
Abstract
The ability to maintain a functional proteome, or proteostasis, declines during the ageing process. Damaged and misfolded proteins accumulate with age, impairing cell function and tissue homeostasis. The accumulation of damaged proteins contributes to multiple age-related diseases such as Alzheimer’s, Parkinson’s or Huntington’s disease. Damaged proteins are degraded by the ubiquitin–proteasome system or through autophagy-lysosome, key components of the proteostasis network. Modulation of either proteasome activity or autophagic-lysosomal potential extends lifespan and protects organisms from symptoms associated with proteostasis disorders, suggesting that protein clearance mechanisms are directly linked to ageing and age-associated diseases.
Similar content being viewed by others
Introduction
The integrity of the proteome, or proteostasis, is challenged during the ageing process. Damaged proteins accumulate as a consequence of ageing and may ensue from the accumulation of reactive oxygen species and a progressive decline in the ability to maintain a functional proteome1. This demise in proteostasis is considered one of the hallmarks of ageing1 and contributes to multiple age-related diseases such as Alzheimer’s (AD)2, Parkinson’s (PD)3 or Huntington’s disease (HD)4. Proteostasis is maintained by a network of cellular mechanisms that monitors folding, concentration, cellular localization and interactions of proteins from their synthesis through their degradation5. Chaperones assure the proper folding of proteins throughout their life cycle and under stress conditions but their activity declines with age (reviewed in refs 6, 7, 8, 9, 10). When the activity of chaperones is insufficient to maintain proteome stability, misfolded proteins may accumulate and impair cell function and tissue homeostasis. Damaged, misfolded, aggregated or unnecessary proteins are scavenged by the proteasome or through autophagy-lysosome11.
The ubiquitin–proteasome system (UPS) is a complex mechanism where proteins are first _targeted for degradation by the ubiquitination machinery and then recognized, unfolded and proteolyzed by the proteasome12. In autophagy-mediated proteolysis, proteins are degraded by the lysosome. Although lysosomal proteolysis was initially considered to be a non-selective system, it has been shown that chaperones and other cargo-recognition molecules such as ubiquitins determine the degradation of specific proteins by the lysosome11,13,14. Therefore, the proteasome and autophagy might be interrelated by using ubiquitin as a common marker for proteolytic degradation11,13,14. Both proteasome functionality and autophagic-lysosomal potential decline during the ageing process and this failure contributes to the development of age-related diseases11,15.
Here we discuss the mechanistic links between protein clearance mechanisms, ageing and neurodegenerative diseases. We further describe how longevity-promoting pathways modulate the proteostasis network, providing increased stability of the proteome and protecting from the symptoms associated with neurodegenerative diseases.
Mechanisms of protein degradation
In this section, we review the molecular mechanisms underlying the two main protein clearance mechanisms of the proteostasis network.
The proteasome
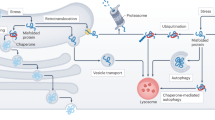
The UPS is the primary selective proteolytic system in eukaryotic cells15, a precise mechanism that maintains the proper concentration of many regulatory proteins involved in multiple biological processes (Fig. 1). In addition, the UPS is a key component of the proteostasis network to terminate damaged proteins11. After sequential conjugation of ubiquitins, proteins are recognized by the proteasome, which is responsible for their degradation. The proteasome is formed by the assembly of several subunits12 (Fig. 1). The core particle (20S) of the proteasome exhibits a barrel-like structure in which the 28 subunits are assembled into four seven-membered rings12. The two outer rings are composed by seven α-subunits (named α1 to α7), while the two inner rings are composed by seven β-subunits (β1-β7)12. β-rings contain the proteolytic active sites: β1, β2 and β5 present caspase-like, trypsin-like and chymotrypsin-like activities, respectively12. A specialized form of the 20S proteasome, known as the immunoproteasome because of its prevalence in antigen-presenting cells, is generated by replacement of the catalytic subunits by β1i, β2i and β5i (ref. 16). β5t, a homologue of β5, is exclusively found in the thymus and its incorporation to the proteasome reduces chymotrypsin-like activity of the proteolytic machine17. α-rings control the entrance of the substrate into the catalytic cavity. Although 20S particles can exist in a free form, its default status is closed and requires the binding of proteasome activators to degrade polyubiquitylated proteins. Therefore, 20S particles are considered to be inactive, unable to degrade polyubiquitylated proteins16. However, free 20S particles have a detectable activity independent of ATP/ubiquitination and could play a significant role in the degradation of oxidized proteins15,18. Oxidative modification is a signal for proteolysis by 20S, a process that could gain relevance with age.

Degradation by the UPS is initiated by the conjugation of ubiquitin, a highly conserved 76 amino-acid residue polypeptide, to the substrate protein12. The attachment of a ubiquitin molecule is achieved through an enzymatic sequential mechanism involving three distinct classes of enzymes12. First, the ubiquitin-activating enzyme (E1) activates the carboxyl-terminal glycine residue of a ubiquitin in an ATP-dependent manner. Activated ubiquitin is next transferred to a cysteine site of a ubiquitin-conjugating enzyme (E2). In the third step, a ubiquitin ligase (E3) links ubiquitin from the E2 enzyme to a lysine residue of the _target protein. While apparently there are only two E1’s, there are several E2 enzymes and many E3 ubiquitin ligases, each of which recognizes one or several of specific protein motifs12. Therefore, E3 ubiquitin ligases are responsible for the selectivity of the process _targeting specific proteins to degradation. The same cascade links additional molecules to the primary ubiquitin via internal ubiquitin lysines, forming a ubiquitin chain. Ubiquitin has seven lysine residues, all of which can form polymer chains. A chain of at least four lysine 48-linked ubiquitins is the primary signal for degradation, while other linear ubiquitin chains participate in different processes such as signal transduction39. After ubiquitination, the polyubiquitylated protein is recognized and degraded by the proteasome. Active proteasomes are formed by the interaction of proteasomal regulatory particles with a core particle (20S), which contains the proteolytic active sites. Free intracellular 20S are normally found in an inactive/closed state and require the binding of 20S activators to degrade proteins. The major assembly of the 20S proteasome is with the 19S regulatory protein, which recognizes the polyubiquitylated substrate, removes the ubiquitin moieties and unfolds the substrate to translocate it into the 20S proteolytic chamber. The 20S can also be activated by the PA28 complex or the Blm10/PA200 protein. Finally, the substrate is cleaved into short peptides. The proteasome is involved in a variety of cellular functions, such as quality control of the proteome, stress response or cell cycle regulation.
Active proteasome exists in several forms, but its major assembly is formed through the assembly of the 20S and the 19S (26S, single capped or 30S, double capped)12. The 19S imparts regulation on the activity of the holo complex by recognizing, unfolding and translocating polyubiquitylated proteins to the 20S for degradation in an ATP-dependent process12,19. The 19S is composed of two subcomplexes: a base adjacent to the 20S and a lid that sits on top of the base12. The base contains six AAA-ATPases (Rpt1–6) and three non-ATPases subunits (Rpn1–2 and Rpn10). The lid, formed by eight subunits (Rpn3, Rpn5–9 and Rpn11–12), is critical for substrate recognition and deubiquitination12. In addition to regulation by 19S, the core particle can be activated by other regulatory particles such as Blm10/PA200 or PA28 (also known as 11S)16. These proteasome activators bind to the core particle opening the channel and promoting ATP- and ubiquitin-independent proteolytic degradation16. Compared with the 19S, their substrates are less clear although their biological functions are emerging. Blm10/PA200, a monomeric protein of 250 kDa, forms hybrid complexes in which it binds to one end of the 20S proteasome and the 19S to the opposite end15. PA28 is formed by hetero-heptameric rings of 28-kDa proteins (PA28α and PA28β) or homo-heptameric rings of PA28γ16. PA28αβ is inducible by interferon-γ and modulates the presentation and generation of specific viral antigens15. PA28γ is involved in cell cycle regulation promoting the degradation of small proteins such as p21 (ref. 15). It has been proposed that PA28 is involved in the degradation of oxidized proteins18. Oxidative stress causes a disassembly of the 26S/30S proteasome towards 20S and induces the PA28 and the immunoproteasome, forms supporting degradation of oxidized proteins. Taken together, proteasome activity is tightly modulated by a vast spectrum of proteasome regulators and conditions.
Autophagy
Autophagy is an intracellular catabolic process in which cytosol fractions, organelles and macromolecules are degraded through the lysosome. One of the main roles for autophagy is to act as a bulk intracellular degradation system that, on metabolic stress, such as nutrient deprivation, DNA damage, hypoxia or oxidative and endoplasmic reticulum (ER) stress, degrades many different substrates to support energy balance20,21. Although autophagy was initially described as a stress-induced mechanism, it exhibits basal activity. In addition, autophagy has a key role as a quality-control mechanism of proteins and organelles to maintain cellular homeostasis. Autophagy is emerging as a selective mechanism to degrade misfolded and aggregated proteins13. This function includes the degradation of aggregates of neurodegenerative-associated proteins such as tau, α-synuclein and polyglutamine-expanded proteins22. Specific receptors and adaptors, such as p62 (also known as sequestosome 1), recognize ubiquitinated proteins and _target these substrates to the autophagy machinery13. Moreover, due to the capacity to engulf whole cellular regions, autophagy is critical in every process that requires extensive cellular remodelling, such as embryogenesis, cellular differentiation or cellular death23. Autophagy has been also implicated in both acquired and innate immunity through antigen presentation and degradation of peptides from external pathogens24.
