
Abstract
Manipulation C-C coupling pathway is of great importance for selective CO2 electroreduction but remain challenging. Herein, two model Cu-based catalysts, by modifying Cu nanowires with Ag nanoparticles (AgCu NW) and Ag single atoms (Ag1Cu NW), respectively, are rationally designed for exploring the C-C coupling mechanisms in electrochemical CO2 reduction reaction (CO2RR). Compared to AgCu NW, the Ag1Cu NW exhibits a more than 10-fold increase of C2 selectivity in CO2 reduction to ethanol, with ethanol-to-ethylene ratio increased from 0.41 over AgCu NW to 4.26 over Ag1Cu NW. Via a variety of operando/in-situ techniques and theoretical calculation, the enhanced ethanol selectivity over Ag1Cu NW is attributed to the promoted H2O dissociation over the atomically dispersed Ag sites, which effectively accelerated *CO hydrogenation to form *CHO intermediate and facilitated asymmetric *CO-*CHO coupling over paired Cu atoms adjacent to single Ag atoms. Results of this work provide deep insight into the C-C coupling pathways towards _target C2+ product and shed light on the rational design of efficient CO2RR catalysts with paired active sites.
Similar content being viewed by others

Introduction
Electrochemical CO2 reduction reaction (CO2RR) offers a promising strategy to achieve carbon neutrality by converting CO2 into value-added carbon-containing fuels or chemicals using renewable electricity1,2,3,4,5. Compared to C1 products, direct conversion of CO2 to multi-carbon (C2+) products (e.g., ethylene, ethanol, etc.) is more attractive6,7,8,9,10. Unfortunately, the catalytic performance of electrochemical CO2 reduction to C2+ products is still far from satisfactory with low selectivity and sluggish reaction kinetics11,12.
The slow carbon-carbon (C-C) coupling reaction is the fundamental issue for poor conversion of CO2 to C2+ products13,14. Mechanistically, the CO2-to-C2+ pathway (eg., ethylene, ethanol) is conventionally considered to proceed via a complex C-C coupling mechanism involving various intermediates such as *COCO, *COCHO, *COCOH, *CH2CO, and *OCHCH2, etc1,15,16,17,18,19,20,21,22,23,24,25. Therefore, regulating the key intermediates involved in the CO2RR over catalytic sites and optimizing their binding energies become critical to adjust the C-C coupling pathways towards _targeted C2+ products. Nevertheless, manipulation of C-C coupling pathways is still highly challenging due to the complexity of the catalytic system, the unclear reaction intermediates and the limited mechanistic understanding.
Cu-based materials have been well established as promising electrocatalysts capable to reduce CO2 to multiple C2+ products11,26,27,28. Till now, a large number of Cu-based tandem catalysts have been developed to realize high selectivity in CO2RR to produce C2+ products, primarily resulting from the improved mass transport of *CO intermediate from CO-formation catalyst and the promoted C-C coupling over Cu surface29,30,31,32. For instance, Cu-Ag catalyst often shows high activity for the formation of C2+ hydrocarbons and oxygenates33,34. However, the tandem approach usually leads to uncontrolled C-C coupling, producing a variety of C2+ products such as ethylene, ethanol, and acetate through cascade catalysis. Grätzel et al.35 and Yang et al.34 suggested that the efficient *CO spillover from Ag to Cu could improves ethylene performance. Qiao et al.23 claimed that significantly boosted ethanol production could be achieved by the tailored introduction of Ag to optimize the coordinated number and oxidation state of surface Cu sites. However, due to the complexity and diversity of the catalyst’s structure, the underlying mechanism of CO2RR to ethylene/ethanol in the Cu-Ag system remains unclear34,36,37. Recently, the rapid development of single-atom catalysis has opened up new possibilities for constructing model catalysts with asymmetric catalytic sites38,39. In particular, modification of copper-based catalysts with single metal atoms, and rational design of catalysts with dual- or multi- catalytic sites have been shown promising to induce asymmetric C-C coupling in CO2RR40,41,42. However, little understanding about the exact asymmetric C-C coupling mechanism is known.
In this work, via a modified galvanic replacement approach, two model Cu-based catalysts decorated with Ag nanoparticles (AgCu NW) and single Ag atoms (Ag1Cu NW) were rationally designed and prepared for CO2RR, which exhibited distinct selectivity in electrochemical CO2 reduction to ethylene (Faradaic efficiency: ca. 54.9%, current density: ca. 156.0 mA cm−2) and ethanol (Faradaic efficiency: ca. 56.3%, current density: ca. 172.8 mA cm−2). Based on isotopic labelling experiments together with various operando/in-situ characterizations including operando X-ray absorption spectroscopy (XAS), quasi in-situ X-ray photoelectron spectroscopy (XPS), operando Raman spectroscopy and operando attenuated total reflectance surface enhanced infrared absorption spectroscopy (ATR-SEIRAS), the dynamic oxidation state evolution of Cu and Ag were discerned, and the key reaction intermediates involved in C-C coupling during CO2RR, such as *COCO and *COCHO, were captured over AgCu NW and Ag1Cu NW. Further coupled with density functional theory (DFT) calculations, the atomically dispersed Ag was identified to boost H2O dissociation to release H+ and reduce the energy barrier for the formation of *CHO over its neighboring Cu site, leading to boosted asymmetric *CO-*CHO coupling over paired Cu atoms adjacent to the single Ag atom.
Results
Catalyst synthesis and characterization
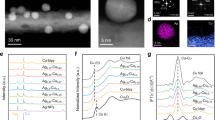
The model Cu-based catalysts of Cu nanowires decorated with Ag nanoparticles and single Ag atoms were synthesized via a galvanic replacement strategy43. In detail, the standard electrode potential difference drives the galvanic replacement reaction of \({2{{{\rm{Ag}}}}}^{+}\left({{{\rm{aq}}}}\right)+{{{\rm{Cu}}}}\left({{{\rm{s}}}}\right)\to 2{{{\rm{Ag}}}}\left({{{\rm{s}}}}\right)+{{{{\rm{Cu}}}}}^{2+}\left({{{\rm{aq}}}}\right)\) (Supplementary Table 1). Figure 1a illustrates the synthesis of AgCu NW and Ag1Cu NW and the experimental details are summarized in the Supplementary Methods. One-dimensional Cu nanowires with an average diameter of 45 ± 3 nm were imaged by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 1b and Supplementary Figs. 1, 2). As shown in Fig. 1c, the AgCu NW exhibits typical characteristics of supported catalyst, with Ag nanoparticles distributed as islands on the surface of the Cu NW (Supplementary Figs. 3, 4), and obvious grain boundaries between Ag nanoparticles and Cu NW can be clearly observed (Supplementary Fig. 4b). In contrast, Ag1Cu NW displays similar microstructural features to Cu NW (Fig. 1d and Supplementary Figs. 5, 6). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), energy-dispersive X-ray spectroscopy (EDX) elemental mapping and electron microscopy-based atom recognition statistics (EMARS) analysis on HAADF images of Ag1Cu NW indicate that isolated Ag atoms are uniformly distributed on the surface of Cu NW matrix in Ag1Cu NW (Fig. 1e and Supplementary Figs. 6c, d and 7)44. The X-ray diffraction (XRD) patterns of Cu NW and Ag1Cu NW exhibit identical diffraction peaks, which can be assigned to fcc Cu (Supplementary Fig. 8), indicating atomic dispersion of Ag atoms in Ag1Cu NW45. While for AgCu NW, diffraction peaks of both metallic Cu and Ag are clearly observable. The contents of Ag in Ag1Cu NW and AgCu NW were measured by inductively coupled plasma emission spectrometer (ICP) and the results are summarized in Supplementary Table 2.

