

Summary
Rare diseases with a low prevalence are a key public health issue because the causes of those diseases are difficult to determine and those diseases lack a clearly established or curative treatment. Thus, investigating the molecular mechanisms that underlie the pathology of rare diseases and facilitating the development of novel therapies using disease models is crucial. Human induced pluripotent stem cells (iPSCs) are well suited to modeling rare diseases since they have the capacity for self-renewal and pluripotency. In addition, iPSC technology provides a valuable tool to generate patient-specific iPSCs. These cells can be differentiated into cell types that have been affected by a disease. These cells would circumvent ethical concerns and avoid immunological rejection, so they could be used in cell replacement therapy or regenerative medicine. To date, human iPSCs could have been generated from multiple donor sources, such as skin, adipose tissue, and peripheral blood. However, these cells are obtained via invasive procedures. In contrast, several groups of researchers have found that urine may be a better source for producing iPSCs from normal individuals or patients. This review discusses urinary iPSC (UiPSC) as a candidate for modeling rare diseases. Cells obtained from urine have overwhelming advantages compared to other donor sources since they are safely, affordably, and frequently obtained and they are readily obtained from patients. The use of iPSC-based models is also discussed. UiPSCs may prove to be a key means of modeling rare diseases and they may facilitate the treatment of those diseases in the future.
Keywords: iPSCs, “urine cells”, rare disease models
1. Introduction
A rare disease is defined by the World Health Organization (WHO) as a disease with a prevalence of less than 0.65‰–1‰, but it is seriously debilitating even life-threatening (1). Because of its large population, China has the largest rare disease population in terms of prevalence. However, the causes of rare diseases have yet to be identified and they are difficult to manage since there are currently no curative or disease-modified treatments available (2). More than 80% of intractable and rare diseases involve a genetic cause (3). Thus, disease models are indispensable tools for studying molecular mechanisms of rare diseases and developing therapeutic approaches. Such models would need to recreate the phenotypic and pathological variants of the disease in vitro.
Human embryonic stem cells (ESCs) can differentiate into all somatic cells and they can be grown indefinitely in culture. This is why human ESC technology has garnered worldwide attention for its therapeutic applications in vivo (4). However, human ESC treatment is plagued by immune rejection and ethical concerns. Prior to 2006, Japanese researchers induced four transcription factors (OCT3/4, SOX2, c-MYC, and KLF4) in murine fibroblasts with retroviruses and they generated the first induced pluripotent stem cells (iPSCs) (5). Human iPSCs are highly similar to ESCs: they display ESC-like morphology, they express pluripotency markers, the share a similar level of gene expression and epigenetic states (6,7), and they possess the capacity to develop into three germ layers in vitro and in vivo (8). Given these facts, iPSCs could provide a valuable model to study germ cell determination in vitro, but they could also serve as a promising way to differentiate stem cells into diseased cells in order to model disease, screen drugs, and examine the effects of cell therapy. Several groups of researchers have indicated that iPSCs might be derived from urine samples (9,10). Compared to the procedures used to obtain other donor cells, urine collection is safe, affordable, ubiquitous, and readily accepted by patients. This article aims to present the benefits of iPSCs derived from urine for the study of rare diseases, and this article also highlights the applications of rare disease models.
2. Urine as an efficient source of cells for generation of iPSCs
Human iPSCs can be generated from a large variety of donor sources (Table1). However, the ideal cell source would be obtained non-invasively, simple and cost-effective to obtain, and obtained universally (from patients of any age, sex, ethnic group, or body type) (28). The first successful culture of urinary tract cells occurred in 1972 (29), and that feat was repeated by many researchers (30–37). Zhou et al. generated iPSCs from urine-derived cells (9). Here, urine-derived cells will be referred to simply as urine cells. UiPSCs are advantageous in many ways. First, UiPSCs can be generated at a low cost. Urine is merely a body waste, so the only cost is the culture medium. Second, obtaining cell samples from patients, and especially young children with rare diseases, is difficult, but this can be avoided by repeated urine collections without medical assistance. Third, UiPSCs may have fewer genetic alterations than iPSCs from the skin due to less direct exposure to radiation (38,39). Moreover, UiPSCs have a high level of reprogramming efficiency, between 0.1% and 4% in general (40). This is because most urinary cells of an epithelial origin do not require mesenchymal-to-epithelial transition (MET) during reprogramming, unlike fibroblasts.
Table 1. Human iPSCs can be generated from different donor sources.