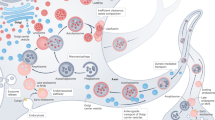
Lysosomes are the essential catalytic component of the autophagic degradation system. Lysosomes are single-membrane vesicles that contain a large variety of cellular hydrolases such as proteases, lipases, nucleotidases and glycosidases. These hydrolases reach their higher enzymatic activity at the acidic pH of the lysosomal lumen. Of particular interest of this review is proteolysis, which results from the combined action of endo- and exoproteases. This proteolytic activity leads to the conversion of proteins into small di- and tripeptides and free aminoacids that are released into the cytosol through permeases at the lysosomal membrane and either used to obtain energy or recycled to synthesize the novo proteins25. Depending on the type of cargo and delivery to lysosomes, autophagy is classified in different modalities. In mammalian cells, the best characterized types of autophagy are macroautophagy (Fig. 2), microautophagy and chaperone-mediated autophagy (CMA)14,23,26,27. Macroautophagy (herein after called autophagy) is a self-catabolic process in which cytoplasm portions are engulfed into double membrane vesicles, which are named autophagosomes. About 35 genes, generically known as autophagy-related genes (ATG) (Table 1), participate in autophagy28. Atg proteins organize into complexes that regulate each of the steps of autophagy. After the autophagosome is formed with the sequestered cargo inside, it is transported to the lysosome, where its outer membrane fuses with the lysosome membrane and both the sequestered material and the inner membrane of the autophagosome are degraded. The fusion is dependent on SNARE proteins VAMP8 and Vti1B29. In microautophagy, a pathway characterized to a much lesser extent, small components of the cytoplasm are engulfed directly by the lysosome membrane14. In CMA, particular cytoplasmic proteins are recognized by chaperones through a consensus sequence and transferred to the lysosome for their degradation via the lysosomal membrane-protein type 2A (LAMP-2A) receptor26. Autophagy and microautophagy can degrade both organelles and proteins, whereas CMA only participates in the degradation of proteins26.

Autophagy, or macroautophagy, starts with the formation of the phagophore, a double membrane that can be either newly synthesized or can be originated from the ER, mitochondria or plasma membrane. The Atg1 complex is key for the formation of the phagophore. The mammalian Atg1 homologues are the kinases ULK1 and ULK2. The ULK complex is formed, among others, by ULK1/2, Atg13 and FIP200. To promote the expansion of the phagophore, mammalian PI3K Vps34 complex—Vps15, Vps34, ATG14, Beclin-1, UVRAG, Rubicon—produces phosphatydilinositol-3-phosphate, which is crucial for the formation of the autophagosome. In the second part of the autophagic process, the cytoplasmic fraction is engulfed into the phagophore. The membrane then elongates until its edges fuse and give rise to the autophagosomes. Maturation of the phagophore comes with the conjugation of LC3 to phosphatidylethanolamine, a process known as LC3 lipidation. LC3 lipidation can occur either via assembly through the Atg12-Atg5-Atg16L complex or via processing of the newly synthesized LC3 through Atg4 to the cytosolic LC3 form (LC3I), and subsequently to the membrane binding form (LC3II). These reactions are catalyzed through Atg7 and Atg3 in a ubiquitin-like reaction. LC3II-positive autophagosomes are trafficked to the lysosomes through the microtubule network in a dynein-dependent manner.
Autophagy is modulated by many signalling pathways such as adenosine monophosphate-activated protein kinase (AMPK), Sirtuin 1 (SIRT1) and mTOR, a serine/threonine kinase which exists in two complexes named mTORC1 and mTORC2. mTORC1, the mammalian _target of rapamycin, can be modulated by several regulators and modulates the assembly of the Atg complex required for the formation of the autophagosomes30. Under fed conditions, mTORC1 is activated on the binding of growth factors to the insulin-like growth factor receptor (IGF1R), in a phosphatidylinositol 3-kinase (PI3K)/AKT-dependent manner21. Activated mTORC1 phosphorylates and inactivates the Atg1 complex. Conversely, under starving conditions mTOR is inhibited and the Atg1 complex consequently triggers autophagosomal vesicle formation21. In contrast, mTORC2 is insensitive to rapamycin inhibition and activates autophagy. Recently, unexpected findings have indicated that IGF1R depletion inhibits mTORC2 and attenuates autophagosome formation31. The conserved sensor of intracellular energy AMPK activates the mammalian orthologues of Atg1, ULK1 and ULK2, when the ratio AMP/ATP rises as a consequence of the energetic demand of the cell20. In addition, AMPK induces autophagy via the inhibition of mTOR21. SIRT1, a deacetylase that regulates cellular metabolism and survival, can also activate autophagy by deacetylation of several Atg proteins32 and transcription factors, such as FOXO1,-3 and -4 (ref. 21).
Loss of clearance mechanisms as a determinant of ageing
With age, post-mitotic cells lose extensive control of the proteostasis equilibrium: widespread, aberrant changes in translation, a generalized downregulation of chaperones and a loss of function in protein degradation machineries often appear in differentiated cells across time. Proteasome dysfunction during ageing can occur at different levels such as decreased expression, alteration and/or replacement of proteasome subunits33,34,35, disassembly of proteasomes36 or inactivation by interacting with protein aggregates37,38. This later mechanism could induce a catastrophic proteostasis feedback during ageing, since inhibition of proteostasis itself can induce the accumulation of protein inclusions, which in turn can obstruct and further inhibit proteasome activity37. A decline in proteasome function during ageing and senescence has been observed in several mammalian tissues and cells such as human skin, epidermal cells, fibroblasts and lymphocytes, bovine eye lens and rat liver, lung, heart, kidney, spinal cord, hippocampus, cerebral cortex and muscle tissues39. Human fibroblasts are a well-established model to define senescence mechanisms. In comparison with fibroblasts that have undergone fewer passages, senescent fibroblasts exhibit a reduction in the levels of all three proteasome activities and proteasome subunit levels40. Several stress responses such as the heat stress response (HSR) induce the expression of chaperones to correct defects in protein folding7,8,9. The HSR declines with age contributing to the accumulation of damaged proteins that may overwhelm the proteolytic machinery7,8,9. In addition, the HSR regulates the expression of specific chaperones, such as HSP70, that bind to unfolded proteins to stimulate their degradation by the proteasome. CHIP, an E3 ubiquitin ligase, interacts with HSP70 and promotes the proteolysis of its unfolded binding partners. CHIP deficiency in mice causes a reduction in lifespan and exacerbation of ageing phenotypes, providing a link between chaperones, proteasome and ageing41.
Similar to proteasome activity, autophagic-lysosomal potential also declines during ageing and senescence21,42. Ageing impairs autophagy of organelles, particularly that of defective mitochondria43, and clearance of autophagosomes with the consequent accumulation of undigested cargos inside the lysosome44. The molecular mechanisms mediating this demise are not fully established, although multiple studies indicate that ATG proteins, autophagy inductors and the cellular response to hormones that trigger autophagic degradation decline with age21. For instance, Atg5, Atg7 and Beclin-1 are downregulated in the aged human brain45. Furthermore, the levels of ULK1, Beclin-1 and LC3 are reduced in age-related pathologies, such as osteoarhrtitis46. In the liver, age-dependent decrease in autophagy may be the consequence of an inefficient clearance of the autophagosome cargo by the lysosome44 and reduced hormonal regulation of the pathway47. The rate of CMA decreases with ageing48 due to a decrease in the LAMP-2A receptor and a consequent reduction in the binding of proteins to the lysosome and their transport into the organelle48,49. The mechanism of LAMP-2A downregulation with ageing seems to be at the post-transcriptional level, since RNA levels of LAMP-2A are unchanged in ageing49. The heat shock protein 90 (Hsp90) is key for the assembly of LAMP-2A receptor50. Thus, the decline in availability of chaperones during ageing, and particularly, in Hsp90 in the aged liver51 could explain the impairment in the trafficking and stability of LAMP-2A. Although merely correlative, these findings indicate that the loss of proteasome and autophagy may contribute to the ageing process. However, further studies in vivo are required to strengthen this potential link such as using ubiquitinated proteasome reporters or examining the autophagic flux rather than autophagosome numbers.
