

Abstract
Objective
Hepatic steatosis is a common complication in patients with lipodystrophies and can lead to cirrhosis. There is no proven effective therapy for hepatic steatosis, but cholic acid (CA), a farnesoid × receptor agonist, has previously been shown to reduce hepatic triglyceride (TG) content in mice and serum triglycerides in humans. Our objective was to assess clinical efficacy and tolerability of CA therapy in patients with lipodystrophy and hepatic steatosis.
Design
A randomized, double-blind, placebo-controlled, crossover study.
Methods
Eighteen patients with genetic or autoimmune lipodystrophies, and elevated hepatic TG content participated in the study. The intervention was CA (15 mg/kg/day) compared with placebo for a period of 6 months each. Hepatic TG content, the primary outcome variable, was measured with 1H magnetic resonance spectroscopy at baseline and at 3 and 6 months during each study period. Levels of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transpeptidase (GGT) and TG were secondary end-points of the study.
Results
Compared to placebo, CA did not reduce hepatic TG content [median (interquartile range) 14.8% (9.4-19.0%) vs. 15.9% (10.5-26.5%), respectively; p = 0.42] or serum TG [340 mg/dL (233 – 433 mg/dL) vs. 390 mg/dL (233 – 595 mg/dL) respectively; p=0.45]. CA therapy also did not change AST, ALT or GGT levels. Two patients developed diarrhea and excessive flatus while taking CA and these symptoms resolved after reducing the dose of CA.
Conclusion
CA was well tolerated but did not reduce hepatic TG content in patients with lipodystrophy.
Keywords: lipodystrophy, cholic acid, farsenoid x receptor, hepatic steatosis
Introduction
Lipodystrophies are rare disorders characterized by selective loss of adipose tissue 1 and predisposition to metabolic complications such as diabetes mellitus, hypertriglyceridemia, and hepatic steatosis. Hepatic steatosis is a common complication in patients with genetic or acquired lipodystrophies 1, 2. Both cirrhosis requiring hepatic transplantation 3 and hepatocellular carcinoma 2, possibly due to long-standing hepatic steatosis, have been reported in patients with lipodystrophies. We and others have previously reported that recombinant leptin therapy reduced liver size and hepatic steatosis in hypoleptinemic patients with lipodystrophies 4-9. However, many patients with lipodystrophy are not hypoleptinemic 10, 11. Therefore, there is a need to develop other therapies for treatment of hepatic steatosis in patients with lipodystrophies.
There is no proven effective therapy for hepatic steatosis 12. A variety of drugs have been investigated including ursodeoxycholic acid 13, insulin sensitizers (metformin and thiazolidinediones) 14, 15, vitamin E 14, betaine 16, and probucol 17. Thus far, only vitamin E has been shown to improve histology in patients with hepatic steatosis 14. However, these results have not yet been replicated and concerns related to an increase in mortality with vitamin E 18 have limited its use.
Bile acids play a role in the metabolism of both serum and hepatic triglycerides (TG). Cholic (CA) acid has been reported to reduce serum TG by as much as 29% in patients with hyperlipoproteinemia 19, 20. Recently, it has been noted that bile acids, which are the endogenous ligands of the farnesoid × receptor (FXR, NR1H4) activate transcription of several genes, particularly the atypical nuclear receptor small heterodimer partner and thus can influence triglyceride metabolism within hepatocytes 21. Both CA and chenodeoxycholic acid (CDCA), the endogenous primary bile acids, are potent ligands for FXR. Giving 0.5% CA for 3 weeks reduced hepatic TG content by over 50% in KK-Ay mice (a model for diet-induced hypertriglyceridemia) fed chow or high fat diet 22. In addition, activation of FXR was shown to robustly attenuate liver steatosis in leptin-receptor mutated Zucker fa/fa rats (a genetic model of insulin resistance and obesity-driven liver injury) 23. Therefore, FXR agonists may be beneficial for treating hepatic steatosis, however, there are no previous studies assessing their efficacy for reducing liver TG content in humans.
Thus, we designed a randomized, double-blind, placebo-controlled, crossover study to evaluate the efficacy and safety of CA therapy in treating hepatic steatosis in patients with lipodystrophy.
Methods
Setting and Participants
All patients were evaluated at the Clinical and Translational Research Center (CTRC) at UT Southwestern Medical Center, Dallas, Texas. A written informed consent was obtained from all participants, and the study was approved by the Institutional Review Board of UT Southwestern. Thirty-seven patients with a clinical diagnosis of genetic or acquired autoimmune lipodystrophy (not HIV-associated) underwent screening with 1H magnetic resonance spectroscopy (MRS) for liver fat. Ten subjects did not qualify after screening (8 had low levels of liver fat, one was too obese for the MRI machine, one had a history of alcohol use), 8 subjects lost interest, and one could not be contacted (Figure 1).
