

Similar content being viewed by others


To the editor:
In childhood and young adolescent B-cell precursor acute lymphoblastic leukemia (B-ALL) PAX5 is one of the most frequent _targets of genetic alterations comprising deletions, intragenic amplifications (PAX5AMP), and point mutations as well as rearrangements (PAX5-r) with multiple partner genes [1]. PAX5-r account for 2–3% of all newly diagnosed B-ALL cases and result in the expression of fusion oncoproteins [1, 2].
Patients with PAX5-r tend to have higher relapse and poorer overall survival (OS) rates compared to good-risk genetic groups [3,4,5]. Recent studies indicate that PAX5-r is associated with the IKZF1plus copy number alteration (CNA) profile and a rather poor event-free survival (EFS) [3, 5]. In small cohorts of infant B-ALL, PAX5-r patients had a worse outcome than those with other non-KMT2A genetic subtypes [6, 7].
Most, but not all, cases with PAX5-r belong to the PAX5-altered (PAX5alt) subtype identified by gene expression profiling [1]. PAX5alt is associated with an intermediate to poor prognosis with a strong dependence on IKZF1 codeletion [1, 8, 9]. Despite sharing a distinctive expression signature, the underlying genetic landscape of PAX5alt is heterogeneous and various types of PAX5 lesions, which differently affect disease biology and outcomes, are merged into a single group [1, 9]. Moreover, PAX5 is fused to a multitude of different partner genes, and since most of these fusions have been detected only in a few cases [1], the impact of individual PAX5-r on outcomes remains to be determined.
In this international study, we collected cases with PAX5::AUTS2 B-ALL diagnosed over the past decades in patients aged 0–18 years, without any time or study protocol restrictions, to evaluate the prognostic relevance of this rare genetic subtype. In accordance with the Declaration of Helsinki, patients were enrolled in respective clinical trials with written informed consent from their parents or legal guardians, and the use of surplus diagnostic material for research purposes was approved by the institutional review boards of the participating centers.
Patients with a confirmed PAX5::AUTS2 fusion detected by RT-PCR or a next-generation sequencing (NGS) approach were eligible for inclusion in the study. Additionally, since PAX5::AUTS2 frequently results from unbalanced der(9)t(7;9)(q11;p13) rearrangements, cases with breakpoints in both PAX5 and AUTS2 detected by single nucleotide polymorphism (SNP) array analysis were included without additional molecular genetic verification, likewise cases identified by optical genome mapping (OGM). All PAX5::AUTS2 fusion transcripts were in frame or at least predicted to be. CNA profiling for IKZF1 deletion and IKZF1plus was performed by SNP array, multiplex ligation-dependent probe amplification (MLPA), digitalMLPA or OGM following standard procedures (Supplementary Methods).
The Kaplan–Meier method was used to determine EFS and OS rates and the analysis was performed in R (version 4.2.2) statistical environment. Adverse events were defined as relapse at any site, the development of a second malignant neoplasm (SMN), or death from any cause. OS was defined as the time from diagnosis to the date of last follow-up or death. The cumulative incidence of relapse (CIR) was calculated using the Kalbfleisch and Prentice method and compared with the Gray’s test considering death and SMN as competing events. Multivariate analysis was conducted using a Cox proportional hazards regression model.
We identified 50 patients diagnosed with PAX5::AUTS2 B-ALL, including 16 previously published cases (Supplementary Table S1). The main demographic and clinical features showed a male predominance (60% vs 40%), a median age of 2.0 years (range 0.6–15.5 years), including eight infants (≤1 year), and highly variable white blood cell counts (WBC) ranging from 1.2–537.6 × 109/L (median 40.1 × 109/L) (Table 1).
We detected IKZF1 deletions in 69.6% (32/46 with available data) of PAX5::AUTS2 cases, most displaying also CDKN2A/B (90.0%, 27/30) and/or PAX5 (87.1%, 27/31) deletions (Supplementary Table S1). Based on this deletion pattern, 62.2% (28/45) showed the IKZF1plus CNA profile [10] (Table 1), which is higher than reported for childhood PAX5alt (20–30% IKZF1-deleted, 20% IKZF1plus) or PAX5AMP (13% IKZF1plus) cases [1, 9, 11].