a Schematic illustration showing synthesis of Ag1Cu NW and AgCu NW (Ag (gray) and Cu (yellow)). TEM images of (b) Cu NW, (c) AgCu NW and (d) Ag1Cu NW. e HAADF-STEM image of Ag1Cu NW. Notes: Ag atoms are not restricted to the positions indicated by the yellow circles. f High-resolution Cu 2p and Ag 3 d XPS spectra of Cu NW, Ag NW, Ag1Cu NW and AgCu NW. Normalized (g) Cu and (h) Ag K-edge XANES spectra of Ag1Cu NW, AgCu NW and reference samples (inset shows the enlarged Cu and Ag K-edge XANES spectra).
The surface valence states of the as-synthesized Cu NW, Ag1Cu NW and AgCu NW were analyzed by X-ray photoelectron spectroscopy (XPS). The survey XPS spectra show a gradual increase of the Ag peak with increasing Ag content (Supplementary Fig. 9 and Supplementary Table 3). The high-resolution O 1 s XPS spectrum of Cu NW, Ag1Cu NW and AgCu NW exhibit similar characteristic peaks at 530.5 and 532.4 eV, which are related to adsorbed surface hydroxide (OHads) and adsorbed water (H2Oads), respectively (Supplementary Fig. 10)46. As shown in Fig. 1f and Supplementary Fig. 11, the binding energy of Ag 3 d displays a positive shift from 368.2 eV (Ag NW) to 368.4 (Ag1Cu NW) and 368.3 eV (AgCu NW), implying a gradually decreased charge density on Ag due to electron transfer from Ag to Cu47.
To further determine the chemical states and local coordination environments of Cu and Ag in Ag1Cu NW and AgCu NW, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were conducted. The Cu K-edge XANES spectra show that the edge features of Ag1Cu NW and AgCu NW (designated as 1 s → 4 p transition) are close to those of the reference commercial Cu foil (Fig. 1g), indicating that Cu in Ag1Cu NW and AgCu NW exist mainly in the form of Cu (0)48. The corresponding EXAFS spectra of Ag1Cu NW and AgCu NW also show the dominant Cu-Cu coordination at ca. 2.21 Å (Supplementary Figs. 12, 13 and Supplementary Table 4)49. As displayed in Fig. 1h, the absorption edges of the Ag K-edge XANES for Ag1Cu NW and AgCu NW are observed shifted towards higher energies compared to that for Ag foil, indicating a higher average valence state of Ag in Ag1Cu NW and AgCu NW, matching well with the XPS results.
CO2RR performance
The electrochemical CO2RR performance of Cu NW, Ag1Cu NW and AgCu NW were first evaluated in an H-type cell filled with CO2-saturated 0.1 M KHCO3 electrolyte (pH = 6.8), and the gas and liquid products were quantified by gas chromatography (GC) and nuclear magnetic resonance (NMR), respectively (Supplementary Figs. 14, 15). Linear sweep voltammetry (LSV) curves shown in Fig. 2a reveal that the current densities of both Ag1Cu NW and AgCu NW outperform Cu NW in the potential range from −0.6 to −1.4 V vs. RHE. Meanwhile, the current densities of Cu NW, Ag1Cu NW and AgCu NW in CO2-saturated electrolyte are obviously higher than those in N2-saturated electrolyte at the same applied potential, indicating that CO2RR could occur readily compared to HER, consistent with the CV results (Supplementary Fig. 16). Figure 2b summarizes the potential-dependent Faradaic efficiencies (FEs) of different CO2RR products over Ag1Cu NW (I), AgCu NW (II) and Cu NW (III). Pure Cu NW electroreduced CO2 to a wide variety of products including CO, HCOOH, C2H4, and ethanol50,51. The AgCu NW and Ag1Cu NW exhibited greatly improved CO2RR performance with distinctive selectivity towards ethylene and ethanol, with CO2-to-ethylene and CO2-to-ethanol Faradaic efficiency of ca. 42.4% and ca. 42.8%, respectively (Fig. 2b). The Faradaic efficiency of total C2 products over Ag1Cu NW could reach up to 65.1% at −1.22 V vs. RHE (Supplementary Figs. 17–19). Furthermore, the Ag1Cu NW achieved the optimal ethanol Faradaic efficiency of 56.3% with a current density of 172.8 mA cm−2 at −1.0 V vs. RHE in a flow cell (Fig. 2c, Supplementary Figs. 20, 21a), while ethylene Faradaic efficiency reach a maximum value of 54.9% over the AgCu NW with a current density of 156.0 mA cm−2 at −1.1 V vs. RHE in a flow cell (Supplementary Figs. 21b, 22, 23 and Supplementary Table 5). Both Ag1Cu NW and AgCu NW catalysts displayed outstanding catalytic stability, evidenced by the tiny decay in current density and ethanol/ethylene Faradaic efficiency during continuous electrolysis (Fig. 2d and Supplementary Fig. 24). The Ag1Cu NW and AgCu NW exhibited similar surface structures, valence states, and Ag species distributions after continuous electrolysis as observed in the fresh catalyst (Supplementary Figs. 25–27 and Supplementary Table 6).

a LSV curves acquired in CO2- and N2-saturated 0.1 M KHCO3 solution on a rotating disc electrode at a rotation speed of 1600 rpm and a scan rate of 5 mV s−1. b Potential dependent Faradaic efficiencies for CO2RR on I: Ag1Cu NW (left), II: AgCu NW (middle) and III: Cu NW (right) (H-type cell). c Faradaic efficiencies for CO2-to-C2 products on I: Ag1Cu NW (left) and II: AgCu NW (right) in 1 M KOH electrolyte (flow cell). Error bars represent the standard deviation of three independent measurements. All voltage was recorded without iR correction. d The stability test of Ag1Cu NW acquired in a flow cell using 1 M KOH as the electrolyte at −1.0 V vs. RHE. The results of each potential in Fig. 2b, c were collected after one hour of continuous electrolysis.
Dynamic evolution of the catalytic sites under CO2RR
To understand the vastly different CO2 reduction to C2 selectivity over AgCu NW and Ag1Cu NW, quasi in-situ XPS was performed to investigate the surface properties of the Ag1Cu NW and AgCu NW (Supplementary Fig. 28 and Supplementary Table 7). The quasi in-situ XPS spectra were collected at the open-circuit condition (OCV, immersed in KHCO3 electrolyte), −0.7 and −1.2 V vs. RHE, respectively. As shown in Fig. 3a and Supplementary Fig. 29a, the high-resolution Cu 2p XPS spectrum of Ag1Cu NW reveals the existence of partially oxidized Cu species under OCV condition. Only metallic Cu could be observed at −1.2 V vs. RHE, suggesting that the oxidized Cu species in Ag1Cu NW could be completely reduced to metallic Cu during CO2RR, which is also evidenced in the Cu Auger spectra collected before and after CO2RR (Supplementary Fig. 30). Meanwhile, the oxidation state of Ag at −0.7 and −1.2 V vs. RHE was observed to be higher than that under OCV condition (Fig. 3b and Supplementary Fig. 29b), which is possibly due to the adsorption of intermediate species on single Ag atoms during CO2RR. As a reference, quasi in-situ XPS spectra for Ag1Cu NW were also collected in Ar atmosphere under identical conditions (Supplementary Fig. 31). The similar variation trend of Ag oxidation state as a function of applied potential suggests that the intermediate species bound to the single Ag atoms of Ag1Cu NW may originate from water rather than CO2. For AgCu NW, more negative shift in binding energies of both Cu and Ag were observed at the applied potential of −0.7 and −1.2 V vs. RHE as compared to OCV condition, verifying that metallic Cu and Ag participated in the CO2RR (Supplementary Figs. 32, 33).

a, b Quasi in-situ XPS spectra of high-resolution Cu 2p3/2 and Ag 3d5/2 for Ag1Cu NW recorded at OCV, −0.7 V and −1.2 V vs. RHE in CO2-saturated 0.1 M KHCO3. c, d Normalized operando Cu and Ag K-edge XANES spectra of Ag1Cu NW recorded at OCV, −0.7 V, −1.2 V vs. RHE and AFT in CO2-saturated 0.1 M KHCO3 solution.