| Donor sources | Cell types | Obtained process | Ref. |
|---|---|---|---|
| Fibroblasts | Facial dermal fibroblasts, periodontal ligament fibroblasts, and gingival fibroblasts | Invasive (biopsy) | (11,12) |
| Keratinocytes | Keratin-dense epithelial cells | Not strictly noninvasive | (13) |
| Melanoma cells | Skin melanocytes | Invasive (skin punch biopsies) | (14) |
| Adipose stem cells | White preadipocytes and adipose-derived mesenchymal stem cells | Invasive (liposuction) | (15–17) |
| Cord blood | Cord blood-derived stem cells and endothelial cells | Noninvasive | (18,19) |
| Peripheral blood | Mononuclear, T-, and myeloid cells | Minimal invasion (venipuncture) | (20–22) |
| Neural cells | Neural stem cells | Invasive | (23) |
| Astrocytes | Human astrocytes | Invasive | (24) |
| Hepatocytes | Primary human hepatocytes | Invasive | (25) |
| Amniocytes | Human amniotic fluid-derived cells | Invasive (amniocentesis) | (26,27) |
Different types of somatic cells are used to generate iPSCs, and different reprogramming techniques with varying levels of efficiency are used to reprogram cells with different origins (41). Avoiding viral integration and accomplishing safe and efficient reprogramming is the main goal. iPSCs can be generated without viral integration for basic applications (Table 2). Taking safety and workload into account, Xue et al. described a useful method of generating 93 iPS cell lines from 20 individuals with various backgrounds; their method reprogrammed human urine-derived cells without using a virus, serum, feeder, or oncogene c-MYC (56). This non-viral iPSC bank with genetic information from different individuals revealed that UiPSCs could serve as a powerful in vitro model for the study of the disease. As their research continued, they found that different batches of cells derived from the same person's urine had dramatically different levels of reprogramming efficiency. In addition, half of the cells isolated from urine proliferated only to a limited extent, posing a major hurdle to the generation of iPSCs. Thus, Xue et al. developed three methods of reprogramming urine cells with diverse properties through the use of small molecules and autologous urine cells as feeders. iPSCs can be generated from almost any batch of cells isolated from urine, resulting in further advances in the banking of patient-specific iPSCs (57).
Table 2. Human iPSCs can be induced without viral integration.
| Integration-free methods | Cell types | Reprogramming factors* | Efficiency (%) | Ref. |
|---|---|---|---|---|
| Sendai viruses | Fibroblasts | OSKM | ∼ 1 | (42) |
| Adenoviruses | Fibroblasts and liver cells | OSKM | ∼ 0.001 | (43) |
| Plasmids | Fibroblasts | OSNL | ∼ 0.001 | (44,45) |
| Episomes | Fibroblasts | OSNL | ∼ 0.1 | (46) |
| Transposons | Fibroblasts | OSKM | ∼ 0.1 | (47) |
| Proteins | Fibroblasts | OS | ∼ 0.001 | (48,49) |
| Chemicals or small Molecules | Umbilical vein endothelial cells | OCT4 + small molecules | ∼ 0.01 | (50–53) |
| mRNA | Fibroblasts | OSKM or OSKML + VPA | ∼ 1–4.4 | (54) |
| MicroRNA | Adipose stromal cells and dermal fibroblasts | miR-200c, miR-302s or miR-369s | ∼ 0.1 | (55) |
Abbreviations represent a combination of reprogramming factors: K, KLF4; L, LIN28; M, c-MYC; N, NANOG; O, OCT4; S, SOX2; VPA, valproic acid.
3. Modeling rare diseases using UiPSCs
Patient-specific iPSCs can differentiate into large numbers of affected cell types with the same genetic background as the patient since they are immortal and pluripotent (58). Using these cells to construct rare disease models will prove highly useful in the discovery of effective and safe drugs and in the study of cell replacement therapies. Many groups of researchers have successfully generated patient-specific UiPSCs. These cells are derived from patients with conditions such as renal disease (59), pediatric disease (60), a bleeding disorder (61), neurological disease (62), and a bone disorder (63,64). Obtaining these cells is the critical first step to elucidating the mechanisms that underlie the pathology of those diseases.
As UiPSC generation has become more efficient, many patient-specific UiPSCs have been successfully generated. Cryptorchidism is the most frequent congenital anomaly in human males. Cryptorchidism involves multiple causes, such as genetic mutations and environmental factors (65,66). Researchers have established cryptorchid-specific iPSC lines with genetic variations and they have found that urine cells may represent a better source for generation of iPSCs, especially in the study of pediatric diseases (60). Most patients with complex disorders are treated with various medications over a prolonged period, which may have an impact on donor cell propagation and nuclear reprogramming. Expansion of fibroblasts from patients with end-stage renal disease was problematic since cell division stopped after several passages (67). In contrast, use of immunosuppressive drugs has little effect on urine cell propagation and generation of iPSCs. Thus, urine-derived cells appeared to be a valuable donor source for patients with systemic lupus erythematosus (SLE) (59). Whether these drugs substantially impact the generation of iPSCs needs to be explored further under different conditions.
Although cells of multiple origins may be involved in the pathogenesis of rare diseases, specific cell types could be used to recreate a phenocopy of the disease in vitro so that the cell type could be identified as where the disease originates. Investigating a rare disorder presents many challenges, but UiPSCs represent a world of potential applications – for recreating the phenotypic and pathological variants and also for identifying drug candidates and transplanting autologous cells into patients. Two studies have posited that new treatments for rare diseases can be developed in the future using UiPSCs-based models with integration-free episomal vectors (61,64).
