCTTGCACCGATGCGTTG | Q-PCR of ald |
| p170 | AACCTGGGCGGCGAGTCGACCGGCGACTTC | Q-PCR of ald |
| p171 | CCATGCAGCAAGCTTAGGATTCTGCGGTCC | Cloning of ald |
| p172 | GCCACTACCGGTCCTATCGGAGCCAATCTG | Cloning of ald |
| p189 | AAGATCAGGGACGCCCGGGTACCTGCGACG | Cloning of ald |
| p190 | GAACAAACCTTTGCCCGGGATCGGCGCACC | Cloning of ald |
| AscI | GTTGTTGGCGCGCCTAATGCGCGTCGGTATTCCGACCGAG | His-Ald |
| NotI | AAGGAAAAAAGCGGCCGCTCAGGCCAGCACTCGGGCGGCTCGG | His-Ald |
Mismatches used to create restriction sites for ald cloning are indicated by underlining.
Q-PCR, quantitative reverse transcriptase PCR. His-Ald, PCR to create the His-tagged ald expression plasmid.
For expression of the histidine-tagged alanine dehydrogenase, 1 liter of Luria broth with ampicillin was inoculated with ER2566(pAld-His1) and grown with shaking at 37°C to an OD600 of between 0.5 and 0.8. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to 500 μM, and incubation was continued at room temperature for an additional 4 h. Cells were harvested by centrifugation and resuspended in 30 ml of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole) with 1 mg/ml of lysozyme. Cells were disrupted by sonication with 3 repeats for 15 s. The sonicate was cleared by centrifugation, and the His-tagged Ald purified was loaded onto a Qiagen 1.5-ml Ni-resin column (Qiagen, Valencia, CA). The column was washed three times with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole) and then eluted with 4 ml of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole) as per the manufacturer's instructions. The purified protein was dialyzed twice against 3 liters of phosphate-buffered saline and frozen at −80°C until analyzed (see Fig. S1 in the supplemental material).
Construction of the ald knockout.
The ald gene of M. tuberculosis H37Rv was amplified from genomic DNA with primers p171 and p172 (Table 1). The fragment was cloned with the TOPO Blunt cloning kit (Invitrogen, Carlsbad, CA). The ald gene was cloned into the suicide vector pJQ200KS (38), followed by the insertion of hyg, a hygromycin resistance marker, into the BamHI and XbaI sites in ald. A 53-bp fragment of ald DNA was deleted in the process. The plasmid was electroporated into M. tuberculosis followed by selection on DTA agar plates with 25 μg/ml hygromycin. Inserts of the plasmid into the chromosomal ald were identified by Southern analysis (Fig. 2). To create the double-crossover knockout mutant RVW7, clones were replated on DTA agar containing 2% sucrose and hygromycin. Southern analysis was used to verify the correct structure. The wild-type strain produced a fragment of 3.8 kb, while the knockout strain RVW7 produced a larger fragment of 4.8 kb.
Fig 2.

Southern blot analyses of the ald insertional mutant and complement. (A) DNA was isolated from each strain, cut with SacI, and analyzed with a probe specific for ald. RVW7, Δald::hyg; Ald21, RVW7(pAld-Gen21). The approximate sizes are indicated on the left. (B) Gene organization of ald, showing the insertion of hyg. Arrows indicate open readings frames and the direction of transcription. X, XbaI site; B, BamHI site; S, SacI site. ald′ is the disrupted ald. The ald probe binding site is shown.
To create the complementing plasmid, ald was amplified with primers p189 and p190 (Table 1) and cloned into the insertional plasmid pUC-GEN-INT (30). After the ald sequence was verified, the plasmid was electroporated into RVW7. Selection was for both gentamicin and hygromycin resistance. Southern analysis was used to verify the presence of the inactive ald gene (band size, 4.8 kb), as well as the insertion of the second complementing ald of pAld-Gen21 (band size, 8.8 kb) (Fig. 2).
Digoxigenin-labeled probes for Southern analysis were created by PCR (Roche Diagnostics, Indianapolis, IN) using p169 and p170 as primers.
Subcellular localization of Ald.
To determine the location of Ald, M. tuberculosis was grown in modified DTA without bovine serum albumin and with Tween 80 replaced by Tyloxapol. Aerobic cultures were grown to mid-log phase (OD580, ∼0.4), and NRP-2 cultures were grown for 300 h in the Wayne model. Cells were harvested by centrifugation, resuspended in 100 mM phosphate buffer (pH 7.2), and disrupted by bead beating. Spent medium was filter sterilized and concentrated over a Centriplus column (Millipore). Phenylmethanesulfonyl fluoride was added to all fractions at 44 μg/ml.
Eighty micrograms of protein from each sample was separated on a 10% Tris–HEPES IGels Long-Life gel (NuSep, Lane Cove, NSW, Australia). HBT-10, the mouse anti-Ald antibody, was the primary antibody (28). The secondary antibody for experiments with culture filtrate was goat anti-mouse IgG (Fab-specific)–alkaline phosphatase–conjugated antibody (Sigma, St. Louis, MO), and detection was with 5-bromo-4-chloro-3-indolylphosphate–nitroblue tetrazolium (Amresco, Solon, OH).
The cell extract was separated into different fractions by centrifugation (19, 33). Cell extracts were centrifuged at 27,000 × g at 4°C for 30 min to pellet the cell wall. The supernatant was centrifuged at 100,000 × g at 4°C for 2 h to pellet the cell membrane components. Both of these fractions were washed with phosphate-buffered saline. The final supernatant was taken as the soluble cytosol fraction. Ten micrograms of protein from each sample was separated by SDS-PAGE and probed with HBT-10. The secondary antibody was goat anti-mouse IgG (Fab-specific)–horseradish phosphatase–conjugated antibody (Sigma, St. Louis, MO). Detection was performed with the Pierce enhanced chemiluminescence Western blotting substrate (Thermo, Rockford, IL).