To further investigate the dynamic evolution of catalytic sites over Ag1Cu NW during CO2RR, operando XAS measurements were conducted. As shown in Fig. 3c, operando XANES spectra of Cu K-edge were recorded at OCV, −0.7 V, -1.2 V vs. RHE, and AFT (after the CO2RR), respectively. All spectra show characteristics of metallic Cu, displaying the similar pre-edge and white-line features (marked (1), (2), (3) in the XANES spectra of Fig. 3c) as those of Cu foil (Fig. 1g), revealing that the valence state of Cu in Ag1Cu NW remained at Cu (0) throughout CO2RR41,47. At −0.7 V and −1.2 V vs. RHE, the Cu K-edge of Ag1Cu NW moved towards lower energies as compared with that under OCV, which is due to the reduction of partially oxidized copper species. The slight increase in Cu valence state of Ag1Cu NW at AFT may result from desorption of CO2RR intermediates from the Cu sites after removal of cathodic potential. The FT-EXAFS fitting profiles of the R space recorded at −0.7 V and −1.2 V vs. RHE indicated that the Cu-Cu coordination number of Ag1Cu NW remained stable during the CO2RR process, suggesting that Ag1Cu NW maintained good stability throughout the reaction (Supplementary Figs. 34, 35 and Supplementary Table 8). On the contrary, the white-line energy of Ag in Ag1Cu NW was observed gradually shifted to higher energies with increasing the applied cathodic potential from OCV to −1.2 V vs. RHE (Fig. 3d), further confirming the increase of valence state of Ag during CO2RR, matching well with the quasi in-situ XPS results. To extract the detailed structural information from EXAFS, we analyzed the Cu–Cu path at −0.7 and −1.2 V vs. RHE. The Cu–Cu coordination number (CN) of Ag1Cu NW during the CO2RR was quantified using the ARTEMIS program of IFEFFIT. The results indicated that the Cu–Cu coordination number of Ag1Cu NW remained stable during the CO2RR process, suggesting that the Ag1Cu NW maintained good stability throughout the reaction.
In-situ capture of reaction intermediates
Operando Raman spectroscopy measurements were carried out to study the intermediate species over AgCu NW and Ag1Cu NW involved in CO2RR (Supplementary Fig. 36). Supplementary Table 9 sumarizes all Raman characteristic peaks observed over Cu NW, AgCu NW and Ag1Cu NW. No signals corresponding to any intermediates could be captured in Ar-saturated 0.1 M KHCO3 electrolyte (Supplementary Fig. 37). As shown in the operando Raman spectra recorded in CO2-saturated electrolyte (Fig. 4a, b and Supplementary Figs. 38, 39), Raman peaks in the wavenumber range from 525 to 630 cm−1 were observed over both Ag1Cu NW and AgCu NW under OCV condition, which can be assigned to the CuxO species52,53,54. With increase in applied cathodic potential, the Raman peaks of CuxO disappeared in all catalysts, attributed to reduction of CuxO to metallic Cu during CO2RR. Over AgCu NW, three characteristic peaks associated with surface-adsorbed *CO were identified at 282, 362 and 1950–2150 cm−1, corresponding to Cu-CO frustrated rotation (denoted as P1), Cu-CO stretching (denoted as P2) and C-O stretching, respectively54. Figure 4c displays the potential-dependent intensity ratios between P2 and P1 over AgCu NW and Ag1Cu NW. Compared to Ag1Cu NW, the higher P2/P1 ratios observed over AgCu NW suggest higher *CO coverages on the AgCu NW surface, which shall benefit formation of *COCO intermediates for ethylene production35,55. Besides, the Raman peaks at 704, 1072 and 1556 cm−1 observed across the potential range from −0.6 to −1.4 V vs. RHE can be assigned to in-plane νCO2-, adsorbed carbonate species (νCO32-) and νasCO2−, respectively53. Over Ag1Cu NW, a characteristic peak at 537 cm−1 that can be assigned to *OH species, was observed at −0.6 V vs. RHE, which gradually grew with increasing the applied cathodic potential (Fig. 4b and Supplementary Fig. 40)56. The peak intensity of *OH species was found positively correlated with the Faradaic efficiency of ethanol, suggesting that the in-situ generated *OH species is crucial for the greatly promoted CO2 reduction to ethanol over Ag1Cu NW.

Operando Raman spectra recorded in CO2-saturated 0.1 M KHCO3 solution at E = -0.6 V ~ -1.4 V vs. RHE over (a) AgCu NW and (b) Ag1Cu NW, respectively. c Potential-dependent Raman peak intensity ratio of Cu-CO stretching (P2) to Cu-CO frustrated rotation (P1) over AgCu NW and Ag1Cu NW, and peak intensity of *OH species versus potential over Ag1Cu NW. d, e Operando ATR-SEIRAS spectra collected in CO2-saturated 0.1 M KHCO3 solution at E = -0.4 V ~ −1.6 V vs. RHE over AgCu NW and Ag1Cu NW, respectively. The inset in Fig. 4c, d and e shows the DFT-optimized structures. (Cu: yellow, Ag: gray, O: red, H: white, C: black gray) f Potential-dependent ATR-SEIRAS peak intensity for *COCHO intermediate over Ag1Cu NW (blue bar) and *COCO intermediate over AgCu NW (black solid line). h 13C NMR and i MS spectra of CO2RR products over Ag1Cu NW, using 12CO + H12CHO (top), 13CO + H12CHO (middle), or 12CO + H13CHO (bottom) as the reactants, obtained at −1.2 V vs. RHE. g Schematic diagram showing the CO2RR pathway over Ag1Cu NW.
Operando ATR-SEIRAS measurements were further conducted to probe the reactive intermediates and provide mechanistic insights in CO2 reduction to ethylene and ethanol over AgCu NW and Ag1Cu NW (Supplementary Fig. 41). Figures 4d, e and Supplementary Figs. 42, 43 show the operando ATR-SEIRAS spectra recorded on AgCu NW, Ag1Cu NW and Cu NW in CO2-saturated 0.1 M KHCO3. A distinct H2O absorption peak at around 1620 cm−1 was observed over all catalysts57. As shown in Fig. 4d, two peaks appeared at around 2050 and 1865 cm−1 over AgCu NW, which are associated with the atop-adsorbed *CO (*COatop) and bridge-adsorbed *CO (*CObridge) on Cu surface, respectively23,58,59. The red-shift of the *CO peaks with increase in applied cathodic potential is due to the Stark effect arising from the decreased resonance frequencies in negative electric fields38. In particular, the absorption peak of the *COCO intermediate at 1561 cm−1 appeared at −0.4 V vs. RHE and reached the maximum intensity at −1.2 V vs. RHE, which is positively correlated with the ethylene Faradaic efficiency, suggesting that *COCO is crucial for ethylene generation over AgCu NW (Fig. 4d, f)12. Additionally, a clear absorption peak could be identified at 1400 cm−1 that can be assigned to the adsorbed *COOH intermediate, whose intensity increased with cathodic potential rising from −0.4 to −1.6 V vs. RHE. The assignments for additional peaks observed in the ATR-SEIRAS spectra at ~2332, ~1265, ~1183 and ~1034 cm−1 are summarized in Supplementary Table 10. Compared to AgCu NW, a blue-shift in *CObridge peak on Ag1Cu NW was noticed at various applied potentials, suggesting enhanced binding strength of *CO on Ag1Cu NW. Additional new peaks at around 1244 cm−1 and 1083 cm−1 were observed on Ag1Cu NW, which can be assigned to adsorbed *CHO and *COCHO intermediates, respectively (Fig. 4e)60. As shown in Fig. 4f, the peak intensity of *COCHO intermediate was found to gradually increase with increasing applied cathodic potential from −0.4 to −1.2 V vs RHE, while the peak intensity of *CHO intermediate was correspondingly decreased, suggesting production of *COCHO intermediate from asymmetric *CO-*CHO coupling.