Figure 1.
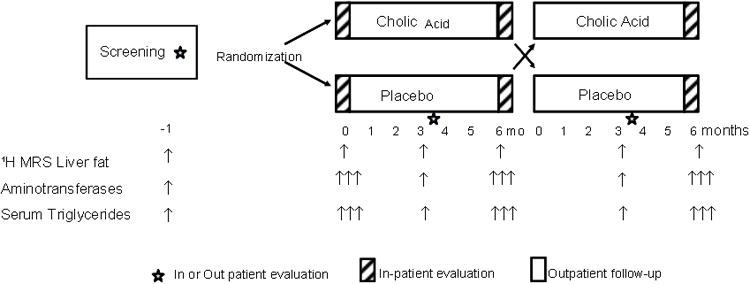
Study design.
Inclusion criteria for the trial were as follows: patients with lipodystrophy as diagnosed by clinical criteria, hepatic steatosis (>5.6% hepatic TG content) as demonstrated by 1H MRS, age 10-70 year, and alcohol intake of less than 40 g per week. Patients with the following were excluded: other liver diseases such as chronic viral hepatitis, autoimmune hepatitis, primary biliary cirrhosis, biliary obstruction, Wilson's disease, hemochromatosis or α-1-antitrypsin deficiency; treatment with drugs associated with steatohepatitis: corticosteroids, high dose estrogens, methotrexate, amiodarone, calcium channel blockers, sulfasalazine, naproxen, or oxacillin in the 6 months prior to the study; decompensated liver disease; hepatocellular carcinoma; congestive heart failure, cerebrovascular disease, respiratory failure, renal failure (serum creatinine >2 mg/dL), acute pancreatitis, organ transplantation, serious psychiatric disease, and malignancy; acute medical illnesses; known HIV infection; current substance abuse; pregnant or lactating women; hematocrit of less than 30%; weight loss during past 3 months; and hypersensitivity or intolerance to CA or any components of its formulation. Additional exclusion criteria included the use of drugs which can potentially decrease hepatic steatosis: ursodeoxycholic acid, high-dose vitamin E, betaine, acetylcysteine and choline. Thiazolidinediones were allowed if the dose had been stable for three months prior to screening.
Design Overview, Randomization and Interventions
This study was a randomized, double-blind, placebo-controlled, cross-over trial (Figure 1). Patients underwent a screening evaluation to determine their eligibility for the trial. For those who were found to be eligible, they continued their usual diet and other lifestyle measures without changing any medications for 1 month in order to establish a baseline state.
All patients were studied during three hospitalizations, each lasting for three days, and two outpatient visits (Figure 1). One month after screening, patients were hospitalized to the CTRC for a period of three days, i.e., the baseline visit. Fasting blood samples were obtained on 3 consecutive days during the inpatient evaluation, and the average of the three measurements was used for data analysis. All patients underwent 1H MRS to determine liver fat during this hospitalization. At 6 and 12 months, patients were again admitted for 3 consecutive days; during these visits, all the studies mentioned in the baseline visit were repeated. The patients returned at 3 and 9 months for fasting blood samples and liver 1H MRS during outpatient visits.
Following the baseline hospitalization, the patients received CA or an identical placebo in the dose of 15 mg/kg/day (divided twice daily) for a period of 6 months and then received the other treatment (CA or placebo) for the next 6 months. The maximum dose of CA was 1500 mg/day. CA and matched placebo were supplied by Global Strategic Connections, LLC (Troy, MI) in 250 mg capsules.
At each study visit, an inquiry was made about any side effects of medication, in particular, gastrointestinal side effects. Medication compliance was assessed by counting the remaining number of pills at the completion of each study period. If a patient was unable to tolerate the full dose, the dose could be reduced by the investigators. Patients were asked to keep doses of their other medications constant for the duration of the study.
Outcomes
The 1H-MRS studies were performed at least four hours post-prandially with the patients lying prone as described previously 24. Briefly, image-guided, proton-localized, MRS and high resolution T1-weighted imaging were performed on a 1.5 T Gyroscan Intera whole body system (Philips Medical Systems, Best, The Netherlands) with the following parameters: repetition time (TR) of 3 s, spin echo time (TE) of 25 ms, and 1024 data points over 1000 kHz spectral width. Volume of interest (voxel 30 mm3) was selected in the upper right hepatic lobe, taking care to avoid vascular structures. Spectra were processed and resonances quantified using a standard analysis package (NUTS; ACORNNMR, Fremont, CA). The hepatic TG content was expressed as the ratio of fat and water resonance peaks.