The first study derived iPSCs from patients with hemophilia A (HA), which is a rare bleeding disorder with a prevalence of 1‰–2‰ caused by clotting factor VIII (FVIII) mutations resulting in deficient production of FVIII protein (68,69). HA-iPSCs are generated from patients' urine cells using an integration-free transfection technique (46,70). Differentiated hepatocyte-like cells derived from HA-iPSCs (HA-iPSC-Hep) display the phenotype of the defective FVIII found in selected patients. At the genetic level, HA-iPSC-Hep carry the mRNA of the defective FVIII gene. FVIII protein is absent on a protein level as well. Furthermore, FVIII activity in the culture supernatant is much lower than that in the reference group. Therefore, this new model is remarkable for two main reasons. Previous researchers investigated the mechanisms underlying HA in animal models. However, different species have differing physiologies, which may partly explain why many novel drugs are not effective in patients when tested in clinical trials (71). Models like HA-iPSC-Hep will help to explain the pathogenesis of disease and facilitate the development of new therapeutics. In patients with a bleeding disorder, an invasive procedure can be life-threatening, so an easily accessible source of donor cells must be obtained in a completely non-invasive manner. Urine cells can be obtained non-invasively and safely, so those cells are a useful source of iPSCs. A novel model using integration-free episomal vectors could be used in cell therapy along with gene editing.
The second study generated iPSCs from patients with fibrodysplasia ossificans progressiva (FOP). FOP is caused by a recurrent heterozygous missense mutation in activin receptor-like kinase 2 (ACVR1/ALK2) (72). FOP is a rare genetic disease characterized by congenital malformation of the halluces and by progressive heterotopic ossification (HO), and FOP is the most catastrophic disorder of HO in humans (73). Endothelial cells (ECs) and pericytes derived from FOP iPSCs can recreate some aspects of the disease phenotype in vitro. Researchers found that ECs were not readily generated from FOP iPSCs and had limited proliferation, which may be related to the crosstalk between bone morphogenetic protein (BMP) signaling and vascular endothelial growth factor (VEGF) signaling. This issue needs to be further explored. Researchers have also found that FOP iPSC-pericytes are prone to mineralization. Moreover, use of ALK2 inhibitor led to a reduction in alkaline phosphatase (ALP) activity. Models such as FOP iPSCs provide a useful tool to determine the underlying mechanisms of endothelial-mesenchymal transition (EndMT), to explore the bioactivity of ALK2 inhibitors, and to screen drugs (64).
The variability between iPSC clones and the intrinsic epigenetic features of donor cells may have an impact on the properties of iPSCs. Reliable control iPSC clones could be generated via gene editing to eliminate any potential confounding effects of variations in genetic background. In other words, isogenic iPSCs may differ only at specific loci while all other locations remain identical (74,75).
4. Applications of iPSC-based models
These two models described indicate that UiPSCs have opened new avenues for modeling rare disorders and they offer proof of principle for basic biological research in the short term. In the long term, the clinical use of UiPSCs in drug discovery and cell replacement therapies will also receive a great deal of attention. With reliable disease models, researchers will be able to dissect the pathophysiological basis of rare diseases and screen drugs in affected cell types. Furthermore, these models will advance the field a step closer to patient-matched cells or tissues for clinical transplantation, which may represent the ultimate goal of stem cell therapy.
4.1. Disease pathogenesis and drug discovery
Many rare human diseases are still poorly understood, with a complicated pathogenesis and multiple causes. Patient-specific iPSC models can recreate the phenotypic and pathological variants of the disease in vitro and can then be used to identify drugs to rescue these phenotypes. Thus, disease models offer an unprecedented opportunity to understand underlying mechanisms of disease and to develop therapeutic candidates.
Some genetic mutations underlying human diseases may affect the generation of patient-specific iPSCs or the maintenance of their pluripotent state (76,77). Nonetheless, iPSCs provide a unique human system for understanding molecular mechanisms of reprogramming and pathogenesis and can thus be used to develop effective drugs. Researchers have recently identified two molecular mechanisms responsible for inhibition of the generation of FOP-iPSCs from patient fibroblasts: incomplete reprogramming of pluripotent and fibroblastic genes and the forced differentiation of cells during and after reprogramming (78). This inhibition was due to the constitutive activation of ALK2 and was mostly overcome by specific suppression of ALK2 expression. While screening a library of chemical substances, the researchers identified a new ALK2 inhibitor candidate to restore generation of FOP-iPSCs. As mentioned earlier, ALP activity was reduced by use of an ALK inhibitor, and this may represent a potential treatment for FOP. Two mechanisms were involved in the pathology of FOP: ligand-independent BMP signaling and ligand-dependent hyper-responsiveness to BMP stimulation. A landmark study by Toguchida's research group generated induced mesenchymal stromal cells (iMSCs) from FOP-iPSCs that, when implanted into immunodeficient mice with Activin-A-expressing cells, induced bone and cartilage formation in vivo (79). In vitro treatment with TGF-β and BMP inhibitors eliminated increased chondrogenesis in FOP-iMSCs. In addition to the two molecular mechanisms that were involved in the pathology of FOP, Toguchida's research group identified a novel third mechanism: Activin-A activates TGF-β and aberrant BMP signaling that results in increased chondrogenesis in FOP-iMSCs. Based on the role and mechanism of action of Activin-A in HO, Activin-A triggers increased chondrogenesis in FOP-iMSCs, but this action can be inhibited by several Activin-A inhibitors. Therefore, Activin-A inhibitors could be a novel therapy for patients with FOP.
Taken together, these findings provide proof of the principle that new effective treatments for FOP can be discovered. In addition, iPSC-based models can be useful in identifying novel drug compounds and in testing drug toxicity and responsiveness (80).