Several genetic and functional studies have supported a casual effect of a decrease in protein clearance mechanisms and ageing. Fibroblasts treated with proteasome inhibitors have a shortened replicative lifespan and a senescent-like phenotype52. Knockdown of 19S and 20S subunits during adulthood shortens lifespan in Caenohabditis elegans53,54. Strikingly, a transgenic mouse with reduced chymotrypsin-like activity by replacement of the β5 subunit with β5t exhibits a shortened lifespan, premature age-related phenotypes and aggravation of age-related metabolic disorders55. Similarly, PA28γ deficiency promotes premature ageing in mice56. In yeast, a genetic screen searching for ageing factors identified multiple short-lived mutants with autophagy defects57. In C. elegans, decreased expression of several autophagic genes (atg1, atg7, atg12, bec1 and atg18) shortens its lifespan, supporting the role of autophagy in longevity58,59. In Drosophila melanogaster, the expression of autophagy genes is decreased in neurons from old flies60 and loss of function of Atg1, Atg8 and Drosophila sestrin (dSesn, a protein required for autophagy) reduces lifespan21. In mice, constitutive knockout of most ATG genes is lethal during the early postnatal period due to the incapacity of energy reserves mobilization24. Tissue-specific deletion of several autophagy genes precipitates ageing and ageing-associated phenotypes (reviewed in refs 21, 24). For instance, the central nervous system-specific knockout of Atg5, Atg7 or Atg17/FIP200 induces the accumulation of ubiquitin-positive inclusion bodies followed by neuronal loss and premature death. Purkinje cell-specific knockout of Atg7 induces axonal dystrophy and degeneration of axon terminals with a subsequent Purkinje cell death and cerebellar ataxia. It is important to highlight these findings provided one of the first direct evidences of an autophagic ubiquitinated-mediated proteolysis mechanism since neurons with compromised autophagy accumulate ubiquitinated aggregates and degenerate even with functional intact proteasomes. Autophagy dysfunction also affects other tissues. Skeletal muscle-specific knockout of Atg7 induces muscle atrophy, age-dependent decrease in force, accumulation of abnormal mitochondria, disorganization of sarcomeres and exacerbates muscle loss during denervation and fasting. Hepatocyte-specific knockout of Atg7 triggers the accumulation of lipid droplets containing triglycerides and cholesterol, induces ER stress and causes insulin stress resistance. β cell-specific deletion of Atg7 induces degeneration of pancreatic islets, impaired glucose tolerance, decreased serum insulin level, accumulation of ubiquitinylated proteins co-localized with p62, mitochondrial swelling and ER distension. Another example is the specific knockout of Atg5 in podocytes that induces spontaneous age-dependent late onset of glomerulosclerosis with accumulation of oxidized and ubiquitinylated proteins, damaged mitochondria, ER stress and podocyte loss.
Protein clearance mechanisms and stem cell maintenance
Different tissues exhibit different proteasome activity61,62. These findings raise the intriguing question whether the proteasome is equally affected in different cell types during ageing. This question can be addressed by using cell-specific photoconvertible reporters to measure proteasome activity in living animals63. A study using this methodology in C. elegans has shown that dorsorectal neurons exhibit a more severe decline in proteasome activity during ageing than body-wall muscle cells63. Tissue-specific differences in autophagic potential and the impact of ageing have not been systematically explored yet.
The specific biological purpose of every cell type may define cellular differences in proteostasis and how ageing impacts on their protein clearance machinery. Indeed, recent findings in embryonic stem cells (ESC) support this hypothesis. ESCs do not undergo replicative senescence—a capacity that necessarily demands avoidance of any imbalance in proteostasis that would otherwise compromise its function during replication. Accordingly, human (hESCs) and mouse ESCs (mESCs) exhibit high proteasome activity compared to their differentiated counterparts and this activity does not differ depending on the passage number64. Increased proteasome activity is correlated with enhanced levels of the 19S subunit PSMD11/Rpn6 (refs 64, 65, 66), which is essential for the activity of the 26S/30S proteasome and stabilizes the otherwise weak interaction between the 20S core and the 19S cap64,67. Moreover, hESCs exhibit increased levels of other proteasome subunits such as α2–α5 (ref. 65) and the immunoproteasome subunits β1i and β5i68. Increased proteasome activity is required for hESC function although the reasons are unknown. Enhanced proteasome activity in these cells could be necessary to avoid senescence and maintain an intact proteome for self-renewal. Since the passage of damaged proteins to daughter cells could compromise the ageing process of an organism, increased proteasome activity could be required to generate a cell lineage with an intact proteome. Notably, the degradation of damaged proteins is triggered on the first signs of mESC differentiation via induction of PA28αβ69,70. Furthermore, both ESCs exhibit higher autophagy activity on early differentiation71. In addition, induced pluripotent stem cells generated from patients with PD show more autophagic vacuoles when differentiated into dopaminergic neurons72 suggesting an active rejuvenation step to generate a pool of ‘healthy’ cells. Taken together, these results raise an intriguing hypothesis: increased proteolytic systems maintain immortality of ESCs but they are further enhanced during the initial steps of differentiation to scavenge damaged proteins that could affect the function and senescence of the new cell lineage and, therefore, organismal ageing. Once the cells are differentiated, their proteolytic potential is diminished and further reduced by the impact of ageing.
Similar to hESCs, the biological function of germ line stem cells (GSCs) suggest that these cells have increased proteostasis. GSCs generate the gametes that will produce the embryos after reproduction and are designed to maintain an unlimited proliferative capacity to fulfil their biological purpose: to be passed from one generation to the next. Human oocytes and hESCs share a common transcriptome signature such as increased expression of PSMD11 (ref. 65). In D. melanogaster, gonads and maturating oocytes have increased 26S proteasome activity and accumulate less damaged proteins compared with ageing somatic tissues61,62. Proteasome activity is already downregulated in middle-aged flies, when signs of ageing first appear61,62. However, maturating oocytes and gonads maintain their high proteasome activity compared with age-matched somatic tissues61. These results support the disposable soma theory of ageing formalized by Thomas Kirkwood73. In this hypothesis, resources are allocated from the soma to the germline under stress conditions and ageing to prevent and repair damage to the germline. By this mechanism, the germline would ensure the fitness of the next generation. Therefore, increased proteasome activity in maturating oocytes and gonads may contribute to ensure the generation a pristine proteome in the ensuing generation. In yeast, aggregated and damaged proteins can be retained in mother cells during division74. In this mechanism, protein aggregates are captured by maternal mitochondria, whereas mitochondria acquired by the daughter cells are free of aggregates75. This asymmetric division enables the generation of a rejuvenated, germ-like, daughter cell lineage76. The results in flies suggest that an evolutionarily conserved mechanism may have evolved to avoid replicative senescence by establishing an ageing (soma) and rejuvenated (germ) lineage, the later with an increased proteasome activity that reduces the accumulation of damaged proteins.
Adult somatic stem cell exhaustion is considered one of the tentative hallmarks of ageing1. These cells persist throughout the lifespan of the organism and rejuvenate tissues. However, somatic stem cell function declines during ageing in tissues such as the brain, skin, blood, bone and skeletal muscle and this failure may contribute to age-related diseases1,77. Whether adult somatic stem cells also have an enhanced proteasome activity remains to be elucidated, but the maintenance of this activity may impact organismal ageing. Experiments with adult somatic stem cells cultured in vitro such as human mesenchymal stem cells (hMSCs)78, haematopoietic stem cells (HSCs), dermal stem cells and epidermal stem cells79 suggest that autophagy activity is increased in these cells compared with their differentiated counterparts. In fetal and postnatal HSCs, a deficiency in essential autophagy genes such as Atg7 or FIP200 deregulates proliferation, suggesting that autophagy is required for stemness of HSCs80. FIP200 is also required for neural stem cell proliferation81. These results suggest that autophagy could play an important role in the maintenance of specific stem cell pools. An intriguing question is whether adult somatic stem cells maintain their higher autophagy levels during organismal ageing and if a decrease in this activity could contribute to their senescence. In old mice, inhibition of mTOR with rapamycin increases lifespan and restores self-renewal of HSCs, thus improving the function of the immune system82. However, the impact of autophagy on the positive regulation of haematopoiesis by rapamycin is unknown. Other studies have shown that old HSCs have higher basal levels of autophagy activity and are able to induce autophagy much like young HSCs83. FoxO3, a forkhead transcription factor linked to stem cell maintenance and longevity84, maintains the expression of proautophagy genes in adult HSCs to allow a quick autophagic response on stress83. That old adult HSCs maintain an autophagy potential similar to young HSCs and exhibit higher levels of autophagy for their survival confronts the prevailing, traditional view where compromised autophagy is seen as a determinant of ageing21. Therefore, further studies are necessary to determine the links between proteolytic systems, homeostasis of stem cell pools and ageing.