To probe the reaction pathway of CO2 reduction to ethanol over Ag1Cu NW, isotopic labelling experiments using 12CO + H12CHO, 12CO + H13CHO or 13CO + H12CHO as the reactants were carried out. As shown in Fig. 4h, 13C NMR signals were detected at chemical shifts of 17.47 and 58.05, respectively, attributing to -CH3 and -CH2OH from ethanol. Meanwhile, mass spectrometry (MS) spectra of liquid products from CO2RR exhibit signals at m/z = 45.03 and m/z = 46.03, which can be attributed to 12CH312CH2O+ and 13CH312CH2O+ or 12CH313CH2O+, respectively (Fig. 4i)61. Based on all these results, Fig. 4g schematically proposes the pathway of CO2RR to ethanol over Ag1Cu NW, where methyl C (-CH3) and methylene C (-CH2OH) in ethanol originate from *CO and *HCHO intermediates, respectively.
Theoretical insights into the catalytic mechanism
Density functional theory (DFT) calculations were performed to gain insights into the underlying reaction mechanisms for the distinctive selectivity of CO2 electroreduction to ethylene and ethanol over AgCu NW and Ag1Cu NW, respectively (Supplementary Figs 44, 45). Supplementary Figs. 46, 47 presents the optimized configurations of the reaction intermediates involved in CO2RR over Ag1Cu NW. As shown in Supplementary Figs. 48, 49, both the Bader- and Hirshfeld-based charge distribution results indicate that the atomically dispersed Ag in Ag1Cu NW can donate electrons to its neighboring Cu atoms, moving the d-band center of Cu from −2.142 to −2.132 eV. Meanwhile, the atomically dispersed Ag atom becomes positively charged, matching well with the XPS and XAS results.
Operando Raman measurements observed in-situ generation of *OH over Ag1Cu NW during CO2RR. To determine the *OH adsorption site, the adsorption and dissociation energies of H2O on Cu NW, AgCu NW and Ag1Cu NW were calculated17. As shown in Fig. 5a, compared to AgCu NW and Cu NW, the H2O adsorption energy over Ag1Cu NW is more negative with more electrons directly transferred from H2O molecule to Ag1Cu NW (Supplementary Fig. 50). Furthermore, H2O tends to more preferably dissociate on the single Ag atomic site over Ag1Cu NW with the lowest H2O dissociation transition state (TS) energy barrier (Fig. 5b and Supplementary Figs. 51–53). The in-situ formation of HO-Ag1Cu NW during CO2RR not only promoted the release of H+ for the subsequent hydrogenation reaction, but also largely reduced the Gibbs free energy for *CO hydrogenation to *CHO (Fig. 5c), which facilitated the asymmetric *CO-*CHO coupling over the adjacent paired Cu atoms (HO-Ag1-Cu-Cu) to preferably reduce CO2 to ethanol (Fig. 5d, e, and Supplementary Figs. 54–57). On the other hand, for AgCu NW, as shown in Supplementary Figs. 58–60, the energy barrier for hydrogenation of *CO2 to *COOH can be greatly reduced from 0.83 to 0.63 eV, revealing preferred production of *CO via the *COOH route over AgCu NW. The increased *CO coverage shall increase the probability of *CO-*CO coupling to form *COCO intermediate, directing CO2 reduction to produce ethylene over AgCu NW (Supplementary Figs. 61, 62).

a, b The adsorption and dissociation energies of H2O on Cu NW, AgCu NW and Ag1Cu NW. c Gibbs free energy diagrams for *CO hydrogenation to *CHO over Cu NW, Ag1Cu NW and HO-Ag1Cu NW. d Calculated Gibbs free energy diagrams for CO2RR to ethanol and ethylene over Ag1Cu NW. e Proposed reaction pathway for CO2 reduction to ethanol over Ag1Cu NW. The inset in Fig. 5a, c and e shows the DFT-optimized structures. (Cu: yellow, Ag: gray, O: red, H: white, C: black gray).
Based on the above results, the reaction mechanisms of CO2RR for the distinctive selectivity towards ethylene and ethanol over AgCu NW and Ag1Cu NW can be clearly evidenced. The ethylene pathway follows the symmetric C-C coupling, arising from the high *CO coverage generated over Ag nanoparticles and the reduced energy barrier for *COCO formation over AgCu NW. While the ethanol pathway follows the asymmetric C-C coupling, triggered by the in-situ generated HO-Ag1-Cu-Cu sites (Fig. 5e), which reduces the energy barrier for the formation of *CHO species and thus promotes asymmetric *CO-*CHO coupling, boosting CO2 reduction to ethanol over Ag1Cu NW.
Discussion
In summary, two well-defined model Cu-based catalysts were developed by modifying Cu nanowires with Ag nanoparticles (AgCu NW) and single Ag atoms (Ag1Cu NW), respectively, and their underlying C-C coupling mechanisms of CO2RR to ethylene and ethanol were successfully elucidated. Operando XAS and quasi in-situ XPS revealed dynamic oxidation state evolution of Cu and Ag over AgCu NW and Ag1Cu NW during CO2RR. Following a combination of operando Raman spectroscopy and operando ATR-SEIRAS measurements, a higher *CO coverage was observed over AgCu NW surface, which would favor symmetric *CO-*CO coupling in CO2RR to produce ethylene. On the contrary, the in-situ formed *OH species via H2O dissociation was dominantly found over Ag1Cu NW. Isotopic labelling experiments indicated that the methyl C (*CH3) and methylene C (*CH2OH) in ethanol product were originated from the *CO and *HCHO intermediates, respectively. Theoretical calculations further demonstrated that the atomically dispersed Ag could not only promote dissociation of H2O to release H+, but also reduce the energy barrier for the formation of *CHO over its neighboring Cu site, resulting in the significantly promoted asymmetric *CO-*CHO coupling over paired Cu atoms adjacent to the single Ag atom, greatly boosting electrochemical CO2 reduction to ethanol.
Methods
Chemicals and materials
Copper (II) chloride dihydrate (CuCl2·2H2O, 99.99%, CAS: 10125-13-0), glucose (C6H12O6, 99.5%, CAS: 50-99-7), hexadecylamine (HAD, 98%, CAS: 143-27-1), hexane (98%, CAS: 110-94-3) and silver nitrate (AgNO3, 99.99%, CAS: 7761-88-8) were purchased from Aladdin. Isopropyl alcohol (C3H8O, 99.9%, CAS: 67-63-0), potassium bicarbonate (KHCO3, 99.95%, CAS: 298-14-6), potassium hydroxide (KOH, 99.99%, CAS: 1310-58-3), ethanol (CH3CH2OH, å 99.9%, CAS: 64-17-5), and formaldehyde (HCHO, 20 wt. % in H2O, 99 atom % 13C, CAS: 3228-27-1) were purchased from Sigma-Aldrich. Nafion solution (D521-1100ew, 5 wt.%, Dupont) was purchased from Alfa Aesar. Carbon paper (AVCarb P75, Product Code: 18070009; AVCarb P75T, 29.8–31.2% Wet Proofed, Product Code: 18070011) was purchased from FuelCell Store. All chemical reagents were used as received without any further purification. Deionized water (18.2 MΩ) was used in all experimental processes.