4.2. Cell therapy
With the recent development of gene editing tools, the idea of patient-personalized therapies and replacement of diseased or damaged organ tissues has come closer to becoming a clinical reality. In the two models mentioned thus far, iPSCs were generated from urine cells using interation-free episomal vectors, so the two models could be used in cell therapy in combination with gene editing. A mutation causing a disease could be corrected in patient-specific iPSCs.
Approximately half of the severe cases of HA are caused by chromosomal inversions in the portion of the FVIII gene. Given this fact, Park et al. used a transcription activator-like effector nuclease (TALEN) to revert the inverted 140-kbp segment in an iPSC-based model of hemophilia (81). Endothelial cells derived from FVIII-corrected iPSCs produced FVIII protein in vitro. Interestingly, the iPSC-based model of hemophilia was created using the same TALEN pair that induced chromosomal inversions affecting the FVIII gene in wild-type iPSCs. This approach could be used in autologous stem cell therapy and to provide a method of disease modeling. However, the challenge is to invert a much larger region that is eight times more prevalent than aforementioned 140-kbp inversion. Park et al. successfully used the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) system to correct these regions in patient-specific iPSCs (82). Combining these gene editing tools with iPSC technology would provide a source of endothelial progenitor cells for transplantation therapies.
A recent study suggested that a one-step procedure can generate gene-corrected ALK2-iPSCs, and the advantages of this approach are that it saves time, labor, and money (83). Activated BMP signaling by an FOP R206H mutation adversely affects the generation of FOP iPSCs since BMPs can induce differentiation of human ESCs (84). Previous studies indicated that iPSCs from FOP-derived skin fibroblasts were generated inefficiently and were also unable to maintain their pluripotent state (85). Therefore, a one-step strategy to concurrently perform reprogramming and gene correction in the CRISPR/Cas9 system would circumvent the iPSC stage. Matsumoto et al. successfully established gene-corrected FOP-iPSCs through the use of bacterial artificial chromosome (BAC)-based homologous recombination (86).
Chronic granulomatous disease (CGD) is a rare genetic disease with a single point mutation (T > G) at the end of intron 1 of CYBB in most patients (87). iPSCs were generated from a patient with mutant CYBB, and the mutations were corrected using a CRISPR-Cas9 site-specific nuclease system (88). Phagocytes derived from these CYBB-corrected iPSCs restored the oxidative burst.
Thus, a combination of iPSC technology and gene editing could provide an autologous source of corrected iPSC-derived cells for transplantation therapies. Natronobacterium gregoryi Argonaute (NgAgo) is the newest tool with which to edit the genes of human cells (89). No studies have used this tool to model disease in iPSCs thus far, but it has potential advantages over Cas9 such as lower off-_targeting levels. Further research is needed to prove that NgAgo can be used effectively with iPSC technology in cell replacement therapy and to expand the clinical use of UiPSCs to model rare diseases.
5. Advantages of and barriers to the study of rare diseases using UiPSCs
Urine is merely a body waste, but it can reproducibly be used as an efficient source of cells with which to generate human iPSCs. A point worth noting is that the isolation of urine cells is completely non-invasive and is readily accepted by patients, and especially young children. In addition, researchers have used a non-viral episome system to compare the reprogramming efficiency of fibroblasts and epithelial cells from foreskin and urine. iPSCs derived from urine were generated with an efficiency of approximately 1.5%, which is two orders of magnitude higher than the efficiency with which fibroblasts were generated (0.01%) (90). This significant difference may be due to the fact that epithelial cells do not have to undergo the MET that fibroblasts must undergo. Nonetheless, researchers have contended that human iPSCs may have remnants of epigenetic memory of donor origin, resulting in biased differentiation towards lineages related to the donor cell type (91,92). iPSCs derived from fibroblasts favor differentiation towards the mesodermal lineage, but an interesting fact is that such a tendency has not been found in UiPSCs.
Rare diseases can profoundly affect humans, so the study of those diseases has garnered worldwide attention. Nonetheless, the causes of those diseases are difficult to determine and those diseases are difficult to diagnose and treatment. The presence of additional complications also increases the difficulty of their treatment. Therefore, a human model of these complex and varying conditions may serve as a useful platform for understanding disease pathogenesis and for developing new therapeutics. Because iPSCs resemble ESCs in terms of pluripotency and the potential for self-renewal, patient-specific iPSCs could represent a promising strategy for investigating the molecular mechanisms of disease and discovery of drugs to treat them (93,94). Recent studies have reported that generating UiPSCs from patients with rare diseases would provide insight into the mechanisms of those diseases (59–64). The availability of limitless amounts of diseased cells from patients with a rare disease could, along with gene editing, prove useful in clinical practice (Figure 1). These cells represent a potential boon to regenerative medicine: cells from a patient's urine sample can be reprogrammed into iPSCs and then differentiated into diseased cells or tissues needed to treat the disease. This customized therapy would overcome the hurdles – immune rejection and ethical concerns – faced by cells derived from embryos. Researchers have indicated that UiPSCs can be used for further regenerative therapies. Urine cells have been used to generate iPSCs in a non-viral episome system and those iPSCs have been differentiated into epithelial sheets. Those sheets developed into ameloblasts in a tooth-like structure with a success rate of up to 30% (95). Moreover, this approach demonstrated that human UiPSCs could be an ideal source for the regeneration of patient-specific teeth.
Figure 1.