Proteasome and autophagy dysfunction in age-related diseases
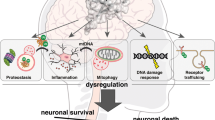
Gain-of-toxic-function diseases, such as AD, HD, PD and amyotrophic lateral sclerosis (ALS), in which aggregation-mediated proteotoxicity exceeds the cellular clearance machinery, are more frequent late in life even though different toxic proteins are involved in their onset2,3,4,10,14,15. The pathophysiological significance of protein aggregates is vaguely known. The accumulation of abnormal proteins may lead to perturbed cellular functions and eventually to neuronal death, ultimately manifesting as neurodegenerative disease. Unfolded monomeric proteins can be degraded through proteasome or CMA; while once aggregated into large macromolecules, bulk degradation through autophagy seems to be the key player in their removal22. The inclusion bodies of AD, PD, HD and ALS contain abnormal amounts of ubiquitin, suggesting a link between neurodegeneration and dysfunction of proteasome and autophagy11,13,85. Changes in the proteolytic machinery may explain why ageing is a risk factor for AD, PD and HD13,39,86. Furthermore, loss-of-function experiments have shown that decreased proteasomal activity and autophagic potential enhance the neurodegenerative phenotype13,39. In addition, there is a crosstalk between proteasome and autophagy: protein aggregation-induced proteasome or CMA impairment can activate macroautophagy, probably as a compensation mechanism87. Sequestration of autophagy repressors such as mTOR in protein aggregates could partially mediate autophagy induction and contribute to the typical late onset of the disease and the survival of cells with aggregates88. After proteasome and/or CMA failure to degrade damaged proteins, autophagy may also end up collapsing and contributing to an enhanced accumulation of aggreggates89. Therefore, therapies that modulate proteolytic potential may reduce aggregation-mediated toxicity.
Alzheimer’s disease
AD is the most common neurodegenerative disorders marked by protein aggregation and results in progressive dementia2. AD is characterized by the deposition of two different protein aggregates: (1) Extracellular amyloid plaques mostly formed by aggregates of β-amyloid (Aβ), which is generated via the proteolytic cleavage of the amyloid precursor protein (APP)2 and (2) Intracellular neurofibrillary tangles mostly composed by hyperphosphorylated tau, a microtubule-associated protein2. Both intracellular Aβ oligomers and aggregated tau can inhibit proteasome activity90,91, which in turn could contribute to the pathology of AD. This mechanism could explain why proteasome activity is decreased in brains from AD patients compared with age-matched controls90. The proteasome may play a key role in delaying the accumulation of neurotoxic tau. In vitro, tau protein can be degraded by the 20S proteasome in an ATP/ubiquitin-independent manner92. However, other studies showed that tau proteasomal degradation is at least partially ubiquitin-dependent in cells39. When tau is not bound to microtubules, it associates with the chaperone Hsp70. The E3 ubiquitin ligase CHIP interacts with Hsp70 and promotes tau ubiquitination and degradation93. Furthermore, the inhibition of the proteasome-associated deubiquitinating enzyme Usp14 promotes tau degradation94. In vitro, the proteasome activator Blm10 accelerates degradation of unstructured tau95. Tau clearance is blocked by FK506 binding protein 51 kDa (FKBP51), which forms a chaperone complex with Hsp90 that prevents tau degradation resulting in tau oligomerization96. In humans, FKBP51 levels increase with age and higher FKBP51 levels are associated with AD progression96.
Autophagy vacuoles are abundant in AD neurons and contain substantial amounts of Aβ97,98. Increased autophagy compartments occurs at early staged of the disease, when amyloid deposits are not visible97. Whether the autophagic flux is enhanced at this stage is unknown, although increased lysosome proliferation and lysosomal enzymes levels were observed99. However, autophagy declines with the disease progression contributing to the deposition of aggregated proteins98. In AD, both the maturation and transport of autophagosomes are impaired, and Aβ-containing autophagosomes consequently accumulate97,100. In addition, impaired lysosomal proteolysis may also contribute to Aβ accumulation. Presenilin-1, an AD-related protein involved in the early onset of the disease, regulates autophagy by acidificating autolysosomes and activating cathepsin. Presenilin-1 hypomorphic mice present impaired clearance of autophagosomes101. Several studies have suggested that reduced autophagy via beclin-1 contributes to the deposition of Aβ39. Beclin-1 expression is reduced in AD brain with age and beclin-1 deficiency reduces autophagy rate and alters APP metabolism102. Beclin-1 heterozygous mice present defective autophagy and neuronal degeneration, while a decrease of beclin-1 in the APP AD model enhances Aβ deposition and neuronal degeneration103.
Parkinson’s disease
PD is a neurodegenerative movement disorder characterized by the accumulation in dopaminergic neurons of Lewy Bodies3, which are mainly composed of α-synuclein, ubiquitin, the E3 ligase parkin and other UPS-related proteins. Soluble α-synuclein is degraded through proteasome or CMA, while once aggregated is scavenged through autophagy104,105. Several studies have suggested that proteasome dysfunction may contribute to the pathology of PD. Rats treated with proteasome inhibitors exhibit characteristics of PD, such as tremor, which is accompanied by α-synuclein and ubiquitin-containing inclusions in dopaminergic neurons106. However, other laboratories could not reproduce these studies85. In an alternative approach, conditional proteasome subunits knockout mouse models were generated since constitutive deletion is embryonically lethal11. Deletion of the ATPase subunit Psmc1/Rpt2 in dopaminergic neurons lead to ubiquitin and α-synuclein positive inclusions which resulted in neuronal death, thus resembling the hallmarks of PD107. In addition, variations in the gene PSMC4/Rpt3 correlate with the age of PD onset in patients108. These results suggest a role for the 26S/30S in α-synuclein degradation although it can also be degraded by free 20S in a ubiquitin-independent process109. Dysfunction of autophagy has also been pointed as a central pathway in the pathogenesis of PD. Mutant α-synuclein binds to the lysosome membrane with high affinity, but cannot be transported into the lysosome. Consequently, the irreversible binding of mutant α-synuclein to the lysosome blocks the binding of other substrates, resulting in a general impairment of the CMA pathway and further α-synuclein accumulation110. Proteins related to familiar PD, such as DJ-1 or LRRK2, significantly alter autophagy111,112.
ALS is another progressive neurodegenerative movement disorder but affects motor neurons3. Impairment of the proteasome in these neurons by conditional knockout of Psmc4/Rpt3 revealed loss of motor neurons, locomotor dysfunction and accumulation of aggregates of TDP-43 and FUS proteins, features typical of ALS113. In contrast, the motor neuron-specific KO of Atg7, results in only subtle cytosolic accumulation of p62 and ubiquitin, with no TDP-43 and FUS pathologies or motor dysfunctions113. Therefore, under these experimental settings, proteasomal degradation appears to govern the development of ALS.
Huntington’s disease
HD is an autosomal dominant disease that affects muscle coordination and leads to progressive cognitive decline and psychosis4. HD displays selective neurodegeneration that occurs preferentially in the brain striatum4. The disorder is caused by the expansion of a CAG triplet repeat region in the Huntingtin gene, which translates into a polyglutamine (PolyQ) stretch in the amino-terminal domain of the protein resulting in fibril formation and aggregation4. An expansion of glutamines of over 40 can trigger the development of HD. Huntingtin inclusions contain ubiquitinated proteins, chaperones and components of the proteasome114. Proteasome activity is reduced in brains from HD patients and mice models115. In contrast to the soluble form of huntingtin, the aggregated form has been found to be ubiquitinated itself, suggesting an impairment of the capacity of the proteasome to degrade aberrant huntingtin115. Nevertheless, whether the formation of inclusion bodies in neurons is protective or toxic is still under debate. Considering the correlation between behavioural deficits and the load of misfolded huntingtin in HD mouse models, the inclusions themselves have been proposed to be on the base of the toxicity in neurons114,116. The exact mechanisms of this toxicity, however, remain unsolved but a general impairment of the proteasomal function resulting in proteostasis collapse and cellular death has been proposed117. An in vitro study suggested that the proteasome is not able to cleave within PolyQ stretches and the occasional failure of these long undegradable sequences to exit the proteasome may interfere with its function118. Accordingly, incubation of proteasomes with mutant huntingtin exerts an inhibitory effect on the proteasome by directly impeding the entrance of other substrates119. However, other studies found that huntingtin aggregates do not affect proteasome activity suggesting that proteasomal dysfunction may be a consequence of a general proteostasis collapse120,121.
Huntingtin aggregates can be degraded through autophagy122 and autophagy inhibition enhances the disease88,123,124. Autophagy dysfunction seems to play an important role in HD but the molecular bases underlying this mechanism have not been fully elucidated. In cells from HD patients and HD mouse models, a defect in the effective cargo recognition by autophagosomes was demonstrated125. In addition, mutant huntingtin can sequester beclin-1, and consequently inhibit autophagy, what further enhances huntingtin accumulation126. Age-induced reduction in beclin-1 expression would also contribute to the major accumulation of huntingtin in old neurons126. Wild-type huntingtin participates in the induction of autophagy on ER stress whereas mutant huntingtin-expressing cells have a perturbed ER function and increase autophagosomes127. Furthermore, decreased dynein function impairs autophagic clearance of aggregate-prone proteins and enhances the toxicity of the mutation that causes HD in fly and mouse models128.