Preparation of Cu nanowires (Cu NW)
Uniform Cu NW were synthesized following a previously reported method with slight improvements62. In a typical synthesis, firstly, glucose (250 mg, 1.4 mmol), CuCl2·2H2O (117 mg, 0.7 mmol), and hexadecylamine (900 mg, 3.8 mmol) were dissolved in 50 mL of deionized water under vigorous stirring at 50 oC for 12 h. Subsequently, the homogeneous solution was kept in a water bath at 75 oC and stirred for 24 h until the solution turned brown. Afterwards, the above solution was transferred into a 150 mL pressure bottle and heated at 120 °C for 24 h. After cooling to room temperature, the products were collected by centrifugation at 16099.2 (×g) for 5 min. The obtained reddish-brown Cu NW was washed with hot deionized water (60 °C), ethanol, and hexane several times to remove excess hexadecylamine and glucose, and dried at 60 °C under vacuum for 12 h.
Synthesis of AgCu NW and Ag1Cu NW
AgCu NW and Ag1Cu NW were synthesized via a modified galvanic replacement approach63. Firstly, Cu NW (32 mg, 0.5 mmol) was dispersed in 50 mL of isopropyl alcohol in a three-necked flask under sonication for 30 min to form a reddish-brown suspension. Immediately afterwards, a certain amount of AgNO3 isopropyl alcohol solution was slowly injected into the above Cu NW suspension via a peristaltic pump at a rate of 0.5 mL min−1 to trigger the galvanic replacement reaction. The added AgNO3 are 0.1 and 0.016 mmol to prepare AgCu NW and Ag1Cu NW. Then, the mixed suspension was kept at room temperature for 30 min with continuous magnetic stirring. The resulting red-grey product was collected by centrifugation at 8000 rpm for 5 min and washed three times with deionized water and ethanol, and dried at 60 °C under vacuum for 12 h. It should be noted that the entire reaction process required Ar as the protective gas to avoid oxidation.
Characterization
The crystal structure of Cu NW, AgCu NW and Ag1Cu NW were characterized by X-ray diffraction (XRD, Bruker AXS D8 Advance) with Cu Kα radiation (λ = 1.5406 Å). The morphology of the catalysts was examined by transmission electron microscopy (HRTEM, JEOL JEM-2100F operated at 200 kV) and field-emission scanning electron microscopy (FESEM, JSM-6700F) equipped with an energy dispersive X-ray spectroscopy (EDX, Aztec X-Max 80 T). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) characterization was conducted on a JEOL JEMARM200F STEM/TEM with a guaranteed resolution of 0.08 nm. Detailed chemical compositions were analyzed by X-ray photoelectron spectroscopy (XPS) on an ESCALAB 250 photoelectron spectrometer (Thermo Fisher Scientific) using a monochromatic Al Kα X-ray beam (1,486.6 eV). All binding energies were referenced to the C1s peak (284.8 eV). The concentration of Ag in AgCu NW and Ag1Cu NW were quantified by inductively coupled plasma optical emission spectroscopy (ICP-OES). For ex-situ XAS measurements, a certain amount of powder samples were placed into a hollow circular mold (made of polytetrafluoroethylene) with a diameter of 1 cm. The filled sample was then fixed and vacuumed to prevent oxidation during the transfer and measurement processes. And the sample was secured on an XAS sample stage, which was positioned at a 45° angle to the X-ray during the measurement, allowing for collection of fluorescence signals.
Quasi in-situ XPS measurements
Quasi in-situ X-ray photoelectron spectroscopy characterizations (XPS) were conducted at the Vacuum Interconnected Nanotech Workstation (Nano-X), Suzhou Institute of Nano-Tech, and Nano-Bionics, Chinese Academy of Sciences (CAS). Quasi in-situ XPS measurements were carried out in a SPECS PHOIBOS 100 analyzer using monochromatic X-ray source of Al Kα radiation (1486.6 eV), and the overall spectrum resolution is Ag 3d5/2, < 0.5 eV FWHM at 20 kcps@UHV. The working electrode (1 × 1 cm2), consisting of carbon paper with drop-casted catalyst, was immersed in CO2-saturated 0.1 M KHCO3 solution. To avoid surface oxidation of the catalyst, the H-type cell was placed in a glove box for the CO2RR. After CO2RR, the sample was rapidly transferred through a chamber with ultrahigh vacuum for XPS testing. Quasi in-situ XPS spectra were collected at open-circuit condition (OCV, immersed in KHCO3 electrolyte), −0.7 and −1.2 V vs. RHE, respectively.
Operando XAS measurements
X-ray Absorption Spectroscopy (XAS) and operando XAS measurements were performed in the total-fluorescence yield (TFY) mode at room temperature. These measurements utilized the TPS 32 A and TLS01C1 beamlines at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. Energy calibration was performed with a Cu and Ag foil standard by shifting all spectra to a glitch in the incident intensity. Fluorescence spectra were recorded using a silicon drift detector. The measurement in a typical three-electrode setup, the same condition as that in electrochemical characterization was performed in a specially designed Teflon container with a window sealed by Kapton tape64,65,66. CO2RR was conducted on a Bio-logic VSP potentiostat in a standard three-electrode setup. The spectral time recording was 40 min for a single spectrum (2 s for the pre-edge region, 2 s for the rising-edge region, and 8 s for the EXAFS region) during CO2RR. Carbon paper (1 × 1 cm2) supported catalyst as the working electrode was used for operando XAS measurements. To prepare the catalyst ink, catalyst (5 mg) and 5 wt.% Nafion solution (40 μL) were introduced into water and isopropyl alcohol solution (960 μL, 1:1 v/v) and ultrasonicated for 3 h. Next, the catalyst ink (400 μL) was applied onto a carbon paper and allowed to dry in air, affording a catalyst loading of 2 mg cm−2. The pre-edge baseline was subtracted, and the spectrum was normalized to the post-edge. EXAFS analysis was conducted using a Fourier transform on k2-weighted EXAFS oscillations to evaluate the contribution of each bond pair to the Fourier transform peak.
Operando Raman measurements
Operando Raman spectra were recorded at controlled electrochemical potentials in CO2-saturated 0.1 M KHCO3 electrolyte in a three-electrode polytetrafluoroethylene cell with Pt wire as the counter electrode and Ag/AgCl as the reference electrode. Operando raman spectra were collected on a Raman spectrometer (Bruker NanoWizard Ultra Speed & inVia Raman) with a 532 nm/785 nm excitation laser (5%), having a power of 2.1 mW measured at the objective. A 50x long working distance objective (Olympus, 0.5 NA) was used, focusing on the sample surface and avoiding contact to the electrolyte. Acquisition time was set as 120 s for the spectral Raman shift ranging from 200 to 3000 cm−1. Operando Raman experiments were performed on a carbon paper as the working electrode with a catalyst loading of 0.5 mg cm−2. CO2-saturated electrolyte was prepared by purging CO2 (99.99%) into 0.1 M KHCO3 aqueous solution for 30 min, and a flow of CO2 was maintained over the electrolyte throughout electrochemical measurements.
Operando ATR-SEIRAS measurements
The operando attenuated total reflectance surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) measurements were performed on a Nicolet iS20 FTIR spectrometer equipped with a MCT detector cooled using liquid nitrogen and PIKE VeeMAX III variable angle ATR sampling accessory. The catalyst was added dropwisely to the surface of Au-plated silicon as the working electrode. A graphite rod and an Ag/AgCl electrode were used as the counter and reference electrode in all tests, respectively. The background was taken at OCV in CO2-saturated 0.1 M KHCO3 electrolyte. Afterwards, potential-dependent spectra were collected during stepping the working electrode potential from −0.4 V to −1.6 V vs. RHE. The step width and duration are 0.1 V and 120 s, respectively.