RNA isolation.
Cultures were quickly harvested by centrifugation, and the cell pellet was resuspended in 1 ml of RNAwiz (Ambion, Austin, TX) before being disrupted in a minibeadbeater by three 1-min pulses. Samples were centrifuged for 5 min at 10,000 × g, and the supernatants was extracted once with CHCl3 followed by precipitation of the RNA with isopropanol. DNA was removed by treatment with DNase (Roche, Indianapolis, IN) for 2 h at 37°C. RNA was purified with the RiboPure bacteria kit (Ambion) and treated for a second time with DNase, which was then inactivated at 70°C for 5 min.
Quantitation of mRNA levels.
cDNA was produced with the RETROscript kit (Ambion) according to the manufacturer's instructions. Approximately 150 ng of RNA was added to a mixture of antisense primers at1 μM. Duplicate samples without Moloney murine leukemia virus reverse transcriptase were prepared as negative controls.
Real-time quantitative PCR was performed with the Brilliant SYBR green QPCR master mix kit (Stratagene, La Jolla, CA). Reactions were performed in a volume of 20 μl containing two gene-specific primers (Table 1) at 0.1 μM, 25 μl of 2× master mix, and 5 μl of cDNA. A 10-fold dilution series of RNA was created, and the 16S RNA was measured to create a standard curve. 16S RNA is stable during long-term persistence in the Wayne model (11). Rv0239 was included as a second standard (21). Amplification was performed in the ICycler (Bio-Rad, Hercules, CA) with sampling during elongation. An initial denaturation of 10 min at 95°C was performed, followed by 30 cycles of 95°C for 30 s and 68°C for 1 min. All samples were run on a 2% agarose gel to verify that only a single band was produced. Each gene was analyzed from 3 independent RNA samples.
RESULTS
Isolation of the putative glycine dehydrogenase.
The putative glycine dehydrogenase was purified from M. tuberculosis H37Rv by two-step purification. First hydrophobic interaction and then anion-exchange chromatography were used. Seven proteins were isolated and identified, including some of unknown function (Table 2). One enzyme with a known function was l-alanine dehydrogenase (Rv2780), which catalyzes the reductive amination of pyruvate to l-alanine with the oxidation of NADH (Fig. 1). It also catalyzes the reverse reaction: the oxidative deaminase reaction of alanine to pyruvate with the reduction of NAD+ (15). The isolation of alanine dehydrogenase was not unexpected, as the procedure to isolate glycine dehydrogenase was based, in part, on the purification of NADH binding proteins.
Table 2.
Proteins identified in a purified GxRA sample from a hypoxic NRP culturea
| Gene | Protein name | Protein description |
|---|---|---|
| Rv0350 | DnaK | Heat shock 70-kDa protein |
| Rv2031c | HspX | α-Crystallin/URB-1 |
| Rv2032 | Acg | Unknown function |
| Rv2780 | Ald | l-Alanine dehydrogenase |
| Rv2626c | Hrp1 | Hypoxic response protein 1 |
| Rv1133c | MetE | Methyltetrahydropteroyltriglutamate-homocysteine methyltransferase |
| Rv1070c | EchA8 | Enoyl-CoA hydratase/isomerase family protein |
Proteins were identified by mass spectrometry after a two-step chromatographic purification for glycine dehydrogenase.
Inactivation of ald.
To determine if the Ald enzyme was also the putative glycine dehydrogenase, two approaches were used. First, the gene was inactivated in M. tuberculosis, and second, in order to biochemically reconstitute the enzyme, it was expressed and purified from E. coli. The gene encoding the alanine dehydrogenase, ald, is located between two genes transcribed in the opposite direction, suggesting it is a monocistronic gene. If ald encodes both the alanine and putative glycine dehydrogenases, then loss of this gene should result in the loss of these activities. To test this, a hygromycin marker was inserted into M. tuberculosis, inactivating ald and creating the strain RVW7 (Fig. 2).
Glycine dehydrogenase has been detected in hypoxic cultures, and alanine dehydrogenase is generally detected in aerobic cultures (2, 24, 58). Both PvRA and GxRA activities were measured in cell extracts from aerobically growing and from nonreplicating persistent cultures of the Wayne model (Fig. 3). Low PvRA and GxRA activities were detected in aerobic cultures. M. tuberculosis from anaerobic cultures showed a 2-fold induction of PvRA activity and 9-fold induction of GxRA activity (Fig. 3B). RVW7 showed neither type of activity under either condition.
Fig 3.

(A and B) NADH breakdown with glyoxylate in aerobic cultures (A) or nonreplicating persistent stage 2 cultures (B). Filled symbols and solid lines show data for complete reactions, while empty symbols and dotted lines are for reactions without ammonium sulfate. Circles, WT; triangles, RVW7; squares, RVW7(pAld-Gen21). The standard deviations are shown. (C and D) PvRA and GxRA activities of the Δald mutant in aerobic cultures (C) or nonreplicating persistent stage 2 cultures (D). PvRA is involved in the reductive amination of pyruvate to alanine. GxRA is involved in the reductive amination of glyoxylate to glycine. RVW7, Δald; Ald21, RVW7(pAld-Gen21). Specific activity is reported in μmoles of NADH oxidized/min/μg of protein. The standard deviations are shown.