Cancer
Ageing is a primary risk factor for cancer1,129. Cancer is the consequence of an aberrant gain of cellular fitness, whereas ageing is characterized by a loss of fitness. Although ageing and cancer may seem to be opposite processes, both can share common origins: the cellular damage accumulated during ageing triggers cellular senescence, but this damage occasionally provides aberrant advantages to specific cells1,129. Several studies have suggested that proteasome activity is elevated in human cancers39. The high degree of cell division and high mutation rates in cancer cells lead to an accumulation of misfolded proteins, which activate stress responses and/or apoptosis. The upregulation of proteasomal activity may eliminate these aberrant proteins that otherwise would be toxic for the cell130. Proteasome activity could have an important role in theories that suggest a relationship between the onset of cancer and ageing of somatic stem cells129. During ageing, these cells accumulate DNA damage from stress such as intracellular oxidants. Typically, these damaged cells undergo cell arrest, apoptosis or senescence decreasing homeostasis of stem cell pools. However, some rare cells can escape from the default pathway, acquiring additional mutations that allow them to proliferate and increase the likelihood of cancer129. Thus, a better knowledge of how somatic adult stem cells regulate their proteasome activity may help to understand the origin of cancer stem cells and find specific treatments. Whether proteasome modulation would result in selective inhibition of malignant cell growth is unclear, but proteasome inhibitors have become valuable tools in the treatment of certain types of cancer39. The underlying mechanisms for this selectivity might be that malignant cells show greater sensitivity to the cytotoxic effects of proteasome inhibition than non-cancer cells39. Likewise, immortal hESCs are more sensitive to proteasome inhibition than their differentiated counterparts64. In contrast, increased autophagy plays an important role in oncogenic suppression since its inhibition increases the frequency of tumours in mice21. Autophagy regulators such as beclin-1, Atg4c and AMBRA1, act as oncosuppressors since their loss increases the frequency of tumours in rodents131. Removal of the oncogenic protein p62 induced by autophagy suppresses oncogenesis132. In addition, autophagy is upregulated during and partially required for oncogenic Ras-induced senescence and apoptosis limiting the oncogenic potential of deregulated Ras signals133,134.
Proteostasis systems determine longevity and healthspan
Enhanced proteasome activity and/or autophagy extend longevity. Here we discuss recent insights into the activation of protein clearance mechanisms induced by longevity-promoting pathways.
Increased protein clearance mechanisms extend healthspan
Several reports suggest that sustained proteasome activity correlates with longevity. Notably, proteasome activity is increased in long-lived humans (centenarians)135. In this study, the levels of proteasome subunits and proteasome activity were analysed in fibroblasts derived from healthy centenarians and compared with fibroblast from young and old control donors. Strikingly, proteasome subunits and activity levels in healthy centenarian-derived fibroblasts are more similar to the younger than the older control donor-derived fibroblasts135. Furthermore, increased proteasome activity was found in extremely long-lived animals such as the naked mole rat136 and the giant clam137. This link between increased proteasome activity and longevity has been further supported by genetic approaches (Fig. 3a). In Saccharomyces cerevisae, overexpression of the proteasome chaperone UMP1/POMP induces an enhanced preservation of proteasome-mediated proteolysis and increased viability during stationary-phase, a model for post-mitotic ageing138. In yeast, Rpn4 acts as a transcription factor regulating the levels of proteasome subunits and promoting proteasome activity139. Rpn4 degradation is promoted by the E3 ubiquitin ligase Ubr2. Loss of UBR2 results in elevated levels of Rpn4 and increased replicative lifespan and resistance to proteotoxic stress in yeast140. Ectopic expression of 19S proteasome subunits extends lifespan in invertebrates. A genetic gain-of-function screen in D. melanogaster showed that Rpn11 overexpression suppresses the age-dependent reduction of 26S/30S proteasome activity and extends lifespan, whereas rpn6 overexpression increases longevity in worms141. Furthermore, increased assembly of active proteasomes induced by the overexpression of β5 or POMP confers resistance to oxidative stress and delays senescence in human fibroblasts142.

(a) Proteasome activity declines with ageing as a consequence of a reduction in the expression of proteasomal subunits, alteration or replacement of several proteasomal subunits, disassembly of the proteasome or inactivation of the proteasome through direct interaction with age-induced protein aggregates. Consequently, interventions that increase and sustain proteasome activity such as the overexpression of proteasome subunits and chaperones can extend lifespan and delay the onset of age-related diseases. Several longevity-promoting pathways (that is, DR or reduced insulin/IGF-1 signaling pathway (IIS)) increase proteasome activity. Likewise, long-lived organisms (that is, the naked mole rat and the giant clam) and centenarians exhibit increased proteasome activity. (b) Autophagy declines with age as a consequence of reduced expression of autophagy-related proteins, decreased clearance of autophagosomes cargo or impaired response to hormonal regulation. Activation of autophagy, either pharmacologically or genetically, promotes longevity and delays the onset of age-related diseases.
Activation of autophagy, either pharmacologically or genetically, promotes longevity in C. elegans143, D. melanogaster144 and mice145 (Fig. 3b). Rapamycin treatment extends longevity in invertebrates and mammals21. However, due to the multiple _targets of mTOR, it cannot be ruled out that other autophagy-independent pathways, such as the immune environment or regulation of protein translation, could be also involved in rapamycin-induced longevity146. Strikingly, enhanced expression of Atg1, Atg8 and dSesn promotes longevity in flies21 and overexpression of Atg5 increases autophagy and extends lifespan in mice147. Furthermore, an increase in the levels of LAMP-2A prevents ageing-associated dysfunctions in the liver of old mice148. In C. elegans, enhanced autophagy via overexpression of the basic helix–loop–helix (HLH) transcription factor EB (TFEB) extends lifespan149. TFEB/HLH-30 induces many autophagy and lysosomal-related genes150. However, the longevity phenotype induced by TFEB/HLH-30 is caused by increased lysosomal lipolysis and might be independent of proteostasis149,151.
Multiple studies have shown that increased clearance mechanisms ameliorate age-related diseases. Enhancement of the proteasome machinery has been proven beneficial in HD models. Increased levels of Rpn6 in C. elegans or Rpn11 in D. melanogaster reduce toxic aggregates and suppress expanded PolyQ-induced neurodegeneration in HD models54,141. Increased expression of PA28γ improves cell survival in a cellular model of HD152. Modulation of autophagy has beneficial effects on neurodegenerative diseases. In flies and mouse models of AD, activation of autophagy by rapamycin treatment increases clearance of tau and Aβ aggregates153,154. Likewise, activation of autophagy by rapamycin treatment increases clearance of α-synuclein aggregates in cells that express mutant α-synuclein105. The overexpression of beclin-1 ameliorates the accumulation of α-synuclein by enhancing lysosomal function and reducing the deficits in the autophagic pathway155. Pharmacological autophagy activation also ameliorates HD phenotype88,123,124.
Reduced insulin/IGF-1 signalling
Reduced insulin/IGF-1 signalling (IIS) promotes longevity and increases stability of the proteome, which delays the onset of age-related diseases10. Reduced IIS extends lifespan in both invertebrates and vertebrates156 and correlates with increased longevity in humans157,158. The insulin/IGF-1 receptor activates a conserved PI3-kinase /PDK/AKT signalling cascade that phosphorylates FOXO transcription factors, thereby preventing their nuclear localization. When IIS signalling is reduced, FOXO accumulates in the nucleus and regulates downstream genes that extend lifespan and increase stress resistance156. In C. elegans, the CUL-1 E3 ubiquitin ligase complex is required for the extended lifespan of IIS mutant worms. This regulation is achieved, at least in part, by promoting the transcriptional activity of the worm FOXO transcription factor DAF-16 (ref. 53). Decreased IIS induces proteasome activity in worms through DAF-16 transcriptional repression of the proteasome-associated deubiquitinating enzyme ubh-4 (ref. 159). UCHL5, the human orthologue of ubh-4, increases degradation of toxic proteins in mammalian cells159. Autophagy also contributes to the longevity phenotype of reduced IIS C. elegans mutants. In fact, IIS worm mutants constituted the first model in which a causative effect between autophagy and longevity was shown160. Since then, the requirement of autophagy for lifespan extension has been demonstrated in practically all long-lived C. elegans strains such as dietary restriction (DR), mitochondrial electron transport chain, reduced TOR and germline-lacking mutant worms149,161. In this context, the transcription factor HLH-30/TFEB, which activates autophagy, may be an universal regulator of longevity since it is required for the lifespan of long-lived mutant worms149. In worms with mutations in the insulin receptor daf-2, longevity is abolished and autophagosomes accumulate when autophagy is inhibited by mutation of essential ATG genes160. Autophagy-dependent lifespan extension requires bec1 (a beclin-1 orthologue) but not DAF-16. Nevertheless, due to the requirement of DAF-16 for lifespan extension in daf-2 mutants, autophagy is most probably not sufficient for the longevity phenotype162.