Blood was obtained after overnight (12 h) fast daily for chemistry profile (SMA-25) and lipoprotein levels while the patients were admitted. Serum chemistry profile including the aminotransferases, lipoproteins, gamma-glutamyl transpeptidase (GGT), electrolytes etc. were measured by autoanalyzer (Quest Diagnostics, Irving, Texas). Glycosylated hemoglobin (HbA1c) levels were measured by an immunoturbidimetric colorimetric assay (Quest Diagnostics, Irving, TX).
Primary and Secondary Endpoint Variables
The primary end-point variable was reduction in the liver TG content as measured by 1H MRS. Reductions in the levels of serum alanine and aspartate aminotransferases (ALT, AST), GGT and TG were secondary end-points of the study.
Statistical Analysis
We determined that a sample of 12 patients completing the 12 month crossover study would provide a power of 0.80 at an alpha of 0.05 to detect an absolute treatment difference in hepatic fat content of 3.5% with the standard deviation of the difference of 4.0%. We therefore planned to enroll at least 16 subjects assuming a maximum 30% rate of loss to follow-up.
A block randomization with block size of two was generated using SAS version 9.2 version 9.2 (SAS Institute, Cary, NC, USA). At the beginning of the study, a randomized treatment sequence was prepared by the study statistician who sent it directly to the investigative pharmacy. The pharmacist assigned participants to the interventions according to the treatment sequence. Patients were recruited by the study personnel. All participants and study personnel and those evaluating outcomes (e.g., 1H-MRS results) were blinded. Only the pharmacist was aware of the intervention sequence. The code was broken upon study completion.
All data on patients who had at least one 3 month measurement of hepatic TG content during the placebo or CA phase were included in the analysis (n=15). For continuous outcome variables, statistical analysis of this two-phase crossover study was performed using mixed linear model repeated measures analysis with the study subject modeled as a random effect. The mixed-effects analysis was also used to assess a possible treatment sequence effect. All hypothesis tests were two-sided and a p-value < 0.05 was considered statistically significant. Results are reported as median and interquartile range. Statistical analyses were conducted using SAS.
Results
Patients
Recruitment began in December 2006, and the last patient finished the 12 month follow-up visit in April 2011. Eighteen patients were enrolled in the study and 12 of them completed both the CA and Placebo phases, whereas an additional 3 patients had 3 month data (Figure 2). The five dropouts included three who were unable to be contacted, two who had hospitalizations and medication changes for unrelated illnesses, and one who had an implantable cardioverter-defibrillator placed for pre-existing cardiomyopathy due to which MRS could not be performed.
Figure 2. CONSORT 2010 Flow Diagram.
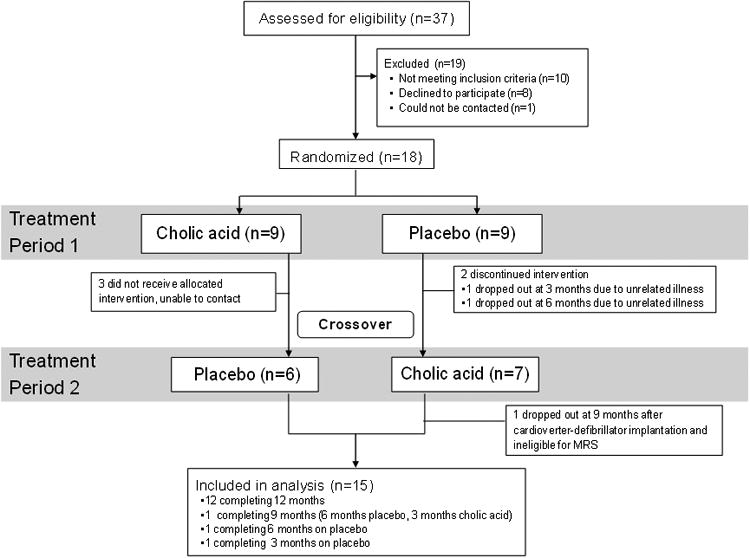
Table 1 describes the baseline characteristics of all patients enrolled in study. At screening, the mean (SD) hepatic TG content was 23.9% (13.4%), and the range was 6.1%-60.3%. Serum ALT concentration ranged from 11 to 168 U/L; AST ranged from 12 to 55 U/L; GGT ranged from 16 to 72 U/L; and serum TG concentration ranged from 155 to 3455 mg/dL. Twelve patients had a previous diagnosis of diabetes mellitus.
Table 1.