To complement the knockout mutant, ald plus 527 bp of upstream DNA was cloned into the insertional vector pUC-GM-INT (30) to make pAld-Gen21. This plasmid was electroporated into RVW7, creating RVW7(pAld21) (Fig. 2). This strain was tested for GxRA and PvRA activities in both aerobic and NRP cultures (Fig. 3). Both were similar to the wild type (WT), suggesting that insertional inactivation of ald was responsible for the loss of PvRA and GxRA activities, which could be complemented with a functional copy of ald.
Enzymatic activity of Ald.
The alanine dehydrogenase of M. tuberculosis was expressed as a histidine-tagged protein and purified from E. coli. The enzymatic activity of the His-tagged protein was first verified by determining the pH dependence for the alanine dehydrogenase reactions (Fig. 4A). For the PvRA activity, the optimal pH was 8.5, similar to the previously reported ranges of 8.5 to 9 (15) and 7.0 to 7.5 (24). For the ALD activity, the optimal pH was 9.0, lower than the reported range of 9.5 to 11 (15, 24).
Fig 4.

Activity of His-tagged Ald at different pH levels. (A) Pyruvate reductive amination activity (circles) and alanine dehydrogenase activity (squares). (B) Glyoxylate reductive amination activity (circles), glycine dehydrogenase activity (squares), and glyoxylate reductive amination activity with glutamine in place of ammonium sulfate (triangles). Specific activity is shown in μmoles of NADH oxidized/min/μg of protein. The standard deviations are shown.
The His-tagged Ald was analyzed for the glyoxylate reductive aminating activity (GxRA) attributable to glycine dehydrogenase. This activity was detected, but GDH activity was not (Fig. 4B). Glutamate was tested for its ability to replace ammonium sulfate, but only weak activity was seen. Therefore, the alanine dehydrogenase enzyme of M. tuberculosis also has the glyoxylate aminase activity, which defines the putative glycine dehydrogenase (16). To compare the enzyme activities, GxRA activity at different pH values was measured. This activity was detected with an optimal pH of 8.5 (Fig. 4B), higher than the reported optimum pH of 6.2 (16). These results, taken together with those from the knockout mutant of M. tuberculosis, prove that ald encodes an enzyme having both PvRA and GxRA activities.
To further characterize this enzyme, the ability to utilize other substrates was tested. In a reductive amination reaction, there was no activity with 2-ketoglutarate, malate, acetate, or glycolate (Fig. 5A). Approximately 60% activity was seen with hydroxypyruvate, but only if ammonium sulfate was included in the reaction mixture, indicating Ald does not have hydroxypyruvate reductase activity. Low activity was detected with oxaloacetate and methylglyoxal. For the oxidative deamination reaction, no activity was seen with proline, aspartate, or serine (Fig. 5B).
Fig 5.

Activity of the Ald enzyme with alternate substrates. (A) Reductive amination reaction with pyruvate (Pyv), glyoxylate (Glx), α-ketoglutarate (aKT), hydroxypyruvate (Hpv), malate (Mal), acetate (Ace), glycolate (Glc), oxaloacetate (Oaa), and methylglyoxal (Mg). (B) Oxidative deamination reaction with alanine (Ala), glutamate (Glu), proline (Pro), aspartate (Asp), serine (Ser), and glycine (Gly). The specific activity of the His-tagged Ald is reported in μmoles of NADH oxidized/min/μg of protein. The standard deviations are shown.
The crystal structure of the M. tuberculosis alanine dehydrogenase indicates that Arg15 and Lys75 stabilize pyruvate in the active site by binding to the carboxyl group by hydrogen bonds (1, 51). The conserved residues His96 and Asp270 are the catalytic groups. Computer modeling suggests that glyoxylate, hydroxypyruvate, and oxaloacetate, but not α-ketoglutarate, fit into the active site of the holo enzyme (data not shown). However, as the sizes of the substrate increased, they possibly put increasing stress on the active site and the stabilizing amino acids, which resulted in a decline in activity (Fig. 5B). Alanine, glycine, and serine also fit into the active site of the holo but not the apo conformation, based on computer modeling. Why alanine serves as a substrate but glycine or serine do not is not clear.
Enzyme kinetics.
The Km values of His-Ald were determined (Table 3). For pyruvate, the Vmax was 5.8 mmol/min/mg of protein, and for glyoxylate it was 2.5 mmol/min/mg of protein. The apparent Km of Ald for pyruvate was 2.8 mM, which was previously reported to range from 0.76 to 1.45 mM (1, 24). The apparent Km for glyoxylate was 5.5 mM, close to the published range of 0.22 to 5.4 mM (16, 59). The ranges for other apparent Km values are the following: ammonium, 35 to 2,900 mM; NADH, 9 to 98 μM; alanine, 14 to 16 mM; NAD+, 310 μM (1, 16, 16, 24, 59). The differences may be due to buffer composition and pH, which can affect substrates. Ammonium concentrations, for example, are affected by pH. At higher pHs the level of NH4+, the actual substrate, is higher. Here, a pH of 8.5 in phosphate buffer was used, which is higher than that reported for other studies.
Table 3.
Apparent Km values for His-tagged alanine dehydrogenase on different substrates
| Substrate | Km (mM) |
|---|---|
| Pyruvate | 2.8 |
| Glyoxylate | 5.5 |
| Ammonium | 2.3 |
| NADH | 0.0384 |
| Alanine | 4.3 |
| NAD+ | 0.1328 |
Subcellular localization of Ald.