According to National Cancer Institute (NCI) criteria (age ≥10 years and/or WBC ≥50 × 109/L; infants), 60.0% (30/50) of patients had high-risk (HR) status. Of the patients with available data, 24.4% (10/41) showed a poor prednisone response (Table 1), which is ~15% higher than in the average population of childhood B-ALL [10]. Measurable residual disease (MRD) data assessed by flow cytometry (FCM) on day 15 were only available for 23 patients, and according to AIEOP-BFM definitions, 30.4% (7/23) showed HR disease with ≥10% residual blast cells [12]. At the end of induction therapy (EOI; days 28-42), PCR- and/or FCM-MRD measurements showed that 79.5% (35/44) of patients were MRD-positive: 31.8% (14/44) ≥5×10−4 and 47.7% (21/44) <5 × 10−4. At the end of consolidation therapy (EOC; mainly day 78; range 71–117 days), 29.7% (11/37) of patients still presented with MRD, but except for two (≥5 × 10−4), with levels ≤1 × 10−4 (Table 1 and Supplementary Table S1).
Overall, 46.0% (23/50) of PAX5::AUTS2 patients experienced a relapse with a median time-to-event of 1.6 years (range 0.6-4.1 years) with the majority (65.2%, 15/23) occurring within two years after diagnosis (Fig. 1A; Supplementary Table S1). Most patients had isolated bone marrow (BM; 60.9%, 14/23) relapses, but CNS disease was detected in 34.8% (8/23) of patients mostly combined with BM recurrence (Table 1). In total, 38.0% (19/50) of patients had CNS disease: 22.0% (11/50) only at diagnosis, 6.0% (3/50) at diagnosis and relapse, and 10.0% (5/50) only at relapse. Among infants, 87.5% (7/8) had CNS involvement, with 62.5% (5/8) already at diagnosis (Supplementary Methods), one patient at both time points, and two additional cases only at relapse (Supplementary Table S1). CNS disease was more frequently detected in infants than in children [87.5% (7/8) vs 40.0% (12/30), Fisher exact test p = 0.0031].

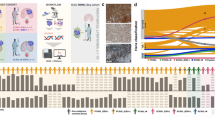
A Swimmer plot illustrating the clinical course of each individual patient. Relapses (orange dots), bone marrow transplantation (BMT; blue triangles), and death (red crosses) as well as age groups <18 months (light brown bars) and ≥18 months (gray bars) are indicated. B–C Cumulative incidence of relapse (CIR) and Kaplan-Meier survival curves of event-free (EFS) and overall survival (OS), (B) of all PAX5::AUTS2 cases (n = 50), (C) based on age group; red, <18 months; blue, ≥18 months. Gray’s test p-value for CIR, Log-ranks test p-values for EFS and OS. D Forrest plot showing results of multivariate Cox regression analysis. HR hazard ratio, CI confidence interval, p Gray’s test p-value, WBC white blood cell count, NCI-HR National Cancer Institute high-risk, MRD-EOI (measurable residual disease) determined by PCR and/or flow cytometry at the end of induction (EOI) therapy.
The 5-year CIR for all patients was 48.0 ± 7.8% (Fig. 1B) and of the patients who relapsed, 17.4% (4/23) experienced a second BM relapse within 3–9 months of the first (Fig. 1A). Among patients experiencing a relapse, 30.4% (7/23) died, three of progressive disease and four of treatment-related mortality (TRM), and in total, 20.0% (10/50) of patients died (n = 3 leukemia, n = 4 TRM, n = 3 other/unknown reasons) (Fig. 1A, Table 1 and Supplementary Table S1).
With a median follow-up of 4.8 years (range 0.3–13.0 years), we observed 5-year EFS and OS rates for all patients of 47.9 ± 7.6% and 76.2 ± 7.1%, respectively, (Fig. 1B). Notably MRD levels (≥5 × 10−4 vs <5 × 10−4 vs negative) at EOI did not significantly impact CIR, EFS or OS (Supplementary Fig. S1A), suggesting that in PAX5::AUTS2 patients EOI-MRD negativity does not predict a favorable outcome. Patients with ≥5 × 10−4 EOI-MRD had a worse EFS compared to cases with <5 × 10−4/negative EOI-MRD, but the result did not reach statistical significance (p = 0.057) (Supplementary Fig. S1B). Furthermore, we did not find any differences in 5-year EFS and OS between IKZF1plus and non-IKZF1plus patients (46.6 ± 9.9% vs 60.6 ± 14.0%, p = 0.67; 78.7 ± 8.5% vs 93.8 ± 6.1%, p = 0.89) indicating that also IKZF1plus has no predictive value (Supplementary Fig. S1C).