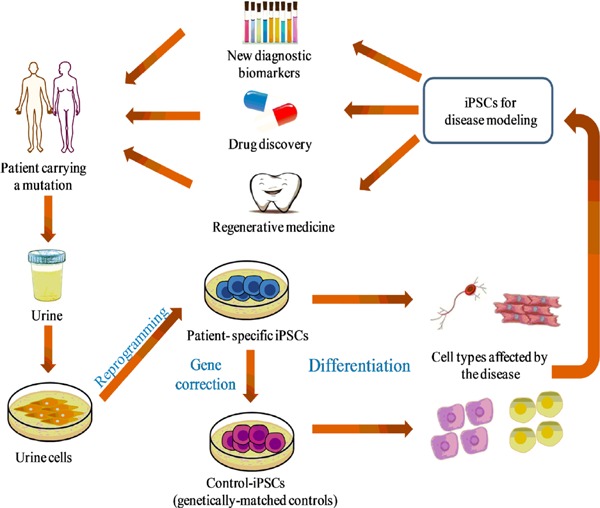
Creation of models and applications of UiPSCs in the study of rare diseases. A diagram showing the creation of models of specific rare diseases using UiPSCs and their applications. Cells are isolated from patient urine samples and reprogrammed into iPSCs. Isogenic controls are created with gene-editing tools and then differentiated into diseased cells that can recreate crucial aspects of the disease in vitro. Patient-specific disease models can be used to identify new diagnostic biomarkers and to screen effective and novel drugs as well as to replace cells or tissues in regenerative medicine.
Clinical studies of rare diseases face severe challenges since patients are often geographically dispersed or are not readily available. One solution to this problem is to use gene editing to introduce mutations associated with a disease into iPSCs from healthy donors. A recent study efficiently introduced specific point mutations with CRISPR/Cas9 and it edited just one copy of a gene, rather than both copies (96). Another crucial point in disease modeling is what to use as a control. In many cases, researchers compare a patient's iPSCs with those from a healthy individual. However, the cells behave too differently in culture because of differences in the genomic background or gene expression. Researchers have turned to a refined method of gene editing – gene _targeting – to produce genetically-matched controls.
There are several issues that need to be addressed in more detail before UiPSCs can be successfully used in clinical practice, but the creation of faithful disease models will aid in further clinical use of UiPSCs. The generation of UiPSCs is universal except in some rare cases, such as renal failure or cancer requiring a cystectomy (40). Some genetic mutations underlying human diseases could affect the generation of patient-specific iPSCs or the maintenance their pluripotent state. Further study is needed to determine what effect these mutations have. In addition, several iPSC clones generated from the same patient's cells should be assessed for consistent functional readout in order to minimize the risk of acquired genetic (or epigenetic) abnormalities. Moreover, differentiation protocols are inefficient and produce only impaired and/or heterogeneous cell populations in many cases. The challenge is to obtain mature cell types. Furthermore, a wide range of growth factors and specific culture conditions need to be used to differentiate iPSCs. Clinical trials must first thoroughly assess whether the use of different molecules is safe (97). Given that undifferentiated iPSCs can cause teratomas in vivo, the crucial step is to remove residual undifferentiated cells before transplantation. Therefore, researchers are now working to ensure the identity and safety of cell lines in terms of genomic variability, patterns of gene expression, and the aspects mentioned earlier. One way to resolve these issues is to devise a set of clinical-grade practices of quality control for the banking of allogeneic iPSCs. These practices would ensure that donor cells in large-scale collections are immunologically compatible and they would provide a foundation for the treatment of rare diseases. Banking of iPSCs would have numerous advantages for patients, no matter when or where those cells are needed.
6. Conclusion
In summary, the use of patient-specific UiPSCs offers a novel strategy for modeling disease, and this approach has already demonstrated its potential to provide a better understanding of the molecular mechanisms of disease. Some exemplary applications mentioned here can be expanded to study countless other rare diseases using UiPSCs. As generation of patient-specific UiPSCs becomes more routine and more scalable, the models they allow will serve as a useful tool to determine the mechanisms of disease, to identify drug candidates, and to develop personalized regenerative medicine. Further study is needed to develop therapeutic applications of UiPSC technology for treatment of rare diseases. Further technical improvements, particularly in generation and differentiation of patient-specific UiPSCs and increased banking of allogeneic iPSCs, will further advance the field. That said, extreme caution is required when considering use of UiPSC technology in clinical practice. Many challenges still need to be overcome and a great deal more knowledge is needed before UiPSC technology can become a routine clinical approach to the treatment of rare diseases.
Acknowledgements
This work was supported by the National Science & Technology Support Program during the Twelve Five-Year Plan Period from Ministry of Science and Technology of the People's Republic of China (2013BAI07B01, 2013BAI07B02) and the Taishan Scholar Programme of Shandong Province.