Alanine dehydrogenase is reported to be a secreted enzyme, with high levels found in the medium (2, 39, 41), while glyoxylate reductive aminase activity is associated with cell extracts (58). The location of Ald for M. tuberculosis was examined in aerobic and NRP cultures by two methods. For the first method, enzyme activity was measured, and for the second an Ald-specific antibody was used. M. tuberculosis H37Rv was grown to either mid-log phase in aerobic cultures or to NRP-2. The cells were concentrated by centrifugation, while the cell-free spent medium was removed and concentrated. Packed cells were disrupted, and both PvRA and GxRA activities were determined for each fraction (Table 4). Neither activity was detected in culture filtrates from aerobic or NRP cultures. Strong activity was measured in NRP cell lysates, with lower levels from the aerobic cell lysates. Differences in Ald location related to virulence have been reported for different strains (2, 39), and so the experiment was repeated with the more virulent Erdman strain (Table 4). Activity was detected only in cell lysates and not in culture filtrates.
Table 4.
Glyoxylate and pyruvate reductive aminase activitiesa
| Strain, growth condition, and fraction analyzed | Sp act (μmol NADH oxidized/min/μg of protein) (mean ± SD) |
|
|---|---|---|
| GxRA | PvRA | |
| H37Rv | ||
| AG, cell lysate | 0.023 ± 0.004 | 0.010 ± 0.002 |
| AG, culture filtrate | 0.003 ± 0.001 | 0 ± 0.004 |
| NRP, cell lysate | 0.110 ± 0.003 | 0.069 ± 0.002 |
| NRP, culture filtrate | 0.001 ± 0 | 0 ± 0.001 |
| Erdman | ||
| AG, cell lysate | 0.046 ± 0.001 | 0.014 ± 0.000 |
| AG, culture filtrate | 0.003 ± 0.003 | 0 ± 0.000 |
| NRP, cell lysate | 0.038 ± 0.005 | 0.054 ± 0.002 |
| NRP, culture filtrate | 0.003 ± 0.001 | 0.001 ± 0.002 |
Glyoxylate and pyruvate reductive aminase activities for H37Rv and Erdman were determined. Each sample was incubated aerobically or anaerobically, and cells and medium were then separated and analyzed for activity.
It was possible the Ald enzyme excreted from the cell was inactive. Therefore, the protein was detected with an anti-Ald antibody (28). Again, alanine dehydrogenase was detected only in the cell extract from NRP cultures (Fig. 6A). Cell extracts were separated into cell wall, membrane, and soluble fractions. Equal amounts of protein were separated by SDS-PAGE and probed with the anti-Ald antibody. Most of the protein (90%) was associated with the cell membrane, with a smaller amount (10%) in the soluble fraction (Fig. 6B).
Fig 6.
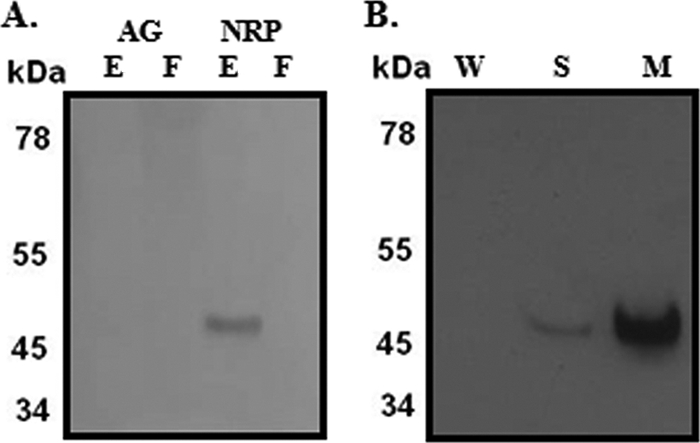
Subcellular location of Ald. Cell extracts and culture filtrates from aerobically growing or NRP-2 cultures were probed with an anti-Ald antibody. (A) AG, aerobically growing cells; NRP, nonreplicating persistent stage 2 cells; F, growth medium filtrate; E, cell extracts. (B) W, cell wall fraction; S, soluble fraction; M, cell membrane.
Role of Ald in utilization of amino acids.
Ald, functioning as an alanine dehydrogenase, is essential for the growth of M. bovis when alanine is the sole nitrogen source (9), but the role of GxRA activity in the synthesis or breakdown of glycine is not known. The roles of the various enzymatic activities of Ald in M. tuberculosis were determined during growth with different nitrogen sources, including ammonium chloride and amino acids. With asparagine as the nitrogen source, wild-type M. tuberculosis grew with a doubling time of 24.7 ± 0.4 h and 25.0 ± 0.4 (means ± standard deviations) for RVW7 (Fig. 7A). Growth of both strains on ammonium chloride was slower, but with no significant difference between strains (WT, 35.4 ± 1.0 h; RVW7, 34.5 ± 1.0 h). RVW7 grew using ammonium as the sole nitrogen source, showing Ald is not essential for the synthesis of either alanine or glycine. With glycine as the nitrogen source there was no difference in growth (WT, 35.4 ± 1.1 h; RVW7, 36.7 ± 3.7 h), indicating that Ald is not essential for the breakdown of glycine. Finally, with alanine as the nitrogen source, wild-type M. tuberculosis grew with a doubling time of 83.2 ± 2.7 h (Fig. 7B). RVW7 was unable to grow, but the complemented strain RVW7(pAld21) was restored to growth (doubling time of 87.5 ± 2.8 h). This indicated that Ald plays an essential role in the utilization of alanine but not of glycine.
Fig 7.

Growth of RVW7. (A) Growth of M. tuberculosis in LIM with different nitrogen sources. Circles, asparagine; squares, NH4Cl; diamonds, glycine. WT samples, solid lines with filled symbols; RVW7, dotted lines with empty symbols. (B) Growth in LIM with alanine. Circles, WT; squares, RVW7; triangles, RVW7(pAld-Gen21). The standard deviations are shown.