Characteristics of all patients enrolled in studya.
| All enrolled (n=18) | Min - Max | |
|---|---|---|
| Age (years) | 46.0 (15.3) | 17-70 |
| BMI (kg/m2) | 30 (6) | 22-44 |
| Male: Female | 7:11 | |
| Diabetes | 67% | |
| Hypertension | 50% | |
| Dyslipidemia | 100% | |
| Hepatic TG (%) | 23.89b (13.4) | 6.1 - 60.3 |
| Weight (kg) | 80.7 (16) | 56.0 - 109.0 |
| Total Cholesterol (mg/dL) | 208.4 (64.9) | 116.0 - 355.0 |
| HDL-C (mg/dL) | 33.0 (7.4) | 18.0 - 51.7 |
| TG (mg/dL) | 389b | 155 - 3455 |
| HbA1C (%) | 6.8 (1.3) | 5.3 - 9.1 |
| Glucose (mg/dL) | 125.6 (33.5) | 80 – 199 |
| AST (U/L) | 29.4 (13.2) | 12.0 – 55.0 |
| ALT (U/L) | 38.5 (35.0) | 11.0 – 168.0 |
| GGT (U/L) | 35.4 (16.7) | 16.0 – 72.0 |
| Lipid-lowering agents (n) | ||
| Statins | 7 | |
| Fibrates | 7 | |
| Fish oil | 7 | |
| Ezetimibe | 3 | |
| Niacin | 1 | |
| Diabetes medications (n) | ||
| Metformin | 10 | |
| Insulin | 7 | |
| Sulfonylurea | 5 | |
| Thiazolidinediones | 2 | |
| GLP-1 agonist | 2 | |
| DPP-4 inhibitor | 1 |
Values are given as mean (SD) unless otherwise noted. Abbreviations: BMI, body mass index; TG, triglycerides; HDL-C, high density lipoprotein cholesterol; HbA1C,hemoglobin A1C; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transpeptidase. GLP, glucagon-like peptide; DPP, dipeptidyl peptidase.
median
Nine patients had familial partial lipodystrophy of the Dunnigan variety (FPLD) due to mutations in the lamin A/C (LMNA) gene. Eight patients had other types of familial partial lipodystrophy. One patient had acquired generalized lipodystrophy.
Primary and Secondary End Points
The sequence in which the patients received CA or placebo had no effect on the results. Compared with placebo, CA therapy did not result in any significant reduction in hepatic TG concentration (geometric mean difference, 13.8%; 95% confidence interval [CI] −19.7% to 61.1%). Similarly, there was no significant change in serum TG levels (11.4%; CI −17.7% to 50.8%). Individual responses for hepatic TG concentration and serum TG levels are shown in Figure 3 and overall results are reported in Table 2. CA therapy did not lower ALT (−3.0%; CI −24.3% to 24.2%), AST (2.5%; CI −17.6% to 27.7%), or GGT (1.6%; CI −18.0% to 26.0%) levels. Mean difference in HbA1C was 0.41% (CI −0.23% to 1.05%); however, it was not statistically significant (p = 0.18). Absolute difference in weight was −0.23 kg (CI, −2.02 to 1.56 kg) for CA relative to placebo; however, it was also not statistically significant.
Figure 3.
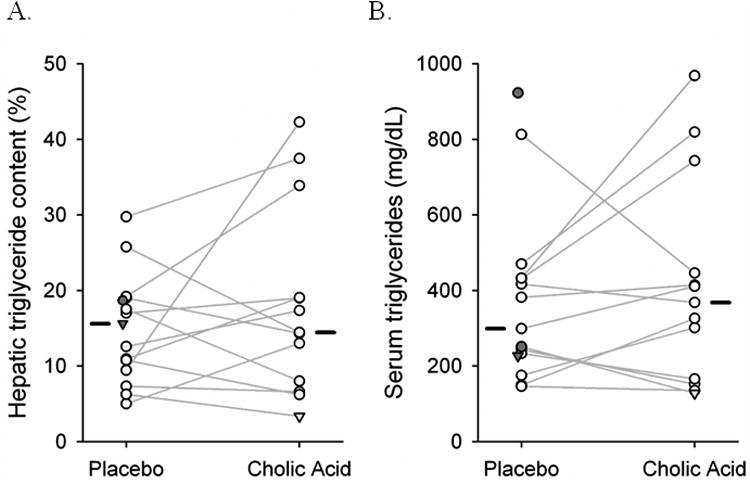
Hepatic triglyceride content (A) and serum triglyceride levels (B) during the study. Connected open circles represent the 12 subjects who completed both the six month study periods. For three subjects with incomplete data, filled symbols represent subjects with placebo data only; inverted triangles represent 3 month data. Median values for each phase are shown as horizontal bars
Table 2.