References
- 1. Song P, Gao J, Inagaki Y, Kokudo N, Tang W. Intractable and rare diseases research in Asia. Biosci Trends. 2012; 6:48-51. [PubMed] [Google Scholar]
- 2. Cui Y, Han J. A proposed definition of rare diseases for China: From the perspective of return on investment in new orphan drugs. Orphanet J Rare Dis. 2015; 10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wuhl E, van Stralen KJ, Wanner C, Ariceta G, Heaf JG, Bjerre AK, Palsson R, Duneau G, Hoitsma AJ, Ravani P, Schaefer F, Jager KJ. Renal replacement therapy for rare diseases affecting the kidney: An analysis of the ERA-EDTA Registry. Nephrol Dial Transplantat. 2014; 29:iv1-iv8. [DOI] [PubMed] [Google Scholar]
- 4. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282:1145-1147. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663-676. [DOI] [PubMed] [Google Scholar]
- 6. Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, Plath K, Hochedlinger K. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007; 1:55-70. [DOI] [PubMed] [Google Scholar]
- 7. Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008; 454:49-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318:1917-1920. [DOI] [PubMed] [Google Scholar]
- 9. Zhou T, Benda C, Dunzinger S, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012; 7:2080-2089. [DOI] [PubMed] [Google Scholar]
- 10. Song B, Niclis JC, Alikhan MA, Sakkal S, Sylvain A, Kerr PG, Laslett AL, Bernard CA, Ricardo SD. Generation of induced pluripotent stem cells from human kidney mesangial cells. J Am Soc Nephrol. 2011; 22:1213-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861-872. [DOI] [PubMed] [Google Scholar]
- 12. Wada N, Wang B, Lin NH, Laslett AL, Gronthos S, Bartold PM. Induced pluripotent stem cell lines derived from human gingival fibroblasts and periodontal ligament fibroblasts. J Periodontal Res. 2011; 46:438-447. [DOI] [PubMed] [Google Scholar]
- 13. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, Edel M, Boue S, Izpisua Belmonte JC. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008; 26:1276-1284. [DOI] [PubMed] [Google Scholar]
- 14. Utikal J, Maherali N, Kulalert W, Hochedlinger K. SOX2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci. 2009; 122:3502-3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun N, Panetta NJ, Gupta DM, Wilson KD, Lee A, Jia F, Hu S, Cherry AM, Robbins RC, Longaker MT, Wu JC. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A. 2009; 106:15720-15725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sugii S, Kida Y, Kawamura T, Suzuki J, Vassena R, Yin YQ, Lutz MK, Berggren WT, Izpisua Belmonte JC, Evans RM. Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc Natl Acad Sci U S A. 2010; 107:3558-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aoki T, Ohnishi H, Oda Y, Tadokoro M, Sasao M, Kato H, Hattori K, Ohgushi H. Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC. Tissue Eng Part A. 2010; 16:2197-2206. [DOI] [PubMed] [Google Scholar]
- 18. Giorgetti A, Montserrat N, Aasen T, Gonzalez F, Rodriguez-Piza I, Vassena R, Raya A, Boue S, Barrero MJ, Corbella BA, Torrabadella M, Veiga A, Izpisua Belmonte JC. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 2009; 5:353-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haase A, Olmer R, Schwanke K, et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009; 5:434-441. [DOI] [PubMed] [Google Scholar]
- 20. Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010; 7:20-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loh YH, Hartung O, Li H, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010; 7:15-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seki T, Yuasa S, Fukuda K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat Protoc. 2012; 7:718-728. [DOI] [PubMed] [Google Scholar]
- 23. Kim JB, Greber B, Arauzo-Bravo MJ, Meyer J, Park KI, Zaehres H, Scholer HR. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009; 461:649-643. [DOI] [PubMed] [Google Scholar]
- 24. Ruiz S, Brennand K, Panopoulos AD, Herrerias A, Gage FH, Izpisua-Belmonte JC. High-efficient generation of induced pluripotent stem cells from human astrocytes. PLoS One. 2010; 5:e15526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010; 51:1810-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cai J, Li W, Su H, et al. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J Biol Chem. 2010; 285:11227-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li C, Zhou J, Shi G, Ma Y, Yang Y, Gu J, Yu H, Jin S, Wei Z, Chen F, Jin Y. Pluripotency can be rapidly and efficiently induced in human amniotic fluid-derived cells. Hum Mol Genet. 2009; 18:4340-4349. [DOI] [PubMed] [Google Scholar]
- 28. Zhou T, Benda C, Duzinger S, et al. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011; 22:1221-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sutherland GR, Bain AD. Culture of cells from the urine of newborn children. Nature. 1972; 239:231. [DOI] [PubMed] [Google Scholar]
- 30. Linder D. Culture of cells from the urine and bladder washings of adults. Somatic Cell Genet. 1976; 2:281-283. [DOI] [PubMed] [Google Scholar]
- 31. Herz F, Schermer A, Koss LG. Short-term culture of epithelial cells from urine of adults. Proc Soc Exp Biol Med. 1979; 161:153-157. [DOI] [PubMed] [Google Scholar]
- 32. Felix JS, Littlefield JW. Urinary tract epithelial cells cultured from human urine. Int Rev Cytol Suppl. 1979;11-23. [DOI] [PubMed] [Google Scholar]
- 33. Herz F. Culture of urinary cells. Birth Defects Orig Artic Ser. 1980; 16:85-93. [PubMed] [Google Scholar]
- 34. Felix JS, Littlefield JW. Human newborn urine as a source of epithelial cells. Birth Defects Orig Artic Ser. 1980; 16:231-237. [PubMed] [Google Scholar]