Expression of ald.
The expression of ald during aerobic growth with different nitrogen sources was analyzed using quantitative real-time PCR. Expression levels were normalized to 16S RNA, with Rv0239 included as an additional control (21). Expression of ald was induced by growth with alanine as the sole nitrogen source but not by the other nitrogen sources (Fig. 8). Expression of the control gene, Rv0239, was relatively unchanged.
Fig 8.

Quantitative real-time PCR of ald grown with different nitrogen sources. RNA levels are expressed relative to stable 16S rRNA. (Insert) Expression of Rv0239. The standard deviations are shown.
Enzyme activities were measured in extracts from cells grown with different nitrogen sources (Table 5). All three enzyme activities were low during growth on ammonium chloride, asparagine, or glycine. There was an approximately 5-fold induction of enzyme activity during growth with alanine.
Table 5.
Enzyme activities after growth on different nitrogen sourcesa
| Nitrogen source | Sp act (μmol/min/μg of protein) (mean ± SD) |
||
|---|---|---|---|
| PvRA | ALD | GxRA | |
| Asparagine | 0.018 ± 0.003 | 0.015 ± 0.001 | 0.095 ± 0.006 |
| Ammonium | 0.032 ± 0.002 | 0.023 ± 0.001 | 0.127 ± 0.013 |
| Glycine | 0.032 ± 0.004 | 0.024 ± 0.001 | 0.117 ± 0.009 |
| Alanine | 0.167 ± 0.003 | 0.131 ± 0.006 | 0.637 ± 0.026 |
Enzyme activities were determined in cell extracts after growth with the indicated nitrogen source.
DISCUSSION
Dehydrogenation of alanine and glycine.
By using both biochemical and genetic approaches, we have shown that ald of M. tuberculosis encodes an enzyme that has both pyruvate and glyoxylate aminase activities. Inactivation of ald in M. tuberculosis resulted in loss of both activities, which were restored by the insertion of a cloned copy of ald into the genome. In addition, a histidine-tagged form of the protein purified from E. coli catalyzed both reactions.
The gcvB gene, annotated as a glycine dehydrogenase in the M. tuberculosis genome, probably encodes the P protein of the glycine cleavage system. This is based on similarity with known P proteins and because gcvB is in an operon with another gene, gcvH, that is involved in glycine catabolism. The P protein decarboxylates glycine and does not have the activity attributed to the glycine dehydrogenase. The glycine cleavage system may be responsible for utilization of glycine as a nitrogen source (Fig. 7).
Alanine dehydrogenase has been characterized from many different bacteria, but not all these enzymes have GxRA activity. This may be due to the large sequence diversity seen among these enzymes (51). There may be two classes of alanine dehydrogenases, those that can also use glyoxylate and those that cannot. Purified alanine dehydrogenases from Anabaena cylindrical (43) and Streptomyces fradiae (54) lack activity with glyoxylate. The alanine dehydrogenase of Pseudomonas aeruginosa has been proposed to be a glycine dehydrogenase with GxRA activity (20). Glycine produced from the enzyme was used to synthesize cyanide under hypoxic conditions (7). An ald knockout mutant of P. aeruginosa had reduced virulence in rats, showing that the enzyme, although not essential, does play a role in pathogenesis (5). In M. tuberculosis no cyanide was detected in growing or NRP cultures (data not shown).
Ald of M. tuberculosis has a greater affinity for pyruvate than for glyoxylate, but there is evidence that the GxRA reaction occurs in vivo. GxRA was detected in whole cells of M. smegmatis by isotopomer analysis (49). This activity was upregulated in response to hypoxia, while the pyruvate reductive aminase reaction was not. Each activity may be differentially regulated. In this model, the conversion of alanine to pyruvate for the purpose of nitrogen assimilation would be by alanine dehydrogenase during aerobic growth. During hypoxic dormancy, the GxRA reaction would recycle NADH for redox balancing.
Role of alanine dehydrogenase.
A great deal of research has been done to characterize alanine dehydrogenases in many bacterial species, among them M. tuberculosis, for which a 3-dimensional structure has been constructed (1, 51). Many roles have been proposed for this enzyme, in part because of uncertainty of the main direction of the reaction. Alanine dehydrogenase may fulfill several roles, depending on the conditions and perhaps even the species.
Pyruvate reductive amination was proposed to be the main role in vivo serving to produce alanine (9, 24, 51) for the synthesis of peptidoglycan (2). However, RVW7 grew with NH4Cl as the nitrogen source, indicating that in M. tuberculosis Ald is not essential for the synthesis of alanine or glycine.
The PvRA reaction of the alanine/glycine dehydrogenase could also be used to incorporate and store ammonia. M. tuberculosis assimilates ammonia by glutamine synthetase and glutamate synthetase. This is a high-affinity system that requires ATP. Glutamate dehydrogenase provides a lower-affinity non-ATP-requiring system, but this activity was not detected in M. tuberculosis (50). Under certain conditions, alanine dehydrogenase may fulfill an ammonium assimilation role, as has been proposed for other bacteria (6, 46). This could be important if glutamine synthetase is inhibited by high levels of alanine or glycine (9). For Anabaena, Ald has been proposed to synthesize alanine to store and transport nitrogen from heterocysts to vegetative cells (37).
The reverse reaction with ALD, involving the oxidative deamination of alanine to pyruvate, clearly has a role during growth with alanine as the nitrogen source in M. tuberculosis (Fig. 7), M. bovis BCG (9), and M. smegmatis (14). In M. tuberculosis, alanine dehydrogenase is required for the utilization of alanine, but not glycine, as a nitrogen source. The release of ammonium from the ALD reaction was also suggested to play a role in blocking phagosome acidification (39).