Given that patients were enrolled in various clinical trials with differing risk stratification criteria and treatment regimens, we compared the outcomes between earlier clinical trials and the two most contemporary ones (i.e., AIEOP-BFM ALL 2017, ALLTogether-1) but did not find any significant improvement (Supplementary Fig. S1D).
When comparing the outcomes of children and infants, we observed a higher CIR and consequently a poorer EFS for infant patients (Supplementary Fig. S2A). As several patients were just over 1 year of age (Table 1), we explored whether age influenced the outcome. Our analysis according to different age groups revealed that patients aged <18 months had a significantly higher 5-year CIR (68.5 ± 12.7% vs 36.2 ± 9.4%, p = 0.006) and worse outcome (Fig. 1C) than other age groups (Supplementary Figs. S2B–D). In multivariate analysis, EOI-MRD ≥5 × 10−4 (hazard ratio 15.55, p = 0.001) and age (<18 months vs older; hazard ratio 16.65, p = 0.001) had an independent impact on EFS (Fig. 1D).
Collectively, our study demonstrates that childhood PAX5::AUTS2 B-ALL is characterized by a high frequency of CNS involvement and is a relapse-prone subtype with poor outcomes. The disease mainly affects infants and toddlers with over 80% being less than three years old, which differs from the childhood PAX5alt group, which is more common in older patients (children 1–18 years, n = 94, median 9.5 years; ST1 cohort [1]) [1, 8, 9]. The observed EFS and OS survival rates for PAX5::AUTS2 B-ALL were lower than those in the overall PAX5alt group but similar to those of patients with PAX5AMP [1, 8, 9, 11], underlining the importance of focusing outcome analysis on genetically homogeneous entities with unique alterations.
Our data also support the notion that PAX5-r is in general recurrent in infants and along with KMT2A-r may represent a further subgroup of infant B-ALL with a potentially dismal outcome, while NUTM1-r has a favorable prognosis [6, 7, 13].
The finding that PAX5::AUTS2 patients enrolled in contemporary trials still exhibit high relapse rates, emphasizes the need to further explore innovative _targeted treatment options [3]. Moreover, strategies to mitigate the risk of relapse and treatment-related mortality associated with salvage therapy are needed. Frontline use of the bi-specific T-cell engager blinatumomab, which has been proven to be safe and effective also in infants [14, 15] may be a worthwhile consideration.
Data availability
The data relevant to this study are provided in Supplementary Table S1. More detailed de-identified patient and genetic data may only be obtained from the relevant clinical trial committees upon reasonable request.
References
Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307.
Nebral K, Denk D, Attarbaschi A, Konig M, Mann G, Haas OA, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:134–43.
Fazio G, Bresolin S, Silvestri D, Quadri M, Saitta C, Vendramini E, et al. PAX5 fusion genes are frequent in poor risk childhood acute lymphoblastic leukaemia and can be _targeted with BIBF1120. EBioMedicine. 2022;83:104224.
Schwab C, Cranston RE, Ryan SL, Butler E, Winterman E, Hawking Z, et al. Integrative genomic analysis of childhood acute lymphoblastic leukaemia lacking a genetic biomarker in the UKALL2003 clinical trial. Leukemia. 2023;37:529–38.
Luhmann JL, Zimmermann M, Hofmann W, Bergmann AK, Moricke A, Cario G, et al. Deciphering the molecular complexity of the IKZF1(plus) genomic profile using optical genome mapping. Haematologica. 2024;109:1582–7.
Fazio G, Bardini M, De Lorenzo P, Grioni A, Quadri M, Pedace L, et al. Recurrent genetic fusions redefine MLL germline acute lymphoblastic leukemia in infants. Blood. 2021;137:1980–4.