Electrochemical measurements
H-type cell
Electrochemical measurements were performed at room temperature and ambient pressure on a glassy carbon electrode in a two-compartment H-cell filled with CO2 saturated 0.1 M potassium bicarbonate (KHCO3) electrolyte using a CHI660e electrochemical workstation. A three-electrode cell configuration was employed, with a glassy carbon rotating disc electrode (RDE) (EE300a1 Rotating Disk Electrode, diameter: 5 mm) as the working electrode, a platinum foil (1.5 × 1.5 cm2) as the counter electrode, and a saturated calomel electrode as the reference electrode (SCE, Hg/Hg2Cl2). To ensure the accuracy of the RHE, we periodically calibrated the reference electrode in H2-saturated 0.1 M HClO4 and 0.1 M KOH electrolytes before conducting experiments. A reversible HOR/HER reaction was performed using two Pt plates as both the working and counter electrodes, with H2 bubbling over the working electrode during the process. For calibration, the clean Pt electrode was cycled at a slow sweep rate of 10 mV s−1 around the expected RHE value in each electrolyte until the CV curve stabilized. The RHE potentials in the two electrolytes were then determined from the corresponding open-circuit potentials. H-cell separated by a proton exchange membrane (Nafion 117, 183 μm, Dupont). And the Nafion membrane needs to be pretreated. The steps are as follows: (1) Place the cut Nafion 117 membrane in a beaker, add an excess of 5 wt% H2O2 solution, and boil it in a water bath at 80 °C for 0.5 h to remove organic and inorganic impurities from the membrane and rinse it 2-3 times with deionized water. (2) Add 0.5 mol L-1 HNO3 solution and boil it in a water bath at 80 °C for 0.5 h and rinse it 2-3 times with deionized water. The same three-electrode system, but with carbon paper (1 × 1 cm2) as the working electrode, was used for stability test. For all the measurements, CO2 (with 1% Ar) was continuously purged into the solution at a flow rate of 20 SCCM by a mass flow controller (CS200A, Beijing Sevenstar Flow Co., LTD). Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) acquired in CO2-saturated 0.1 M KHCO3 solution on a RDE at a rotating speed of 1600 rpm with a scan rate of 5 mV s−1. All potentials were calculated with respect to the reversible hydrogen electrode (RHE) scale according to the Nernst equation: \({{{\rm{E}}}}\,\left({{{\rm{vs}}}}.{{{\rm{RHE}}}}\right)={{{\rm{E}}}} \, \left({{{\rm{vs}}}}.{{{\rm{SCE}}}}\right)+0.2415+0.0591{{\times }}{{{\rm{pH}}}}\). The voltage was recorded without iR correction. The pH was determined by a pH meter (S220 SevenCompact™ pH/Ion). Supplementary Fig. 63 shows the ohmic drop during CO2RR. To prepare the working electrode, catalyst (5 mg) and 5 wt.% Nafion solution (40 μL) were introduced into water and isopropyl alcohol solution (960 μL, volume ratio of 1:1) and ultrasonicated for 3 h. Next, the catalyst ink (20 μL) was applied onto a glassy carbon RDE (diameter: 5 mm) and allowed to dry in air, affording a catalyst loading of 0.5 mg cm−2. For stability test, catalyst ink (100 μL) was brushed onto carbon paper (AVCarb P75T, 29.8–31.2% Wet Proofed, Product Code: 18070011, Purchased from FuelCellStore, 1 ×1 cm2) at 60 °C, affording a catalyst loading of 0.5 mg cm−2.
Flow cell
The electrochemical measurements were performed in a flow cell (manufactured by Dioxide Materials Company) separated by an anion exchange membrane (FAA-3-PK-130, Fumasep, Soak the membrane in 1 M KOH for 24 h before use.) using a CHI660E electrochemical workstation at room temperature. The flow cell was equipped with three electrodes, in which the Ag/AgCl electrode as the reference electrode, Ni-foam (200–250 μm, Youveim) as the counter electrode and the obtained GDE as the working electrode. CV and LSV curves were recorded on a CHI660E electrochemical workstation at a scan rate of 5 mV s−1 from −1.0 V to −2.5 V vs. SCE. 1 M KOH electrolyte was used as the anolyte and was circulated using a peristaltic pump (F01A-STP, Kameor) at 2.5 mL min−1. The flow rate of the CO2 gas flowing into the cathode flow field was kept at 20 SCCM by a mass flow controller (CS200A, Beijing Sevenstar Flow Co., LTD). All potentials were converted to the reversible hydrogen electrode (RHE) scale by the following equation: \({{{\rm{E}}}}({{{\rm{vs}}}}.{{{\rm{RHE}}}})={{{\rm{E}}}}({{{\rm{vs}}}}.{{{\rm{Ag}}}}/{{{\rm{AgCl}}}})+0.197{{{\rm{V}}}}+0.0591{{{\rm{\times }}}}{{{\rm{pH}}}}\). The voltage was recorded without iR correction.
Preparation of gas diffusion layer (GDL)
Carbon paper (AVCarb P75, Product Code: 18070009, Purchased from FuelCellStore) was repeatedly soaked in a polytetrafluoroethylene (PTFE, DISP 40LX, CAS: 25667-42-9) solution until the weight of PTFE reached 25 wt.%. The carbon paper was heated in air at 350 °C for 2 h. A carbon black (Vulcan XC-72R, Sigma) ink containing 40 wt.% PTFE was drop-casted on one side of the above carbon paper. After further heating in air at 350 °C for 2 h, the GDL was prepared.
Preparation of gas diffusion electrode (GDE)
The catalyst (4 mg) and Nafion solution (DuPont, ~5 wt.% in a mixture of isopropyl alcohol and deionized water) were firstly mixed and dispersed into isopropyl alcohol (1 mL) under ultrasonication for 30 min to form a uniform catalyst ink. Then, the catalyst ink (1 mL) was spray-casted onto a GDL (2 × 2 cm2), affording a catalyst loading of 1 mg cm-2, and dried at 60 °C to form the GDE.
Faradaic efficiency evaluation
The products of electrochemical CO2 reduction reaction (CO2RR) were measured using chronoamperometry at each fixed potential in a CO2RR electrolysis cell. The electrolyte in the cathodic compartment was stirred at a rate of 500 rpm during electrolysis. CO2 gas was introduced into the cathodic compartment at a rate of 20 SCCM and was routed into a gas chromatograph (GC, Agilent 7890). The gas chromatograph was equipped with a Molecular Sieve 5 A capillary column and a packed Carboxen-1000 column. The gaseous products (H2, CO, CH4, and C2H4) were quantified by gas chromatograph equipped with a flame ionization detector (FID) for CO, CH4 and C2H4, and a thermal conductivity detector (TCD) for H2. Ultrapure nitrogen (N2, 99.9999%) was used as the carrier gas. The liquid products (HCOOH and CH3CH2OH) were examined by nuclear magnetic resonance spectroscopy (NMR). 1H NMR and 13C NMR spectra were collected on a Bruker Avance III HD 400 NMR spectroscope in 10% D2O using water suppression mode, with DMSO as an internal standard.
The Faradaic efficiency (FE) was calculated using the following equation:
where Qi is the quantity of electric charge needed to produce product i at a certain time interval (\({Q}_{i}={z}_{i}{{\times }}F{{\times }}{N}_{i}\)); Qtotal is the quantity of electric charge needed to produce all products at a certain time interval (\({Q}_{{total}}=I{{\times }}t\)); zi is the number of electrons required to produce one i molecule, which is 2, 2, 8, and 12 for CO, H2, CH4, and C2H4, respectively; F is the Faradaic constant (96485.3 C mol−1); Ni is the number of moles of product i in the GC sampling loop (\({N}_{i}={N}_{{total}}{{\times }}{V}_{i}\)); Ntotal: moles of all gases in the GC sampling loop (\({N}_{{total}}=\frac{{P}_{o}{{\times }}{V}_{o}}{R{{\times }}{T}_{o}}\)); Vo: volume of the GC sampling loop (\({V}_{o}={{{\rm{G}}}}{{\times }}{{{\rm{t}}}}\)); I is the measured current; t is the time needed for gas to fill the GC sampling loop; vi is the volume ratio of product i obtained from GC; G is the volumetric flow rate; Po is atmospheric pressure (1.01 × 105 Pa); T0 is the sample loop temperature (298 K); and R is the ideal gas constant (8.314 J mol K−1).