Enzyme activity and RNA levels were induced by growth with alanine as the nitrogen source (Table 5 and Fig. 8). Ammonium suppresses Ald protein levels in M. bovis BCG (9), while in M. smegmatis, ald transcription and enzyme activity were upregulated in rich media by the presence of alanine (14). Expression of alanine dehydrogenase was also upregulated by hypoxia (Table 4), which appears to be independent of the DosR/DevR regulator (56). The regulators in both cases are unknown, but a regX3 mutant showed increased expression of ald during aerobic growth and reduced expression under hypoxic conditions, suggesting a role for the two-component senX3-regX3 system (36, 40).
A role in recycling of NADH during the interruption of aerobic respiration has been proposed for Ald of M. tuberculosis (59, 60). Both the GxRA and PvRA activities were induced by hypoxia (Fig. 5). A similar role was proposed for alanine dehydrogenase in M. smegmatis (23), and indeed, an ald knockout mutant was unable to grow anaerobically (14). The slower growth of an alanine dehydrogenase mutant in Anabaena could also be due to impairment of NADH recycling (37).
Additional roles for alanine dehydrogenase are possible. These include the utilization and detoxification of propionate or glyoxylate. The main energy source for M. tuberculosis in vivo is thought to be fatty acids. β-Oxidation of fatty acids containing an even number of carbons produces acetyl coenzyme A (CoA), while β-oxidation of fatty acids with odd numbers of carbons and cholesterol produces both acetyl-CoA and propionyl-CoA. Propionyl-CoA is converted to methylisocitrate in the methylcitrate cycle and then to pyruvate by isocitrate lyase (18). Acetyl-CoA enters the glyoxylate cycle and is converted by isocitrate lyase to glyoxylate. Both of these products, pyruvate and glyoxylate, are substrates for alanine dehydrogenase. This enzyme may connect these two cycles, regulating glyoxylate and propionate levels.
Location.
Alanine dehydrogenase was detected only in association with the cells of M. tuberculosis, not in the extracellular medium. Specifically, most of the protein was isolated with the membrane and a lesser amount in the cytosol. Neither alanine dehydrogenase protein (Fig. 6) nor enzymatic activity (Table 4) was detected in the culture filtrate. In contrast, others have detected protein (2, 28, 34, 42) and activity (39) in the extracellular medium of growing cultures of M. tuberculosis. In Mycobacterium marinum and in M. bovis expressing the M. tuberculosis ald, alanine dehydrogenase was secreted (9). We used DTA medium while the other reports utilized Sautons medium for growth, but the difference is probably not due to the medium, as a comprehensive analysis of proteins exported from H37Rv also cultured in Sautons medium did not detect alanine dehydrogenase (31).
GxRA and PvRA activities were detected in cell filtrates, but not culture filtrates, of both the H37Rv and Erdman strains. Proteome analysis had detected this enzyme only in H37Rv and not the Erdman strain (25). The Erdman strain expressed higher levels during aerobic growth (Table 4).
Malate synthase, which is involved in the glyoxylate shunt, and the glycolytic enzyme fructose-1,6-bisphosphate aldolase are exported to the cell surface of M. tuberculosis (10, 26). Both of these enzymes and alanine dehydrogenase lack signal sequences. In addition to their metabolic function, they are involved in host-pathogen interactions. Alanine dehydrogenase may also play such a role, which would explain its membrane location. Alternatively, GxRA and ALD activities may be regulated by an interaction with membrane components of the respiratory chain.
Importance of Ald in pathogenesis.
It is intriguing that alanine dehydrogenase is important in other bacteria that have specialized persistence programs, such as sporulation. In Myxococcus xanthus, an ald mutant showed delayed aggregation and reduced levels of sporulation (57). An ald mutant of Bacillus subtilis was defective for sporulation (47). This was partially complemented by the addition of pyruvate, suggesting that the role of alanine dehydrogenase is to provide pyruvate as an energy source.
The ald gene of M. bovis contains a single nucleotide deletion and therefore lacks this enzyme (9). Also absent due to a point mutation is pyruvate kinase, suggesting there are major differences in central metabolism between M. bovis and M. tuberculosis (8, 55). Despite these changes, M. bovis can produce disease in humans. M. bovis BCG expressing the M. tuberculosis ald showed similar survival in both macrophages and mice (44). Alanine dehydrogenase is probably not essential for pathogenesis in M. tuberculosis but may still play an important role.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sylors Chem and Karen Kelley for valuable discussions and review of the manuscript, Sandy Sudberg for technical help, and Sheldon Broedel at AthenaES for protein purification.
The following reagent was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH: monoclonal anti-Mycobacterium tuberculosis Ald (gene Rv2780), clone IT-46 (HBT10) (produced in vitro) (NR-13651). This study was supported by the Medical Research Services of the U.S. Department of Veterans Affairs.