Results of cholic acid versus placeboa.
| Cholic Acid | Placebo | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Measure | Baseline | 3 months | 6 months | 3 months | 6 months | p-value | |
| Hepatic TG (%) | 18.7 [10 - 25.5] | 11.6 [7.6 - 17] | 15.9 [10.5 - 26.5] | 15.6 [7.2 - 24.7] | 14.8 [9.4 - 19] | 0.42 | |
| HbA1C (%) | 6.4 [5.6 - 7.4] | 6.2 [5.8 - 7.1] | 7.0 [5.4 - 8.3] | 6.7 [5.8 - 7.7] | 6.6 [5.6 - 7.6] | 0.18 | |
| Weight (kg) | 77 [67.1 - 90.9] | 77 [65.8 - 94.5] | 75 [66.2 - 89.6] | 80.4 [68 - 91.1] | 76.9 [65.7 - 84.5] | 0.77 | |
| Glucose (mg/dL) | 115 [91 - 134] | 112 [99 - 152] | 122 [87 - 152] | 105 [90 - 131] | 108 [92 - 140] | 0.91 | |
| TG (mg/dL) | 278 [238 - 489] | 314 [217 - 532] | 390 [233 - 595] | 283 [220 - 400] | 340 [233 - 433] | 0.45 | |
| Total Cholesterol (mg/dL) | 214 [160 - 280] | 176 [167 - 227] | 196 [173 - 242] | 186 [169 - 206] | 200 [164 - 234] | 0.31 | |
| HDL-C (mg/dL) | 36 [31.9 - 42] | 30 [28 - 37] | 32.2 [24.7 - 36] | 33 [28.6 - 37] | 30.7 [24.7 - 37] | 0.72 | |
| AST (U/L) | 23 [18 - 31] | 20 [15 - 25] | 22 [15.5 - 28.5] | 18 [14 - 34] | 19 [14.7 - 32.3] | 0.80 | |
| ALT (U/L) | 26 [17.7 - 39.3] | 25 [18 - 29] | 25.1 [18.9 - 48] | 24.5 [18 - 49] | 22.8 [19.7 - 37.3] | 0.79 | |
| GGT (U/L) | 26.7 [21.7 - 30.3] | 30 [21 - 38] | 27.2 [21.8 - 41.1] | 26 [22 - 44] | 29.8 [23 - 47.3] | 0.87 | |
Values are given as median [interquartile range] unless otherwise noted. Abbreviations: BMI, body mass index; TG, triglycerides; HDL-C, high density lipoprotein cholesterol; HbA1C,hemoglobin A1C; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transpeptidase.
Adverse Events
CA was well tolerated except for two patients who developed diarrhea and excessive flatus while taking CA. Their symptoms resolved after reducing the dosage of CA to 5-10 mg/kg/day. Three other patients reported mild diarrhea which subsided within one to two weeks without any dose adjustment.
Four patients had adverse events during the trial. One patient developed gastroenteritis requiring intravenous fluids. Two patients developed flu-like symptoms with vomiting and diarrhea and also required intravenous fluids for volume depletion. One patient with multiple co-morbidities had four hospitalizations for fluid accumulation due to heart failure, chronic obstructive pulmonary disease, and pneumonia. All of these adverse events occurred while the patients were taking placebo.
Discussion
We hypothesized that CA would improve hepatic TG content in humans based on the report of >50% reduction in hepatic TG content in mice upon feeding 0.5% CA for three weeks 22. CA therapy restored liver morphology to normal with lower levels of unstained inclusions on hematoxylin and eosin staining, and less accumulation of neutral lipids on Oil Red O staining. These effects seem to occur through SHP and liver X-receptor (LXR) mediated down-regulation of sterol regulatory element binding protein 1c (SREBP-1c) expression 22. These findings were recently replicated in rats 25. However, we were unable to observe any significant improvement in hepatic steatosis with CA therapy in lipodystrophic patients with markedly elevated hepatic TG content.
Recent data support the activation of FXR, a nuclear hormone receptor regulated by bile acids, for treatment of hepatic steatosis 26. FXR-deficient mice exhibit a hepatic phenotype similar to steatohepatitis patients with significant hepatic TG accumulation, hepatic inflammation and injury and development of hepatocellular carcinoma 27, 28. WAY-362450, a potent, selective and orally active synthetic FXR agonist has been shown to protect against hepatic steatosis in mice fed a methionine and choline deficient diet 29. This hepatoprotection by WAY-362450 is abolished in FXR-deficient mice, demonstrating the requirement for functional FXR 29.
FXR activation alters the expression of many genes involved in lipid metabolism. While Apolipoprotein C-1, Apolipoprotein C-II, Apolipoprotein CIV, Apolipoprotein E, Fatty Acid Synthase, and Peroxisome Proliferator-Activated Receptor (PPAR) α are upregulated, angiopoietin like protein (ANGPTL) 3, Apolipoprotein A-1, Apolipoprotein C-III, Sterol regulatory element binding protein (SREBP)-1c 22, 30. All these changes may lead to reduction of hepatic steatosis and serum triglycerides upon FXR activation.