All Faradaic efficiencies and error bars reported in this work were determined from the average of at least three replicates for each potential studied.
Computational methods
All spin-polarized DFT calculations were implemented in the Vienna ab initio Simulation Package (VASP 6.3.0)67. The exchange-correlation interactions were modeled by the functional of Perdew, Burke, and Ernzerhof (PBE) within the generalized gradient approximation (GGA), and the projector augmented-wave method was carried out to describe the ion-electron interactions68,69. A Cu (111) (4 × 4) supercell with a lattice constant of 10.22 Å (α = β = 90°, γ = 120°) was employed to anchor the Ag atoms or support the Ag13 nanoclusters. The Brillouin zone was sampled by 3 × 3 × 1 k-points by means of Monkhorst-Pack scheme with the cutoff of 400 eV during the structural relaxation, while 5 × 5 × 1 k-points for electronic structure calculation such as projected density of states (PDOS) and charge density difference. The convergence criteria for energy and force were set as 10−5 eV and 0.01 eV Å−1, respectively. Along the perpendicular direction, a vacuum space at least 18 Å was added to eliminate the effects of periodic structure, and the dispersion corrected density functional theory (DFT-D3) was utilized to include van der Waals (vdWs) interaction between adsorbate and interface70.
The adsorption energies of intermediates such as *CO2, *COOH and *CO were calculated based on the following equation:
where Etotal, E*, and Eadsorbate represent the DFT energy of adsorption system, substrate, and adsorbate, respectively.
Based on the theory proposed by Nørskov et al.71, the change of Gibbs free energy for each CO2RR elementary reaction was calculated based on computational hydrogen electrode (CHE) model72, referred as \(\varDelta G=\,\varDelta E+\varDelta {ZPE} - {{\mbox{T}}}\varDelta S\), where ΔE is the adsorption electronic energy, while ΔZPE and TΔS represent zero-point energy correction and entropy contribution at 298 K. For the gas molecules, the entropy was taken from NIST database. For the adsorbed intermediates, only vibrational entropy was considered, which was calculated from the DFT calculated vibrational frequencies. The activation barrier of each reaction was obtained from climbing-image nudged elastic band (CI-NEB) calculations73.
Data availability
The ex-situ XPS data, ex-situ XAS data, product quantification, quasi in-situ XPS data, operando XAS data, operando Raman data, operando ATR-SEIRAS data, isotopic labelling experiment data and theoretical calculations data generated in this study have been deposited in the Figshare database under accession code (https://doi.org/10.6084/m9.figshare.27199515)74. Source data are provided with this paper.
References
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Jordaan, S. M. & Wang, C. Electrocatalytic conversion of carbon dioxide for the Paris goals. Nat. Catal. 4, 915–920 (2021).
Sullivan, I. et al. Coupling electrochemical CO2 conversion with CO2 capture. Nat. Catal. 4, 952–958 (2021).
Amirbeigiarab, R. et al. Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions. Nat. Catal. 6, 837–846 (2023).
Yang, H. B. et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 3, 140–147 (2018).
Pang, Y. et al. Efficient electrocatalytic conversion of carbon monoxide to propanol using fragmented copper. Nat. Catal. 2, 251–258 (2019).
Zhu, P. & Wang, H. High-purity and high-concentration liquid fuels through CO2 electroreduction. Nat. Catal. 4, 943–951 (2021).
Lin, Y. et al. Tunable CO2 electroreduction to ethanol and ethylene with controllable interfacial wettability. Nat. Commun. 14, 3575 (2023).
Wang, X. et al. Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag–Ru–Cu catalyst. Nat. Energy 7, 170–176 (2022).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Yang, Y. et al. Operando studies reveal active Cu nanograins for CO2 electroreduction. Nature 614, 262–269 (2023).
Kim, Y. et al. Time-resolved observation of C–C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energ. Environ. Sci. 13, 4301–4311 (2020).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Hu, J. et al. Edge-rich molybdenum disulfide tailors carbon-chain growth for selective hydrogenation of carbon monoxide to higher alcohols. Nat. Commun. 14, 6860 (2023).
Wu, Z.-Z. et al. Identification of Cu(100)/Cu(111) Interfaces as Superior Active Sites for CO Dimerization During CO2 Electroreduction. J. Am. Chem. Soc. 144, 259–269 (2021).
Xiao, H. et al. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Nat. Acad. Sci. 114, 6685–6688 (2017).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Zhang, J. et al. Steering CO2 electroreduction pathway toward ethanol via surface-bounded hydroxyl species-induced noncovalent interaction. Proc. Nat. Acad. Sci. 120, e2218987120 (2023).
Ding, J. et al. A tin-based tandem electrocatalyst for CO2 reduction to ethanol with 80% selectivity. Nat. Energy 8, 1386–1394 (2023).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2019).
Zhang, B. et al. Highly Electrocatalytic Ethylene Production from CO2 on Nanodefective Cu Nanosheets. J. Am. Chem. Soc. 142, 13606–13613 (2020).
Feng, S. et al. In situ spectroelectrochemical probing of CO redox landscape on copper single-crystal surfaces. Proc. Nat. Acad. Sci. 119, e2118166119 (2022).
Wang, P. et al. Boosting electrocatalytic CO2–to–ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Kortlever, R. et al. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett 6, 4073–4082 (2015).
Schouten, K. et al. A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci 2, 1902–1909 (2011).
Wang, X. et al. Morphology and mechanism of highly selective Cu(II) oxide nanosheet catalysts for carbon dioxide electroreduction. Nat. Commun. 12, 794 (2021).
Luo, Y. et al. Selective electrochemical synthesis of urea from nitrate and CO2 via relay catalysis on hybrid catalysts. Nat. Catal. 6, 939–948 (2023).
Xu, A. et al. Copper/alkaline earth metal oxide interfaces for electrochemical CO2-to-alcohol conversion by selective hydrogenation. Nat. Catal. 5, 1081–1088 (2022).
Wang, X. et al. Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2–CO co-feeds on Cu and Cu-tandem electrocatalysts. Nat. Nanotechnol. 14, 1063–1070 (2019).
Chen, X. et al. Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20–27 (2020).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Wang, X. et al. Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat. Energy 5, 478–486 (2020).
Choi, C. et al. Highly active and stable stepped Cu surface for enhanced electrochemical CO2 reduction to C2H4. Nat. Catal. 3, 804–812 (2020).
Chen, C. et al. Cu-Ag Tandem Catalysts for High-Rate CO2 Electrolysis toward Multicarbons. Joule 4, 1688–1699 (2020).
Gao, J. et al. Selective C-C Coupling in Carbon Dioxide Electroreduction via Efficient Spillover of Intermediates As Supported by Operando Raman Spectroscopy. J. Am. Chem. Soc. 141, 18704–18714 (2019).
Duan, G. Y. et al. Highly Efficient Electrocatalytic CO2 Reduction to C2+ Products on a Poly(ionic liquid)‐Based Cu0–CuI Tandem Catalyst. Angew. Chem. Int. Ed. 61, e2021106 (2022).
Li, F. et al. Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces. Nat. Catal. 3, 75–82 (2019).
Zhang, J. et al. Molecular tuning for electrochemical CO2 reduction. Joule 7, 1700–1744 (2023).
Wang, Q. et al. Atomic metal–non-metal catalytic pair drives efficient hydrogen oxidation catalysis in fuel cells. Nat. Catal. 6, 916–926 (2023).
Jiao, J. et al. Constructing asymmetric double-atomic sites for synergistic catalysis of electrochemical CO2 reduction. Nat. Commun. 14, 6164 (2023).
Lin, S. C. et al. Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction. Nat. Commun. 11, 3525 (2020).