Footnotes
Published ahead of print 30 December 2011
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Agren D, et al. 2008. Three-dimensional structures of apo- and holo-L-alanine dehydrogenase from Mycobacterium tuberculosis reveal conformational changes upon coenzyme binding. J. Mol. Biol. 377:1161–1173 [DOI] [PubMed] [Google Scholar]
- 2. Andersen ÅB, Andersen P, Ljungqvist L. 1992. Structure and function of a 40,000-molecular-weight protein antigen of Mycobacterium tuberculosis. Infect. Immun. 60:2317–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Awasthy D, et al. 2009. Inactivation of the ilvB1 gene in Mycobacterium tuberculosis leads to branched-chain amino acid auxotrophy and attenuation of virulence in mice. Microbiology 155:2978–2987 [DOI] [PubMed] [Google Scholar]
- 4. Beste DJV, et al. 2011. 13C metabolic flux analysis identifies an unusual route for pyruvate dissimilation in mycobacteria which requires isocitrate lyase and carbon dioxide fixation. PLoS Pathog. 7:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boulette ML, et al. 2009. Characterization of alanine catabolism in Pseudomonas aeruginosa and its importance for proliferation in vivo. J. Bacteriol. 191:6329–6334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caballero FJ, Cárdenas J, Castillo F. 1989. Purification and properties of L-alanine dehydrogenase of the phototrophic bacterium Rhodobacter capsulatus E1F1. J. Bacteriol. 171:3205–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Castric PA. 1983. Hydrogen cyanide production by Pseudomonas aeruginosa at reduced oxygen levels. Can. J. Microbiol. 29:1344–1349 [DOI] [PubMed] [Google Scholar]
- 8. Chavadi S, et al. 2009. Global effects of inactivation of the pyruvate kinase gene in the Mycobacterium tuberculosis complex. J. Bacteriol. 191:7545–7553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen JM, Alexander DC, Behr MA, Liu J. 2003. Mycobacterium bovis BCG vaccines exhibit defects in alanine and serine catabolism. Infect. Immun. 71:708–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de la Paz Santangelo M, et al. 2011. Glycolytic and non-glycolytic functions of the fructose-1,6-bisphosphate aldolase of Mycobacterium tuberculosis, an essential enzyme produced by replicating and non-replicating Bacilli. J. Biol. Chem. 286:40219–40231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DesJardin LE, Hayes LG, Sohaskey CD, Wayne LG, Eisenach KD. 2001. Microaerophilic induction of the alpha-crystallin chaperone protein homologues (hspX) mRNA of Mycobacterium tuberculosis. J. Bacteriol. 183:5311–5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eng JK, McCormack AL, Yates JR., III 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spec. 5:976–989 [DOI] [PubMed] [Google Scholar]
- 14. Feng Z, Cáceres NE, Sarath G, Barletta RG. 2002. Mycobacterium smegmatis L-alanine dehydrogenase (Ald) is required for proficient utilization of alanine as a sole nitrogen source and sustained anaerobic growth. J. Bacteriol. 184:5001–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goldman DS. 1959. Enzyme systems in the mycobacteria. Bioch. Biophys. Acta 34:527–539 [DOI] [PubMed] [Google Scholar]
- 16. Goldman DS, Wagner MJ. 1962. Enzyme systems in the mycobacteria XIII glycine dehydrogenase and the glyoxylic acid cycle. Biochim. Biophys. Acta 65:297–306 [DOI] [PubMed] [Google Scholar]
- 17. Gordhan BG, et al. 2002. Construction and phenotypic characterization of an auxotrophic mutant of Mycobacterium tuberculosis defective in L-arginine biosynthesis. Infect. Immun. 70:3080–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gould TA, van de Langemheen H, McKinney JD, Sacchettini JC. 2006. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol. Microbiol. 61:940–947 [DOI] [PubMed] [Google Scholar]
- 19. Gu S, et al. 2003. Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell. Proteomics 2:1284–1296 [DOI] [PubMed] [Google Scholar]
- 20. Hagins JM, Locy R, Silo-Suh L. 2009. Isocitrate lyase supplies precursors for hydrogen cyanide production in a cystic fibrosis isolate of Pseudomonas aeruginosa. J. Bacteriol. 191:6335–6339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Honaker RW, Leistikow RL, Bartek IL, Voskuil MI. 2009. Unique roles of DosT and DosS in DosR regulon induction and Mycobacterium tuberculosis dormancy. Infect. Immun. 77:3258–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hondalus MK, et al. 2000. Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect. Immun. 68:2888–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hutter B, Dick T. 1998. Increased alanine dehydrogenase activity during dormancy in Mycobacterium smegmatis. FEMS Microbiol. Lett. 167:7–11 [DOI] [PubMed] [Google Scholar]
- 24. Hutter B, Singh M. 1999. Properties of the 40 kDa antigen of a Mycobacterium tuberculosis, a functional L-alanine dehydrogenase. Biochem. J. 343:669–672 [PMC free article] [PubMed] [Google Scholar]
- 25. Jungblut PR, et al. 1999. Comparative proteome analysis of Mycobacterium tuberculosis and Mycobacterium bovis BCG strains: towards functional genomics of microbial pathogens. Mol. Microbiol. 33:1103–1117 [DOI] [PubMed] [Google Scholar]
- 26. Kinhikar AG, et al. 2006. Mycobacterium tuberculosis malate synthase is a laminin-binding adhesin. Mol. Microbiol. 60:999–1013 [DOI] [PubMed] [Google Scholar]
- 27. Lim A, Eleuterio M, Hutter B, Murugasu-Oei B, Dick T. 1999. Oxygen depletion induced dormancy in Mycobacterium bovis BCG. J. Bacteriol. 181:2252–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ljungqvist L, Worsaae A, Heron I. 1988. Antibody responses against Mycobacterium tuberculosis in 11 strains of inbred mice: novel monoclonal antibody specificities generated by fusions, using spleens from BALB. B10 and CBA/J mice. Infect. Immun. 56:1994–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lyon RH, Hall WH, Costas-Martinez C. 1970. Utilization of amino acids during growth of Mycobacterium tuberculosis in rotary cultures. Infect. Immun. 1:513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mahenthiralingam E, et al. 1998. Site-directed mutagenesis of the 19-kilodalton lipoprotein antigen reveals no essential role for the protein in the growth and virulence of Mycobacterium intracellulare. Infect. Immun. 66:3626–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malen H, Berven FS, Fladmark KE, Wiker HG. 2007. Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv. Proteomics 7:1702–1718 [DOI] [PubMed] [Google Scholar]