Popov A, Tsaur G, Permikin Z, Henze G, Verzhbitskaya T, Plekhanova O, et al. Genetic characteristics and treatment outcome in infants with KMT2A germline B-cell precursor acute lymphoblastic leukemia: results of MLL-Baby protocol. Pediatr Blood Cancer. 2023;70:e30204.
Jeha S, Choi J, Roberts KG, Pei D, Coustan-Smith E, Inaba H, et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021;2:326–37.
Li Z, Lee SHR, Chin WHN, Lu Y, Jiang N, Lim EH, et al. Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. 2021;5:5226–38.
Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1(plus) defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36:1240–9.
Schwab C, Nebral K, Chilton L, Leschi C, Waanders E, Boer JM, et al. Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv. 2017;1:1473–7.
Basso G, Veltroni M, Valsecchi MG, Dworzak MN, Ratei R, Silvestri D, et al. Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol. 2009;27:5168–74.
Boer JM, Valsecchi MG, Hormann FM, Antic Z, Zaliova M, Schwab C, et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia. 2021;35:2978–82.
van der Sluis IM, de Lorenzo P, Kotecha RS, Attarbaschi A, Escherich G, Nysom K, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. 2023;388:1572–81.
Lyons KU, Gore L. Bispecific T-cell engagers in childhood B-acute lymphoblastic leukemia. Haematologica. 2024;109:1668–76.
Acknowledgements
This study was in part supported by The European Union’s H2020 Research and Innovation Program through Grant number 825749 “CLOSER: Childhood Leukemia: Overcoming Distance between South America and Europe Regions” to AVM, JT, LC, SS; the ERA-NET TRANSCAN/European Commission under the 7th Framework Programme (FP7), granted by Fondation ARC (www.fondation-arc.org) to HC; a grant from the Italian Association for Cancer Research (AIRC; IG-2023 n. 29191) to GF; a grant from the Deutsche Kinderkrebsstiftung (DKS 2021.14) to perform Optical Mapping in IKZF1 deleted ALL to DS; co-funded by the EU and the State Budget of Czechia OP JAC, project SALVAGE, No. CZ.02.01.01/00/22_008/0004644 to JT, MZ; AP and WM were supported by funding from the “Label-free and rapid optical imaging, detection and sorting of leukemia cells” project carried out within the Team-Net programme (POIR.04.04.00-00-16ED/18-00) of the Foundation for Polish Science co-financed by the European Union; BAL received a return research grant (1193718 – BRA – HFSTCAPES-P) from the Alexander von Humboldt Foundation and the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro—FAPERJ (E_13/2023); ME is supported by Brazilian National Counsel of Technological and Scientific Development-CNPq (PQ-310982/2023-5) and Fundação Carlos Chagas Filho de Amparo à Pesquisa-FAPERJ (E-26/201.113/2021) research grants; AVM was supported by a Blood Cancer UK programme grant (15036). The authors gratefully acknowledge the Center for Biological Resources (CRBcancer; BB-0033-00076) of the Robert Debré Hospital and the VIVO biobank in the UK.
Author information
Authors and Affiliations
Contributions
SS conceived the study, coordinated the data collection, analyzed data, and wrote the manuscript; ACE, GF, AP, JMB, DS, DG, ES, LD, JB, BAL, MZ, GE, IS, ME, JT, LC, MP, AVM, AKB, MLB, WM, GC, and HC contributed genetic and clinical patient data; SH, AI, and KN analyzed, reviewed and interpreted SNP array data; MK, performed FISH analysis and interpreted data; KF conducted molecular genetic analysis of patient samples; DS interpreted data and conducted outcome analysis. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted in accordance with the ethical standards of the institutional and/or national research committees and with the ethical standards as laid down in the Declaration of Helsinki, and all methods were performed in accordance with the relevant guidelines and regulations. Patients were enrolled in an approved international or national clinical trial with written informed consent from their parents or legal guardians. The institutional review boards of the participating study groups approved the use of anonymized patient data for research purposes.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Caye-Eude, A., Fazio, G., Pastorczak, A. et al. PAX5::AUTS2 childhood B-ALL: a relapse-prone genetic subtype with frequent central nervous system involvement and a poor outcome. Leukemia (2024). https://doi.org/10.1038/s41375-024-02502-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41375-024-02502-5