Delayed ageing, by IIS reduction, protects from protein aggregation toxicity163,164. Experiments using C. elegans expressing polyQ in body-wall muscle showed that toxic protein aggregation is age dependent165. IIS reduction delays polyQ aggregation and toxicity suggesting that this pathway modulates the proteostasis network to enable cells to deal with toxic proteins to a much later age165. Whether reduced IIS ameliorates polyQ aggregation by modulating, at least partially, protein clearance mechanisms has not been examined. In a C. elegans model of AD, expression of Aβ1–42 peptide in the body-wall muscle cells produces a progressive paralysis phenotype166. In a DAF-16-dependent manner, reduced function of the IIS can protect from the toxicity of Aβ1–42 by inducing the aggregation of small toxic Aβ1–42 oligomers into larger, less toxic structures167. These results suggest the activation of an aggregation mechanism and have been substantiated in mice, in which a heterozygous mutation in the IGF-1 receptor is protective in a mouse model of AD164. Autophagic degradation of Aβ is also required for the protective effect of reduced IIS in Aβ1–42-expressing worms, whereas the role of proteasome activity has not been examined168. In mice, IIS effects could be independent of autophagy since IGF1R inhibition results in decreased autophagy31. Besides IIS, other growth factors regulate ageing169. Epidermal growth factor signalling promotes longevity in C. elegans. Upregulation of both proteasome activity and polyubiquitination, and a consequent decrease in protein aggregation are required for the lifespan extension induced by epidermal growth factor signalling170.
Signals from the reproductive system
Hypothetically, the need for repairing and preventing damage to the germline dominates resource allocation strategies, while the somatic tissues age and deteriorate73. In support of such theory, modulations of reproduction that eliminate germ cells in C. elegans and D. melanogaster extend lifespan171, a phenotype that may be caused by heightened proteome stability within the post-mitotic soma54,172. Inhibiting germline proliferation induces an increased in proteasome activity and RPN6 levels in the somatic tissues and protects from polyQ-dependent aggregation54,172. Similar to immortal hESCs, increased proteasome activity, rpn6 expression and longevity are modulated by DAF-16 in these long-lived animals64,173. Interestingly, DNA damage in C. elegans germ cells induces a systemic response that protects somatic tissues by increasing their proteasome activity174. Furthermore, autophagy and lipase-4-dependent lypolisis modulate the longevity phenotype of germline-lacking worms in an interconnected manner175. This interplay occurs through a coordinated action of mTOR and PHA-4/FOXA, a transcription factor that regulates the expression of several autophagy genes.
Dietary restriction
Reduced food intake without malnutrition extends lifespan in multiple species176. DR modulates translational rates inducing a decrease in the load of nascent polypeptides to the proteostasis machinery177, which may allow more efficient protein folding and degradation. In mice, DR induces the expression of the 19S proteasome subunit Psmc3/Rpt5 and the proteasome activator PA28α34. In C. elegans, the HECT E3 ubiquitin ligase WWP-1 is a positive regulator of lifespan in response to DR178. This lifespan extension induced by WWP-1 is dependent on pha-4 (ref. 178). In addition, the oxidative stress-responsive transcription factor skn-1 is also required for DR-induced longevity in C. elegans179. skn-1 promotes the expression of aip-1/AIRAP (arsenic-inducible RNA-associated protein) under metabolic stress180. aip-1/AIRAP binds to the 19S increasing proteasome activity and clearance of damaged proteins181. Notably, aip-1/AIRAP ameliorates Aβ and polyQ toxicity182,183. Moreover, skn-1 and its mammalian orthologues Nrf1 and Nrf2 upregulate the expression of proteasomal genes in response to proteasome inhibition and oxidative stress39. Inducible Nrf2 activation in D. melanogaster promotes youthful expression of proteasome subunits in aged somatic tissues62. This result suggests that age-dependent Nrf2 dysfunction may contribute to the decrease in proteasome subunit expression with age. Interestingly, Nrf2-mediated proteasome activation delays senescence in human fibroblasts184. Autophagy is required for the anti-ageing effects of DR in all the species investigated21. In addition, DR is sufficient to increase autophagic potential through the activation of either AMPK or SIRT1 depending on how it is applied (that is, intermittent feeding versus chronically reduced intake), its intensity and its onset21. DR also induces autophagy through the inhibition of IIS143. Similar to germline-lacking worms, DR-induced autophagy requires pha-4, which increases the expression of ATGs162,185.
Future directions
Longevity-promoting pathways increase functionality of protein clearance mechanisms, which contributes to the amelioration of age-related diseases. Although related studies have been performed mostly in invertebrates, their results could be translated into a valuable therapeutic approach for the treatment of progressive, age-related neurodegenerative diseases. However, further studies in mammals are required to strengthen the potential link between increased protein clearance mechanisms and the delayed onset of age-related disorders.
Additional information
How to cite this article: Vilchez, D. et al. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5:5659 doi: 10.1038/ncomms6659 (2014).
References
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013). A review of the nine temptative hallmarks of aging.
Selkoe, D. J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 3, pii: a004457 (2011).
Bosco, D. A., LaVoie, M. J., Petsko, G. A. & Ringe, D. Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Biol. 3, a007500 (2011).
Finkbeiner, S. Huntington's Disease. Cold Spring Harb. Perspect. Biol 3, a007476 (2011).
Powers, E. T., Morimoto, R. I., Dillin, A., Kelly, J. W. & Balch, W. E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 (2009).
Calderwood, S. K., Murshid, A. & Prince, T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging—a mini-review. Gerontology 55, 550–558 (2009).
Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011).
Morimoto, R. I. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 91–99 (2011).
Soti, C. & Csermely, P. Aging and molecular chaperones. Exp. Gerontol. 38, 1037–1040 (2003).
Taylor, R. C. & Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3, a004440 (2011).
Tanaka, K. & Matsuda, N. Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 1843, 197–204 (2014).
Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 (2009).
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Wong, E. & Cuervo, A. M. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2, a006734 (2010).
Schmidt, M. & Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 1843, 13–25 (2014).
Stadtmueller, B. M. & Hill, C. P. Proteasome activators. Mol. Cell 41, 8–19 (2011).
Murata, S. et al. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 316, 1349–1353 (2007).
Jung, T., Höhn, A. & Grune, T. The proteasome and the degradation of oxidized proteins: partII—proteinoxidationandproteasomaldegradation. Redox Biol. 2, 99–104 (2013).
Tanaka, K. The proteasome: from basic mechanisms to emerging roles. Keio J. Med. 62, 1–12 (2013).
Egan, D., Kim, J., Shaw, R. J. & Guan, K. L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644 (2011).
Rubinsztein, D. C., Marino, G. & Kroemer, G. Autophagy and aging. Cell 146, 682–695 (2011).
Martinez-Vicente, M. & Cuervo, A. M. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 6, 352–361 (2007).
Mizushima, N., Levine, B., Cuervo, A. M. & Klionsky, D. J. Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008).
Mizushima, N. & Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 12, 823–830 (2010).
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Cuervo, A. M. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol. Metab. 21, 142–150 (2010).
He, C. & Klionsky, D. J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 (2009).
Klionsky, D. J. et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5, 539–545 (2003).
Furuta, N., Fujita, N., Noda, T., Yoshimori, T. & Amano, A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell 21, 1001–1010 (2010).
Rubinsztein, D. C. et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1, 11–22 (2005).
Renna, M. et al. IGF-1 receptor antagonism inhibits autophagy. Hum. Mol. Genet. 22, 4528–4544 (2013).
Lee, I. H. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl Acad. Sci. USA 105, 3374–3379 (2008).
Ferrington, D. A., Husom, A. D. & Thompson, L. V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 19, 644–646 (2005).
Lee, C. K., Klopp, R. G., Weindruch, R. & Prolla, T. A. Gene expression profile of aging and its retardation by caloric restriction. Science 285, 1390–1393 (1999). Through gene expression analysis of skeletal muscle of aged and caloric restricted mice several proteasome-related genes are found to be upregulated.
Ly, D. H., Lockhart, D. J., Lerner, R. A. & Schultz, P. G. Mitotic misregulation and human aging. Science 287, 2486–2492 (2000).
Vernace, V. A., Arnaud, L., Schmidt-Glenewinkel, T. & Figueiredo-Pereira, M. E. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J. 21, 2672–2682 (2007).
Andersson, V., Hanzen, S., Liu, B., Molin, M. & Nystrom, T. Enhancing protein disaggregation restores proteasome activity in aged cells. Aging 5, 802–812 (2013).
Grune, T., Jung, T., Merker, K. & Davies, K. J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and 'aggresomes' during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 36, 2519–2530 (2004).
Saez, I. & Vilchez, D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 15, 38–51 (2014).
Chondrogianni, N. et al. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 278, 28026–28037 (2003).
Min, J. N. et al. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 28, 4018–4025 (2008).
Cuervo, A. M. Autophagy and aging: keeping that old broom working. Trends Genet. 24, 604–612 (2008).
Yen, W. L. & Klionsky, D. J. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 23, 248–262 (2008).
Terman, A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology 41, (Suppl 2): 319–326 (1995).
Lipinski, M. M. et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 14164–14169 (2010).
Carames, B., Taniguchi, N., Otsuki, S., Blanco, F. J. & Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis. Rheum. 62, 791–801 (2010).