- 32. McAdam RA, et al. 1995. In vivo growth characteristics of leucine and methionine auxotrophic mutants of Mycobacterium bovis BCG generated by transposon mutagenesis. Infect. Immun. 63:1004–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDonough JA, et al. 2008. Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J. Bacteriol. 190:6428–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ortalo-Magné A, et al. 1995. Molecular composition of the outermost capsular material of the tubercle bacillus. Microbiology 141:1609–1620 [DOI] [PubMed] [Google Scholar]
- 35. Parish T. 2003. Starvation survival response of Mycobacterium tuberculosis. J. Bacteriol. 185:6702–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parish T, Smith DA, Roberts G, Betts J, Stoker NG. 2003. The senX3-regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology 149:1423–1435 [DOI] [PubMed] [Google Scholar]
- 37. Pernil R, Herrero A, Flores E. 2010. Catabolic function of compartmentalized alanine dehydrogenase in heterocyst-forming cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 192:5165–5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Quandt J, Hynes MF. 1993. Versatile suicide vectors which allow direct selection for gene replacement in Gram-negative bacteria. Gene 127:15–21 [DOI] [PubMed] [Google Scholar]
- 39. Raynaud C, Etienne G, Peyron P, Lanéelle M-A, Daffé M. 1998. Extracellular enzyme activities potentially involved in the pathogenicity of Mycobacterium tuberculosis. Microbiology 144:577–587 [DOI] [PubMed] [Google Scholar]
- 40. Roberts G, Vadrevu I, Madiraju MV, Parish T. 2011. Control of CydB and GltA1 expression by the SenX3 RegX3 two component regulatory system of Mycobacterium tuberculosis. PLoS One 6:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rosenkrands I, et al. 2002. Hypoxic response of Mycobacterium tuberculosis studied by metabolic labeling and proteome analysis of cellular and extracellular proteins. J. Bacteriol. 184:3485–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosenkrands I, et al. 2000. Mapping and identification of Mycobacterium tuberculosis proteins by two-dimensional gel electrophoresis, microsequencing and immunodetection. Electrophoresis 21:948. [DOI] [PubMed] [Google Scholar]
- 43. Rowell P, Stewart DP. 1976. Alanine dehydrogenase of the N2-fixing blue green alga, Anabaena cylindrica. Arch. Microbiol. 107:115–124 [DOI] [PubMed] [Google Scholar]
- 44. Scandurra GM, Ryan AA, Pinto R, Britton WJ, Triccas JA. 2006. Contribution of L-alanine dehydrogenase to in vivo persistence and protective efficacy of the BCG vaccine. Microbiol. Immunol. 50:805–810 [DOI] [PubMed] [Google Scholar]
- 45. Schaefer WB, Marshak A, Burkhart B. 1949. The growth of Mycobacterium tuberculosis as a function of its nutrients. J. Bacteriol. 58:549–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schuffenhauer G, Schräder T, Andreesen JR. 1999. Morpholine-induced formation of L-alanine dehydrogenase activity in Mycobacterium strain HE5. Arch. Microbiol. 171:417–423 [DOI] [PubMed] [Google Scholar]
- 47. Siranosian KJ, Ireton K, Grossman AD. 1993. Alanine dehydrogenase (ald) is required for normal sporulation in Bacillus subtilis. J. Bacteriol. 175:6789–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sohaskey CD, Wayne LG. 2003. Role of narK2X and narGHJI in hypoxic upregulation of nitrate reduction by Mycobacterium tuberculosis. J. Bacteriol. 185:7247–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang YJ, et al. 2009. Central metabolism in Mycobacterium smegmatis during the transition from O2-rich to O2-poor conditions as studied by isotopomer-assisted metabolite analysis. Biotechnol. Lett. 31:1233–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian J, Bryk R, Itoh M, Suematsu M, Nathan C. 2005. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of α-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 102:10670–10675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tripathi SM, Ramachandran R. 2008. Crystal structures of the Mycobacterium tuberculosis secretory antigen alanine dehydrogenase (Rv2780) in apo and ternary complex forms captures “open” and “closed” enzyme conformations. Proteins 72:1089–1095 [DOI] [PubMed] [Google Scholar]
- 52. Tullius MV, Harth G, Horwitz MA. 2003. Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect. Immun. 71:3927–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Usha V, Jayaraman R, Toro JC, Hoffner SE, Das KS. 2002. Glycine and alanine dehydrogenase activities are catalyzed by the same protein in Mycobacterium smegmatis: upregulation of both activities under microaerophilic adaptation. Can. J. Microbiol. 48:7–13 [DOI] [PubMed] [Google Scholar]
- 54. Vancura A, et al. 1989. Alanine dehydrogenase from Streptomyces fradiae. Eur. J. Biochem. 179:221–227 [DOI] [PubMed] [Google Scholar]
- 55. van Keulen G, Alderson J, White J, Sawers RG. 2005. Nitrate respiration in the actinomycete Streptomyces coelicolor. Biochem. Soc. Trans. 33:210–212 [DOI] [PubMed] [Google Scholar]
- 56. Voskuil MI, et al. 2002. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis persistence program. J. Exp. Med. 198:705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ward MJ, Lew H, Zusman DR. 2000. Disruption of aldA influences the developmental process in Myxococcus xanthus. J. Bacteriol. 182:546–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64:2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wayne LG, Lin K-Y. 1982. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect. Immun. 37:1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wayne LG, Sohaskey CD. 2001. Nonreplicating Persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55:139–163 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.