Studies in FXR knockout mice show that FXR is involved in the regulation of insulin signaling pathways and appears to have a beneficial role in decreasing insulin resistance and gluconeogenesis, as well as in regulating triglyceride, free fatty acid, and lipid levels 28, 31. This adds to the rationale for clinical testing of cholic acid in subjects with chronic liver disease where insulin resistance is a major risk factor for progression to cirrhosis; e.g., nonalcoholic fatty liver disease (NAFLD) and its more clinically significant subtype nonalcoholic steatohepatitis (NASH) 26.
CA has been reported to reduce serum TG levels in patients with mild hypertriglyceridemia 19, 20. Administration of 15 mg/kg body weight/day of CA for three months decreased plasma TG in six out of eight patients with moderate hypertriglyceridemia. However, plasma TG levels (mean ± SEM) declined by only 16% from 283 ± 27 mg/dL to 239 ± 18 mg/dL (p < 0.05) 20, and in the study subjects with type 2a hyperlipoproteinemia, there was no change in plasma TG levels. Einarsson et al. 19 also reported a 29% reduction in serum TG from 411 ± 180 mg/dL (mean ± SD) to 291± 55 mg/dL after 2 - 3 weeks of therapy with 0.8 to 1.0 g of CA. However, this study involved only five men with hypertriglyceridemia, and the results were not statistically significant. In addition, neither of these studies was a randomized, double blind, placebo-controlled trial. More recently, Woollett et al 32 administered CA (15 mg/kg/day) vs no bile acid supplement (control) to 12 healthy, normotriglyceridemic, subjects who were being fed a controlled diet. They failed to find any lowering of plasma TG (78.5 ± 8.6 vs 73.9 ± 8.6 mg/dL, NS). Interestingly, although most of our patients with lipodystrophies were hypertriglyceridemic, we did not observe any significant lowering of serum TG with CA therapy, as compared to placebo therapy. Our results do not confirm the TG lowering effects of CA that were previously reported.
Although CDCA is a more potent FXR agonist than CA and can also lower serum TG 33-36, CA was chosen over CDCA because of better tolerance and less hepatotoxicity 37. CA has been used, without any side effects, since 1994 at the Cincinnati Children's Hospital Medical Center for the treatment of inborn errors of bile acid metabolism 38. In addition, CA has previously been studied at doses as high as 20 mg/kg/day in adults to dissolve gallstones 39, to treat mild hypertriglyceridemia 19, 20, to study bile acid kinetics 40-43, and cholesterol absorption 32, 44. The only adverse effects reported in these studies were of mild diarrhea. In our study, no major adverse events were attributed to CA, although a few patients complained of diarrhea. Our results confirm that CA is not hepatotoxic and overall well tolerated.
The major strength of this study is the robust study design. In view of the rare prevalence of lipodystrophies, we decided to use the randomized, double-blind, placebo-controlled, crossover design. This allowed us to achieve more power with fewer patients compared to the parallel design. We selected six-month duration of treatment, which is long enough to detect effects of CA therapy on hepatic steatosis. Despite that, no effects of sequence in which the patients received CA or placebo-therapy were observed. We also avoided using low dose of cholic acid and selected to administer 15 mg/kg/day for a maximum of 1500 mg/day.
Several possibilities could explain why CA failed to reduce hepatic TG content as measured by 1H MRS. First, hepatic steatosis in lipodystrophy patients may have different pathophysiology 45 and may not involve down regulation of FXR and thus may not respond to CA therapy. Second, since we did not perform liver biopsies, it is possible that some histological improvements occurred but did not result in an overall reduction of hepatic TG content. Finally, CA may not be a potent enough FXR agonist and other more potent FXR agonists, such as obeticholic acid (Intercept Pharmaceuticals, New York), which is ∼100 times more potent than CDCA, GW4064 (GlaxoSmithKline, North Carolina), MFA-1 (Merck, New Jersey) and fexaramine, may prove to be efficacious in treating hepatic steatosis 46.
In conclusion, CA was well tolerated but did not reduce hepatic TG content in patients with lipodystrophy.
Acknowledgments
We thank Sarah Masood and Rashmin Asher for technical assistance. We thank Claudia Quittner for help in conducting this clinical trial.
Funding: Supported by research grant FD003085 by the Food & Drug Administration and M01-RR00633 and UL1-RR-024982 from the National Institutes of Health, and from the Southwest Medical Foundation.
Footnotes
Reprint requests should be addressed to corresponding author
Declaration of Interest: The authors have nothing to disclose.