Donati, A. et al. Age-related changes in the regulation of autophagic proteolysis in rat isolated hepatocytes. J. Gerontol. A Biol. Sci. Med. Sci. 56, B288–B293 (2001).
Cuervo, A. M. & Dice, J. F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513 (2000). There is an age-dependent decline in the rates of chaperone-mediated autophagy, caused by reduced levels of the chaperone receptor LAMP2a.
Kiffin, R. et al. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 120, 782–791 (2007).
Bandyopadhyay, U., Kaushik, S., Varticovski, L. & Cuervo, A. M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 28, 5747–5763 (2008).
Nardai, G., Csermely, P. & Soti, C. Chaperone function and chaperone overload in the aged. A preliminary analysis. Exp. Gerontol. 37, 1257–1262 (2002).
Torres, C., Lewis, L. & Cristofalo, V. J. Proteasome inhibitors shorten replicative life span and induce a senescent-like phenotype of human fibroblasts. J. Cell. Physiol. 207, 845–853 (2006).
Ghazi, A., Henis-Korenblit, S. & Kenyon, C. Regulation of Caenorhabditis elegans lifespan by a proteasomal E3 ligase complex. Proc. Natl Acad. Sci. USA 104, 5947–5952 (2007).
Vilchez, D. et al. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489, 263–268 (2012). Ablation of germline in C. elegans results in an increased, rpn-6 dependent, proteasome activity and proteotoxic resistance in somatic tissues.
Tomaru, U. et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 180, 963–972 (2012). A transgenic mouse with decreased proteasomal chymotrypsin-like activity exhibits a shortened lifespan, premature age-related phenotypes and aggravation of age-related metabolic disorders.
Li, L. et al. REGgamma deficiency promotes premature aging via the casein kinase 1 pathway. Proc. Natl Acad. Sci. USA 110, 11005–11010 (2013). PA28γ deficiency promotes premature aging in mice.
Matecic, M. et al. A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 6, e1000921 (2010).
Hars, E. S. et al. Autophagy regulates ageing in C. elegans. Autophagy 3, 93–95 (2007).
Toth, M. L. et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4, 330–338 (2008).
Simonsen, A. et al. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184 (2008). Enhancement of autophagy in the nervous system of D. melanogaster extends lifespan and confers protection against oxidative stress and protein accumulation.
Fredriksson, A. et al. Effects of aging and reproduction on protein quality control in soma and gametes of Drosophila melanogaster. Aging cell 11, 634–643 (2012). Although proteasome activity declines in somatic tissues during the ageing process, maturating oocytes and gonads maintain their high activity compared to age-matched somatic tissues.
Tsakiri, E. N. et al. Differential regulation of proteasome functionality in reproductive vs. somatic tissues of Drosophila during aging or oxidative stress. FASEB J. 27, 2407–2420 (2013).
Hamer, G., Matilainen, O. & Holmberg, C. I. A photoconvertible reporter of the ubiquitin-proteasome system in vivo. Nat. Methods 7, 473–478 (2010).
Vilchez, D. et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 489, 304–308 (2012). hESCs have increased proteasome activity.
Assou, S. et al. A gene expression signature shared by human mature oocytes and embryonic stem cells. BMC Genomics 10, 10 (2009).
Buckley, S. M. et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell 11, 783–798 (2012).
Pathare, G. R. et al. The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl Acad. Sci. USA 109, 149–154 (2012).
Atkinson, S. P. et al. A putative role for the immunoproteasome in the maintenance of pluripotency in human embryonic stem cells. Stem Cells 30, 1373–1384 (2012).
Hernebring, M., Brolen, G., Aguilaniu, H., Semb, H. & Nystrom, T. Elimination of damaged proteins during differentiation of embryonic stem cells. Proc. Natl Acad. Sci. USA 103, 7700–7705 (2006).
Hernebring, M. et al. Removal of damaged proteins during ES cell fate specification requires the proteasome activator PA28. Sci. Rep. 3, 1381 (2013).
Tra, T. et al. Autophagy in human embryonic stem cells. PLoS ONE 6, e27485 (2011).
Sanchez-Danes, A. et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol. Med. 4, 380–395 (2012).
Kirkwood, T. B. Evolution of ageing. Nature 270, 301–304 (1977).
Aguilaniu, H., Gustafsson, L., Rigoulet, M. & Nystrom, T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753 (2003).
Zhou, C. et al. Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 159, 1–13 (2014).
Erjavec, N., Cvijovic, M., Klipp, E. & Nystrom, T. Selective benefits of damage partitioning in unicellular systems and its effects on aging. Proc. Natl Acad. Sci. USA 105, 18764–18769 (2008).
Signer, R. A. & Morrison, S. J. Mechanisms that regulate stem cell aging and life span. Cell Stem Cell 12, 152–165 (2013).
Oliver, L., Hue, E., Priault, M. & Vallette, F. M. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 21, 2779–2788 (2012).
Salemi, S., Yousefi, S., Constantinescu, M. A., Fey, M. F. & Simon, H. U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 22, 432–435 (2012).
Vilchez, D., Simic, M. S. & Dillin, A. Proteostasis and aging of stem cells. Trends Cell Biol. 24, 161–170 (2014).
Wang, C., Liang, C. C., Bian, Z. C., Zhu, Y. & Guan, J. L. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 16, 532–542 (2013).
Chen, C., Liu, Y. & Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2, ra75 (2009).
Warr, M. R. et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327 (2013).
Eijkelenboom, A. & Burgering, B. M. FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 (2013).
Matsuda, N. & Tanaka, K. Does impairment of the ubiquitin-proteasome system or the autophagy-lysosome pathway predispose individuals to neurodegenerative disorders such as Parkinson’s disease? J. Alzheimers Dis. 19, 1–9 (2010).
Zabel, C. et al. Proteasome and oxidative phoshorylation changes may explain why aging is a risk factor for neurodegenerative disorders. J. Proteomics 73, 2230–2238 (2010).
Massey, A. C., Kaushik, S., Sovak, G., Kiffin, R. & Cuervo, A. M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl Acad. Sci. USA 103, 5805–5810 (2006).
Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 (2004). Rapamycin-mediated mTOR inhibition promotes aggregated huntingtin clearance through enhanced autophagy and protects against neurodegeneration in a fly model of Huntington’s Disease.
Cuervo, A. M. Autophagy in neurons: it is not all about food. Trends Mol. Med. 12, 461–464 (2006).
Keck, S., Nitsch, R., Grune, T. & Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 85, 115–122 (2003).
Tseng, B. P., Green, K. N., Chan, J. L., Blurton-Jones, M. & LaFerla, F. M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29, 1607–1618 (2008).
Grune, T. et al. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch. Biochem. Biophys. 500, 181–188 (2010).
Dickey, C. A. et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl Acad. Sci. USA 105, 3622–3627 (2008).
Lee, M. J., Lee, J. H. & Rubinsztein, D. C. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 105, 49–59 (2013).
Dange, T. et al. Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem. 286, 42830–42839 (2011).
Blair, L. J. et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 123, 4158–4169 (2013).
Nixon, R. A. et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122 (2005). Brains from humans with Alzheimer’s Disease present enhanced accumulation of autophagosomes even in early stages of the disease.
Yu, W. H. et al. Macroautophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98 (2005).
Cataldo, A. M. et al. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14, 671–680 (1995).
Boland, B. et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937 (2008).
Rozmahel, R. et al. Normal brain development in PS1 hypomorphic mice with markedly reduced gamma-secretase cleavage of betaAPP. Neurobiol. Aging 23, 187–194 (2002).
Jaeger, P. A. et al. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS ONE 5, e11102 (2010).
Pickford, F. et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190–2199 (2008).
Vogiatzi, T., Xilouri, M., Vekrellis, K. & Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556 (2008).
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N. & Rubinsztein, D. C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013 (2003).
McNaught, K. S., Perl, D. P., Brownell, A. L. & Olanow, C. W. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 56, 149–162 (2004). Proteasome inhibition in rats causes Parkinson’s Disease-like phenotype, establishing a correlation between impaired proteasome inhibition and the onset of the disease.
Bedford, L. et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 28, 8189–8198 (2008).
Wahl, C. et al. A comprehensive genetic study of the proteasomal subunit S6 ATPase in German Parkinson’s disease patients. J. Neural Transm. 115, 1141–1148 (2008).
Tofaris, G. K., Layfield, R. & Spillantini, M. G. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 509, 22–26 (2001).
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004).
Irrcher, I. et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 19, 3734–3746 (2010).
Plowey, E. D., Cherra, S. J. 3rd, Liu, Y. J. & Chu, C. T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105, 1048–1056 (2008).
Tashiro, Y. et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 287, 42984–42994 (2012).
Holmberg, C. I., Staniszewski, K. E., Mensah, K. N., Matouschek, A. & Morimoto, R. I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO. J. 23, 4307–4318 (2004). Aggregated mutant huntingtin cannot be efficiently degraded by the proteasome and irreversibly sequesters, in turn, the proteasome itself.
Finkbeiner, S. & Mitra, S. The ubiquitin-proteasome pathway in Huntington's disease. Sci. World J. 8, 421–433 (2008).