- 35. Felix JS, Sun TT, Littlefield JW. Human epithelial cells cultured from urine: Growth properties and keratin staining. In Vitro. 1980; 16:866-874. [DOI] [PubMed] [Google Scholar]
- 36. Dorrenhaus A, Muller JI, Golka K, Jedrusik P, Schulze H, Follmann W. Cultures of exfoliated epithelial cells from different locations of the human urinary tract and the renal tubular system. Arch Toxicol. 2000; 74:618-626. [DOI] [PubMed] [Google Scholar]
- 37. Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005; 54:3427-3434. [DOI] [PubMed] [Google Scholar]
- 38. Gore A, Li Z, Fung HL, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011; 471:63-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hussein SM, Batada NN, Vuoristo S, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011; 471:58-62. [DOI] [PubMed] [Google Scholar]
- 40. Benda C, Zhou T, Wang X, Tian W, Grillari J, Tse H-F, Grillari-Voglauer R, Pei D, Esteban MA. Urine as a Source of Stem Cells. Adv Biochem Eng Biotechnol. 2013; 129:19-32. [DOI] [PubMed] [Google Scholar]
- 41. Gonzalez F, Boue S, Izpisua Belmonte JC. Methods for making induced pluripotent stem cells: Reprogramming a la carte. Nat Rev Genet. 2011; 12:231-242. [DOI] [PubMed] [Google Scholar]
- 42. Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009; 85:348-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008; 322:945-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Si-Tayeb K, Noto FK, Sepac A, Sedlic F, Bosnjak ZJ, Lough JW, Duncan SA. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol. 2010; 10:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008; 322:949-953. [DOI] [PubMed] [Google Scholar]
- 46. Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009; 324:797-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hamalainen R, Cowling R, Wang W, Liu P, Gertsenstein M, Kaji K, Sung HK, Nagy A. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009; 458:766-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Scholer HR, Duan L, Ding S. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009; 4:381-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009; 4:472-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ichida JK, Blanchard J, Lam K, et al. A small-molecule inhibitor of tgf-Beta signaling replaces SOX2 in reprogramming by inducing NANOG. Cell Stem Cell. 2009; 5:491-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu S, Li W, Zhou H, Wei W, Ambasudhan R, Lin T, Kim J, Zhang K, Ding S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell. 2010; 7:651-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008; 26:795-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lyssiotis CA, Foreman RK, Staerk J, et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of KLF4. Proc Natl Acad Sci U S A. 2009; 106:8912-8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010; 7:618-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Miyoshi N, Ishii H, Nagano H, et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell. 2011; 8:633-638. [DOI] [PubMed] [Google Scholar]
- 56. Xue Y, Cai X, Wang L, Liao B, Zhang H, Shan Y, Chen Q, Zhou T, Li X, Hou J, Chen S, Luo R, Qin D, Pei D, Pan G. Generating a non-integrating human induced pluripotent stem cell bank from urine-derived cells. PLoS One. 2013; 8:e70573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li D, Wang L, Hou J, et al. Optimized approaches for generation of integration-free iPSCs from human urine-derived cells with small molecules and autologous feeder. Stem Cell Reports. 2016; 6:717-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sterneckert JL, Reinhardt P, Scholer HR. Investigating human disease using stem cell models. Nat Rev Genet. 2014; 15:625-639. [DOI] [PubMed] [Google Scholar]
- 59. Chen Y, Luo R, Xu Y, Cai X, Li W, Tan K, Huang J, Dai Y. Generation of systemic lupus erythematosus-specific induced pluripotent stem cells from urine. Rheumatol Int. 2013; 33:2127-2134. [DOI] [PubMed] [Google Scholar]
- 60. Zhou J, Wang X, Zhang S, Gu Y, Yu L, Wu J, Gao T, Chen F. Generation and characterization of human cryptorchid-specific induced pluripotent stem cells from urine. Stem Cells Dev. 2013; 22:717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jia B, Chen S, Zhao Z, et al. Modeling of hemophilia A using patient-specific induced pluripotent stem cells derived from urine cells. Life Sci. 2014; 108:22-29. [DOI] [PubMed] [Google Scholar]
- 62. Zhang SZ, Li HF, Ma LX, Qian WJ, Wang ZF, Wu ZY. Urine-derived induced pluripotent stem cells as a modeling tool for paroxysmal kinesigenic dyskinesia. Biol Open. 2015; 4:1744-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hildebrand L, Rossbach B, Kühnen P, Gossen M, Kurtz A, Reinke P, Seemann P, Stachelscheid H. Generation of integration free induced pluripotent stem cells from fibrodysplasia ossificans progressiva (FOP) patients from urine samples. Stem Cell Res. 2016; 16:54-58. [DOI] [PubMed] [Google Scholar]
- 64. Cai J, Orlova Valeria V, Cai X, Eekhoff Elisabeth MW, Zhang K, Pei D, Pan G, Mummery Christine L, ten Dijke P. Induced pluripotent stem cells to model human fibrodysplasia ossificans progressiva. Stem Cell Reports. 2015; 5:963-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Foresta C, Zuccarello D, Garolla A, Ferlin A. Role of hormones, genes, and environment in human cryptorchidism. Endocr Rev. 2008; 29:560-580. [DOI] [PubMed] [Google Scholar]
- 66. Bay K, Main KM, Toppari J, Skakkebaek NE. Testicular descent: INSL3, testosterone, genes and the intrauterine milieu. Nat Rev Urol. 2011; 8:187-196. [DOI] [PubMed] [Google Scholar]