References
- 1.Garg A. Acquired and genetic lipodystrophies. N Engl J Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 2.Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine. 2003;82:129–146. doi: 10.1097/00005792-200303000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Cauble MS, Gilroy R, Sorrell MF, Mailliard ME, Sudan DL, Anderson JC, Wisecarver JL, Balakrishnan S, Larsen JL. Lipoatrophic diabetes and end-stage liver disease secondary to nonalcoholic steatohepatitis with recurrence after liver transplantation. Transplantation. 2001;71:892–895. doi: 10.1097/00007890-200104150-00012. [DOI] [PubMed] [Google Scholar]
- 4.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 5.Simha V, Szczepaniak LS, Wagner AJ, DePaoli AM, Garg A. Effect of leptin replacement on intrahepatic and intramyocellular lipid content in patients with generalized lipodystrophy. Diabetes Care. 2003;26:30–35. doi: 10.2337/diacare.26.1.30. [DOI] [PubMed] [Google Scholar]
- 6.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54:1994–2002. doi: 10.2337/diabetes.54.7.1994. [DOI] [PubMed] [Google Scholar]
- 8.Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, Kleiner DE, Gorden P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41:753–760. doi: 10.1002/hep.20672. [DOI] [PubMed] [Google Scholar]
- 9.Simha V, Subramanyam L, Szczepaniak L, Quittner C, Adams-Huet B, Snell P, Garg A. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. 2011;97:785–792. doi: 10.1210/jc.2011-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87:2395. doi: 10.1210/jcem.87.5.8624. [DOI] [PubMed] [Google Scholar]
- 11.Wong SP, Huda M, English P, Bargiotta A, Wilding JP, Johnson A, Corrall R, Pinkney JH. Adipokines and the insulin resistance syndrome in familial partial lipodystrophy caused by a mutation in lamin A/C. Diabetologia. 2005;48:2641–2649. doi: 10.1007/s00125-005-0038-x. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen TA, Sanyal AJ. Pathophysiology guided treatment of nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 2012;27(2):58–64. doi: 10.1111/j.1440-1746.2011.07018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rossle M, Cordes HJ, Zeuzem S, Hein J, Berg T. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–479. doi: 10.1002/hep.23727. [DOI] [PubMed] [Google Scholar]
- 14.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Angelico F, Burattin M, Alessandri C, Del Ben M, Lirussi F. Drugs improving insulin resistance for non-alcoholic fatty liver disease and/or non-alcoholic steatohepatitis. Cochrane Database Syst Rev. 2007:CD005166. doi: 10.1002/14651858.CD005166.pub2. [DOI] [PubMed] [Google Scholar]
- 16.Abdelmalek MF, Sanderson SO, Angulo P, Soldevila-Pico C, Liu C, Peter J, Keach J, Cave M, Chen T, McClain CJ, Lindor KD. Betaine for nonalcoholic fatty liver disease: results of a randomized placebo-controlled trial. Hepatology. 2009;50:1818–1826. doi: 10.1002/hep.23239. [DOI] [PubMed] [Google Scholar]
- 17.Merat S, Malekzadeh R, Sohrabi MR, Sotoudeh M, Rakhshani N, Sohrabpour AA, Naserimoghadam S. Probucol in the treatment of non-alcoholic steatohepatitis: a double-blind randomized controlled study. Journal of Hepatology. 2003;38:414–418. doi: 10.1016/s0168-8278(02)00441-5. [DOI] [PubMed] [Google Scholar]
- 18.Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 19.Einarsson K, Hellstrom K, Kallner M. Effect of cholic acid feeding on bile acid kinetics and neutral fecal steroid excretion in hyperlipoproteinemia (types II and IV) Metabolism. 1974;23:863–873. doi: 10.1016/0026-0495(74)90120-6. [DOI] [PubMed] [Google Scholar]
- 20.Angelin B, Leijd B. Effects of cholic acid on the metabolism of endogenous plasma triglyceride and on biliary lipid composition in hyperlipoproteinemia. J Lipid Res. 1980;21:1–9. [PubMed] [Google Scholar]