Diaz-Hernandez, M. et al. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 98, 1585–1596 (2006).
Ardley, H. C., Hung, C. C. & Robinson, P. A. The aggravating role of the ubiquitin-proteasome system in neurodegeneration. FEBS Lett. 579, 571–576 (2005).
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N. & Goldberg, A. L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104 (2004).
Bennett, E. J., Bence, N. F., Jayakumar, R. & Kopito, R. R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351–365 (2005).
Hipp, M. S. et al. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington's disease. J. Cell Biol. 196, 573–587 (2012).
Pratt, G. & Rechsteiner, M. Proteasomes cleave at multiple sites within polyglutamine tracts: activation by PA28gamma(K188E). J. Biol. Chem. 283, 12919–12925 (2008).
Qin, Z. H. et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244 (2003).
Ravikumar, B., Duden, R. & Rubinsztein, D. C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 (2002).
Sarkar, S. et al. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat. Chem. Biol. 3, 331–338 (2007).
Martinez-Vicente, M. et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 13, 567–576 (2010).
Shibata, M. et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 281, 14474–14485 (2006).
Atwal, R. S. & Truant, R. A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 4, 91–93 (2008).
Ravikumar, B. et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 37, 771–776 (2005).
Finkel, T., Serrano, M. & Blasco, M. A. The common biology of cancer and ageing. Nature 448, 767–774 (2007).
Whitesell, L. & Lindquist, S. L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 (2005).
Maiuri, M. C. et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 16, 87–93 (2009).
Mathew, R. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075 (2009).
Elgendy, M., Sheridan, C., Brumatti, G. & Martin, S. J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 42, 23–35 (2011).
Young, A. R. et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 23, 798–803 (2009).
Chondrogianni, N., Petropoulos, I., Franceschi, C., Friguet, B. & Gonos, E. S. Fibroblast cultures from healthy centenarians have an active proteasome. Exp. Gerontol. 35, 721–728 (2000).
Perez, V. I. et al. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl Acad. Sci. USA 106, 3059–3064 (2009).
Ungvari, Z. et al. Testing predictions of the oxidative stress hypothesis of aging using a novel invertebrate model of longevity: the giant clam (Tridacna derasa). J. Gerontol. A Biol. Sci. Med. Sci. 68, 359–367 (2013).
Chen, Q., Thorpe, J., Dohmen, J. R., Li, F. & Keller, J. N. Ump1 extends yeast lifespan and enhances viability during oxidative stress: central role for the proteasome? Free Radic. Biol. Med. 40, 120–126 (2006).
Dohmen, R. J., Willers, I. & Marques, A. J. Biting the hand that feeds: Rpn4-dependent feedback regulation of proteasome function. Biochim. Biophys. Acta 1773, 1599–1604 (2007).
Kruegel, U. et al. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 7, e1002253 (2011).
Tonoki, A. et al. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell. Biol. 29, 1095–1106 (2009).
Chondrogianni, N. & Gonos, E. S. Proteasome activation as a novel antiaging strategy. IUBMB Life 60, 651–655 (2008).
Kenyon, C. J. The genetics of ageing. Nature 464, 504–512 (2010).
Bjedov, I. et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 11, 35–46 (2010).
Harrison, D. E. et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009).
Kapahi, P. et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 11, 453–465 (2010).
Pyo, J. O. et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4, 2300 (2013). Ubiquitous autophagy enhancement in mice extends lifespan and alleviates the age-associated phenotypes.
Zhang, C. & Cuervo, A. M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965 (2008).
Lapierre, L. R. et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 4, 2267 (2013).
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011).
O'Rourke, E. J. & Ruvkun, G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 15, 668–676 (2013).
Seo, H., Sonntag, K. C., Kim, W., Cattaneo, E. & Isacson, O. Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2, e238 (2007).
Berger, Z. et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 15, 433–442 (2006).
Caccamo, A., Majumder, S., Richardson, A., Strong, R. & Oddo, S. Molecular interplay between mammalian _target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J. Biol. Chem. 285, 13107–13120 (2010).
Spencer, B. et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29, 13578–13588 (2009).
Panowski, S. H. & Dillin, A. Signals of youth: endocrine regulation of aging in Caenorhabditis elegans. Trends Endocrinol. Metab. 20, 259–264 (2009).
Flachsbart, F. et al. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl Acad. Sci. USA 106, 2700–2705 (2009).
Suh, Y. et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl Acad. Sci. USA 105, 3438–3442 (2008).
Matilainen, O., Arpalahti, L., Rantanen, V., Hautaniemi, S. & Holmberg, C. I. Insulin/IGF-1 signaling regulates proteasome activity through the deubiquitinating enzyme UBH-4. Cell Rep. 3, 1980–1995 (2013).
Melendez, A. et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391 (2003).
Gelino, S. & Hansen, M. Autophagy—an emerging anti-aging mechanism. J. Clin. Exp. Pathol Suppl 4, doi:pii.006 (2012).
Hansen, M. et al. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4, e24 (2008).
Cohen, E. & Dillin, A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat. Rev. Neurosci. 9, 759–767 (2008).
Cohen, E. et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139, 1157–1169 (2009).
Morley, J. F., Brignull, H. R., Weyers, J. J. & Morimoto, R. I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 99, 10417–10422 (2002).
Volovik, Y., Marques, F. C. & Cohen, E. The nematode Caenorhabditis elegans: a versatile model for the study of proteotoxicity and aging. Methods 68, 458–464 (2014).
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W. & Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610 (2006).
Florez-McClure, M. L., Hohsfield, L. A., Fonte, G., Bealor, M. T. & Link, C. D. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy 3, 569–580 (2007).
Bartke, A. Growth hormone, insulin and aging: the benefits of endocrine defects. Exp. Gerontol. 46, 108–111 (2011).
Liu, G., Rogers, J., Murphy, C. T. & Rongo, C. EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J. 30, 2990–3003 (2011).
Kenyon, C. A pathway that links reproductive status to lifespan in Caenorhabditis elegans. Ann. NY Acad. Sci. 1204, 156–162 (2010).
Shemesh, N., Shai, N. & Ben-Zvi, A. Germline stem cell arrest inhibits the collapse of somatic proteostasis early in Caenorhabditis elegans adulthood. Aging Cell 12, 814–822 (2013).
Vilchez, D. et al. FOXO4 is necessary for neural differentiation of human embryonic stem cells. Aging Cell 12, 518–522 (2013).
Ermolaeva, M. A. et al. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 501, 416–420 (2013).
Lapierre, L. R., Gelino, S., Melendez, A. & Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 21, 1507–1514 (2011).
Fontana, L., Partridge, L. & Longo, V. D. Extending healthy life span—from yeast to humans. Science 328, 321–326 (2010).
Zid, B. M. et al. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 139, 149–160 (2009).
Carrano, A. C., Liu, Z., Dillin, A. & Hunter, T. A conserved ubiquitination pathway determines longevity in response to diet restriction. Nature 460, 396–399 (2009).
Bishop, N. A. & Guarente, L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447, 545–549 (2007).
Ferguson, A. A., Springer, M. G. & Fisher, A. L. skn-1-Dependent and -independent regulation of aip-1 expression following metabolic stress in Caenorhabditis elegans. Mol. Cell. Biol. 30, 2651–2667 (2010).
Stanhill, A. et al. An arsenite-inducible 19S regulatory particle-associated protein adapts proteasomes to proteotoxicity. Mol. Cell 23, 875–885 (2006).
Hassan, W. M., Merin, D. A., Fonte, V. & Link, C. D. AIP-1 ameliorates beta-amyloid peptide toxicity in a Caenorhabditis elegans Alzheimer’s disease model. Hum. Mol. Genet. 18, 2739–2747 (2009).
Yun, C. et al. Proteasomal adaptation to environmental stress links resistance to proteotoxicity with longevity in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 105, 7094–7099 (2008).
Kapeta, S., Chondrogianni, N. & Gonos, E. S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 285, 8171–8184 (2010).
Jia, K. & Levine, B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3, 597–599 (2007).
Acknowledgements
This work was supported by the Howard Hughes Medical Institute (HHMI), the Deutsche Forschungsgemeinschaft (DFG) (CECAD), the University of Cologne Advanced Postdoc Grant and the European Commission (FP7-PEOPLE-2013-CIG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Vilchez, D., Saez, I. & Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun 5, 5659 (2014). https://doi.org/10.1038/ncomms6659
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6659
This article is cited by
-
Derailed protein turnover in the aging mammalian brain
Molecular Systems Biology (2024)
-
The HSP40 family chaperone isoform DNAJB6b prevents neuronal cells from tau aggregation
BMC Biology (2023)
-
Mitochondrial malfunction and atrophy of astrocytes in the aged human cerebral cortex
Nature Communications (2023)
-
Cold temperature extends longevity and prevents disease-related protein aggregation through PA28γ-induced proteasomes
Nature Aging (2023)
-
In planta expression of human polyQ-expanded huntingtin fragment reveals mechanisms to prevent disease-related protein aggregation
Nature Aging (2023)