Jiao, J. et al. Copper atom-pair catalyst anchored on alloy nanowires for selective and efficient electrochemical reduction of CO2. Nat. Chem. 11, 222–228 (2019).
Li, J. et al. Enhanced multi-carbon alcohol electroproduction from CO via modulated hydrogen adsorption. Nat. Commun. 11, 3685 (2020).
Liu, S. et al. Identify the Activity Origin of Pt Single-Atom Catalyst via Atom-by-Atom Counting. J. Am. Chem. Soc. 143, 15243–15249 (2021).
Li, J. et al. Weak CO binding sites induced by Cu-Ag interfaces promote CO electroreduction to multi-carbon liquid products. Nat. Commun. 14, 698 (2023).
Yan, Y. et al. Electrocatalytic Upcycling of Biomass and Plastic Wastes to Biodegradable Polymer Monomers and Hydrogen Fuel at High Current Densities. J. Am. Chem. Soc. 145, 6144–6155 (2023).
Greiner, M. T. et al. Free-atom-like d states in single-atom alloy catalysts. Nat. Chem. 10, 1008–1015 (2018).
Sun, H. et al. Promoting ethylene production over a wide potential window on Cu crystallites induced and stabilized via current shock and charge delocalization. Nat. Commun. 12, 6823 (2021).
Jennifer, L. D. et al. A Systematic K-edge X-ray Absorption Spectroscopic Study of Cu(III) Sites. J. Am. Chem. Soc. 122, 5775–5787 (2000).
Ma, W. et al. Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts. Chem. Soc. Rev. 50, 12897–12914 (2021).
Zhao, J. et al. Modulation of *CHxO Adsorption to Facilitate Electrocatalytic Reduction of CO2 to CH4 over Cu-Based Catalysts. J. Am. Chem. Soc. 145, 6622–6627 (2023).
Lim, C. Y. J. et al. Surface charge as activity descriptors for electrochemical CO2 reduction to multi-carbon products on organic-functionalised Cu. Nat. Commun. 14, 335 (2023).
de Ruiter, J. et al. Probing the Dynamics of Low-Overpotential CO2-to-CO Activation on Copper Electrodes with Time-Resolved Raman Spectroscopy. J. Am. Chem. Soc. 144, 15047–15058 (2022).
Zhao, Y. et al. Elucidating electrochemical CO2 reduction reaction processes on Cu(hkl) single-crystal surfaces by in situ Raman spectroscopy. Energ. Environ. Sci. 15, 3968–3977 (2022).
Zhan, C. et al. Revealing the CO Coverage-Driven C-C Coupling Mechanism for Electrochemical CO2 Reduction on Cu2O Nanocubes via Operando Raman Spectroscopy. ACS Catal 11, 7694–7701 (2021).
Herzog, A. et al. Operando Raman spectroscopy uncovers hydroxide and CO species enhance ethanol selectivity during pulsed CO2 electroreduction. Nat. Commun. 15, 3986 (2024).
Wang, Q. et al. Attenuating metal-substrate conjugation in atomically dispersed nickel catalysts for electroreduction of CO2 to CO. Nat. Commun. 13, 6082 (2022).
Sun, W. et al. V-Doped Cu2Se Hierarchical Nanotubes Enabling Flow-Cell CO2 Electroreduction to Ethanol with High Efficiency and Selectivity. Adv. Mater. 34, e2207691 (2022).
Ding, L. et al. Over 70 % Faradaic Efficiency for CO2 Electroreduction to Ethanol Enabled by Potassium Dopant‐Tuned Interaction between Copper Sites and Intermediates. Angew. Chem. Int. Ed. 61, e202209268 (2022).
Zheng, M. et al. Electrocatalytic CO2-to-C2+ with Ampere-Level Current on Heteroatom-Engineered Copper via Tuning *CO Intermediate Coverage. J. Am. Chem. Soc. 144, 14936–14944 (2022).
Shi, H. et al. Atomically Dispersed Indium‐Copper Dual‐Metal Active Sites Promoting C−C Coupling for CO2 Photoreduction to Ethanol. Angew. Chem. Int. Ed. 61, e202208904 (2022).
Kim, M. J. et al. Isotropic Iodide Adsorption Causes Anisotropic Growth of Copper Microplates. Chem. Mater. 33, 881–891 (2020).
Lyu, Z. et al. Controlling the Surface Oxidation of Cu Nanowires Improves Their Catalytic Selectivity and Stability toward C2+ Products in CO2 Reduction. Angew. Chem. Int. Ed. 60, 1909–1915 (2020).
Hung, S. F. et al. Identification of Stabilizing High-Valent Active Sites by Operando High-Energy Resolution Fluorescence-Detected X-ray Absorption Spectroscopy for High-Efficiency Water Oxidation. J. Am. Chem. Soc. 140, 17263–17270 (2018).
Hung, S.-F. et al. In Situ Spatially Coherent Identification of Phosphide-Based Catalysts: Crystallographic Latching for Highly Efficient Overall Water Electrolysis. ACS Energy Lett. 4, 2813–2820 (2019).
Wang, N. et al. Boride-derived oxygen-evolution catalysts. Nat. Commun. 12, 6089 (2021).
Kresse, G. et al. Efficient Iterative Schemes for ab initio Total-energy Calculations Using a Plane-wave Basis Set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P. et al. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865 (1996).
Hammer, B. et al. Improved Adsorption Energetics within Density-functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 59, 7413 (1999).
Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction. J. Comput. Chem. 27, 1787–1799 (2006).
Chan, K. et al. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem 6, 1899–1905 (2014).
Norskov, J. K. et al. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Henkelman, G. et al. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 113, 9901–9904 (2000).
Wang, S. et al. Asymmetric C-C Coupling for Highly Selective Electrochemical CO2 Reduction to Ethanol over Paired Cu Atoms Adjacent to Single Ag Atom. figshare. Dataset. https://doi.org/10.6084/m9.figshare.27199515 (2024).
Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0600200 (X.L.)), the National Key Research and Development Program of China (No. 2021YFA1500502 (X.L.)), the NSFC Center for Single-Atom Catalysis (grant No. 22388102 (X.L.)), the National Natural Science Foundation of China (22102176 (X.L.)) and 21925803 (Y.H.)), CAS Project for Young Scientists in Basic Research (YSBR-051 (X.L.)), the DICP.CAS-Cardiff Joint Research Units (121421ZYLH20230008 (X.L.)), the City University of Hong Kong Startup fund (B.L.), and the National Science and Technology Council (NSTC) (110-2112-M-213-019-MY3 (Y.Lu.)). The authors gratefully acknowledge the support of Photon Science Center for Carbon Neutrality.
Author information
Authors and Affiliations
Contributions
S.W., X.L., Y.H., and B.L. designed and conceived the experiment. S.W. performed the catalyst synthesis, structural characterization, and CO2RR electrocatalytic measurements. F.L. conducted the theoretical calculations. S.W. performed the operando electrochemical ATR-SEIRAS measurements and analyses. Z.L., T.L., S.H., and Y.Lu. contributed to the operando XAS spectroscopy measurements and analyses. S.W., Y.Z., and X.R. performed the operando Raman spectroscopy measurements and analyses. S.W., Y.Li., and Y.C., performed the quasi in-situ XPS measurements and analyses. F.L., Y.C., J.Z. and X.Y. carried out DFT data analysis. W.W. performed the HAADF-STEM measurements and analyses. S.L. performed EMARS method based on frequency filtering over HAADF-STEM images. S.W., X.L., and B.L. wrote and revised the manuscript with inputs from all authors. The project was supervised by X.L., Y.H., and B.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Antonia Herzog, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Li, F., Zhao, J. et al. Manipulating C-C coupling pathway in electrochemical CO2 reduction for selective ethylene and ethanol production over single-atom alloy catalyst. Nat Commun 15, 10247 (2024). https://doi.org/10.1038/s41467-024-54636-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54636-w