- 21.Fujino T, Une M, Imanaka T, Inoue K, Nishimaki-Mogami T. Structure-activity relationship of bile acids and bile acid analogs in regard to FXR activation. J Lipid Res. 2004;45:132–138. doi: 10.1194/jlr.M300215-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, Dobbins RL. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288:E462–468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 25.Gabbi C, Bertolotti M, Anzivino C, Macchioni D, Del Puppo M, Ricchi M, Carubbi F, Tagliafico E, Romagnoli D, Odoardi MR, Loria P, Losi L, Carulli N. Effects of bile duct ligation and cholic acid treatment on fatty liver in two rat models of non-alcoholic fatty liver disease. Dig Liver Dis. 2012 doi: 10.1016/j.dld.2012.07.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 26.Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor _targeting to treat nonalcoholic steatohepatitis. Drug Discov Today. 2012;17:988–997. doi: 10.1016/j.drudis.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Edwards PA. FXR signaling in metabolic disease. FEBS Lett. 2008;582:10–18. doi: 10.1016/j.febslet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 28.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. _targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 30.Kast HR, Nguyen CM, Sinal CJ, Jones SA, Laffitte BA, Reue K, Gonzalez FJ, Willson TM, Edwards PA. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol. 2001;15:1720–1728. doi: 10.1210/mend.15.10.0712. [DOI] [PubMed] [Google Scholar]
- 31.Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, Gonzalez FJ, Fruchart JC, Kuipers F, Staels B. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem. 2005;280:29971–29979. doi: 10.1074/jbc.M501931200. [DOI] [PubMed] [Google Scholar]
- 32.Woollett LA, Buckley DD, Yao L, Jones PJ, Granholm NA, Tolley EA, Tso P, Heubi JE. Cholic acid supplementation enhances cholesterol absorption in humans. Gastroenterology. 2004;126:724–731. doi: 10.1053/j.gastro.2003.11.058. [DOI] [PubMed] [Google Scholar]
- 33.Carulli N, Ponz de Leon M, Podda M, Zuin M, Strata A, Frigerio G, Digrisolo A. Chenodeoxycholic acid and ursodeoxycholic acid effects in endogenous hypertriglyceridemias. A controlled double-blind trial. J Clin Pharmacol. 1981;21:436–442. doi: 10.1002/j.1552-4604.1981.tb01746.x. [DOI] [PubMed] [Google Scholar]
- 34.Camarri E, Marcolongo R, Fici F. Hypotriglyceridemic effect of chenodeoxycholic acid after a short time of administration. Int J Clin Pharmacol Biopharm. 1978;16:527–528. [PubMed] [Google Scholar]
- 35.Camarri E, Fici F, Marcolongo R. Influence of chenodeoxycholic acid on serum triglycerides in patients with primary hypertriglyceridemia. Int J Clin Pharmacol Biopharm. 1978;16:523–526. [PubMed] [Google Scholar]
- 36.Bateson MC, Maclean D, Evans JR, Bouchier IA. Chenodeoxycholic acid therapy for hypertriglyceridaemia in men. Br J Clin Pharmacol. 1978;5:249–254. doi: 10.1111/j.1365-2125.1978.tb01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schoenfield LJ, Lachin JM. Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann Intern Med. 1981;95:257–282. doi: 10.7326/0003-4819-95-3-257. [DOI] [PubMed] [Google Scholar]
- 38.Setchell KD, Heubi JE. Defects in bile acid biosynthesis--diagnosis and treatment. J Pediatr Gastroenterol Nutr. 2006;43(1):S17–22. doi: 10.1097/01.mpg.0000226386.79483.7b. [DOI] [PubMed] [Google Scholar]
- 39.Adler RD, Bennion LJ, Duane WC, Grundy SM. Effects of low dose chenodeoxycholic acid feeding on biliary lipid metabolism. Gastroenterology. 1975;68:326–334. [PubMed] [Google Scholar]
- 40.Thistle JL, Hofmann AF. Efficacy and specificity of chenodeoxycholic acid therapy for dissolving gallstones. N Engl J Med. 1973;289:655–659. doi: 10.1056/NEJM197309272891303. [DOI] [PubMed] [Google Scholar]
- 41.Toouli J, Jablonski P, Watts JM. Gallstone dissolution in man using cholic acid and lecithin. Lancet. 1975;2:1124–1126. doi: 10.1016/s0140-6736(75)91008-9. [DOI] [PubMed] [Google Scholar]
- 42.LaRusso NF, Hoffman NE, Hofmann AF, Northfield TC, Thistle JL. Effect of primary bile acid ingestion on bile acid metabolism and biliary lipid secretion in gallstone patients. Gastroenterology. 1975;69:1301–1314. [PubMed] [Google Scholar]
- 43.Thistle JL, Schoenfield LJ. Induced alterations in composition of bile of persons having cholelithiasis. Gastroenterology. 1971;61:488–496. [PubMed] [Google Scholar]
- 44.Sama C, LaRusso NF. Effect of deoxycholic, chenodeoxycholic, and cholic acids on intestinal absorption of cholesterol in humans. Mayo Clin Proc. 1982;57:44–50. [PubMed] [Google Scholar]
- 45.Agarwal AK, Sukumaran S, Cortes VA, Tunison K, Mizrachi D, Sankella S, Gerard RD, Horton JD, Garg A. Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2(-/-) gene lipodystrophic mice. J Biol Chem. 2011;286:37676–37691. doi: 10.1074/jbc.M111.250449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindor KD. Farnesoid X receptor agonists for primary biliary cirrhosis. Current opinion in gastroenterology. 2011;27:285–288. doi: 10.1097/MOG.0b013e32834452c8. [DOI] [PubMed] [Google Scholar]