- 67. Thatava T, Armstrong AS, De Lamo JG, Edukulla R, Khan YK, Sakuma T, Ohmine S, Sundsbak JL, Harris PC, Kudva YC, Ikeda Y. Successful disease-specific induced pluripotent stem cell generation from patients with kidney transplantation. Stem Cell Res Ther. 2011; 2:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roth DA, Tawa NE, Jr., O'Brien JM, Treco DA, Selden RF. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med. 2001; 344:1735-1742. [DOI] [PubMed] [Google Scholar]
- 69. Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003; 361:1801-1809. [DOI] [PubMed] [Google Scholar]
- 70. Liao B, Bao X, Liu L, et al. MicroRNA cluster 302-367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J Biol Chem. 2011; 286:17359-17364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Saha K, Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009; 5:584-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Billings PC, Fiori JL, Bentwood JL, O'Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J Bone Miner Res. 2008; 23:305-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: Mechanisms and models of skeletal metamorphosis. Dis Model Mech. 2012; 5:756-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim HS, Bernitz JM, Lee DF, Lemischka IR. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014; 23:2673-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sterneckert JL, Reinhardt P, Schöler HR. Investigating human disease using stem cell models. Nat Rev Genet. 2014; 15:625-639. [DOI] [PubMed] [Google Scholar]
- 76. Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nature. 2009; 460:1132-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, Blasco MA, Serrano M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009; 460:1136-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hamasaki M, Hashizume Y, Yamada Y, Katayama T, Hohjoh H, Fusaki N, Nakashima Y, Furuya H, Haga N, Takami Y, Era T. Pathogenic mutation of ALK2 inhibits induced pluripotent stem cell reprogramming and maintenance: Mechanisms of reprogramming and strategy for drug identification. Stem Cells. 2012; 30:2437-2449. [DOI] [PubMed] [Google Scholar]
- 79. Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, Toguchida J. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A. 2015; 112:15438-15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC, Wu JC. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013; 127:1677-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Park CY, Kim J, Kweon J, Son JS, Lee JS, Yoo JE, Cho SR, Kim JH, Kim JS, Kim DW. _targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc Natl Acad Sci U S A. 2014; 111:9253-9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Park CY, Kim Duk H, Son Jeong S, Sung Jin J, Lee J, Bae S, Kim JH, Kim DW, Kim JS. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell. 2015; 17:213-220. [DOI] [PubMed] [Google Scholar]
- 83. Kim BY, Jeong S, Lee SY, Lee SM, Gweon EJ, Ahn H, Kim J, Chung SK. Concurrent progress of reprogramming and gene correction to overcome therapeutic limitation of mutant ALK2-iPSC. Exp Mol Med. 2016; 48:e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xu RH, Peck RM, Li DS, Feng X, Ludwig T, Thomson JA. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods. 2005; 2:185-190. [DOI] [PubMed] [Google Scholar]
- 85. Barruet E, Hsiao EC. Using human induced pluripotent stem cells to model skeletal diseases. Methods Mol Biol. 2016; 1353:101-118. [DOI] [PubMed] [Google Scholar]
- 86. Matsumoto Y, Ikeya M, Hino K, Horigome K, Fukuta M, Watanabe M, Nagata S, Yamamoto T, Otsuka T, Toguchida J. New protocol to optimize iPS cells for genome analysis of fibrodysplasia ossificans progressiva. Stem Cells. 2015; 33:1730-1742. [DOI] [PubMed] [Google Scholar]
- 87. Jiang Y, Cowley SA, Siler U, Melguizo D, Tilgner K, Browne C, Dewilton A, Przyborski S, Saretzki G, James WS, Seger RA, Reichenbach J, Lako M, Armstrong L. Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells. 2012; 30:599-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Flynn R, Grundmann A, Renz P, Hänseler W, James WS, Cowley SA, Moore MD. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol. 2015; 43:838-848.e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gao F, Shen XZ, Jiang F, Wu Y, Han C. DNA-guided genome editing using the Natronobacterium gregoryi Argonaute. Nat Biotechnol. 2016; 34:768-773. [DOI] [PubMed] [Google Scholar]
- 90. Drozd AM, Walczak MP, Piaskowski S, Stoczynska-Fidelus E, Rieske P, Grzela DP. Generation of human iPSCs from cells of fibroblastic and epithelial origin by means of the oriP/EBNA-1 episomal reprogramming system. Stem Cell Res Ther. 2015; 6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Huo H, Loh YH, Aryee MJ, Lensch MW, Li H, Collins JJ, Feinberg AP, Daley GQ. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011; 29:1117-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010; 467:285-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008; 134:877-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kim C. Disease modeling and cell based therapy with iPSC: Future therapeutic option with fast and safe application. Blood Res. 2014; 49:7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cai J, Zhang Y, Liu P, et al. Generation of tooth-like structures from integration-free human urine induced pluripotent stem cells. Cell Regen (Lond). 2013; 2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016; 533:125-129. [DOI] [PubMed] [Google Scholar]
- 97. Bilic J, Izpisua Belmonte JC. Concise review: Induced pluripotent stem cells versus embryonic stem cells: Close enough or yet too far apart? Stem Cells. 2012; 30:33-41. [DOI] [PubMed] [Google Scholar]