

Abstract
STAT transcription factors play a critical role in mediating the effects of cytokines on myeloid cells. As STAT _target genes control key processes such as survival, proliferation and self-renewal, it is not surprising that constitutive activation of STATs, particularly STAT3 and STAT5, are common events in many myeloid tumors. STATs are activated both by mutant tyrosine kinases as well as other pathogenic events, and continued activation of STATs is common in the setting of resistance to kinase inhibitors. Thus, the _targeting of STATs, alone or in combination with other drugs, will likely have increasing importance for cancer therapy.
Keywords: STATs, leukemia, myeloproliferative neoplasms, oncology, therapy, transcription factors
Introduction
In the two decades since the STAT signaling pathway was first described, enormous strides have been made in understanding the critical role that this pathway plays in diverse hematopoietic cancers. STATs are typically oncogenic through the constitutive activation of tyrosine kinases, and through the years, a number of mutant kinases have been characterized that activate STAT signaling. Because of the critical role of tyrosine kinases in many cancers, much effort has gone into the search for inhibitors of these kinases that may be effective for cancer therapy. However, because of the critical role that STATs play in mediating the effect of kinases, they may also be directly _targeted and may be effective anti-cancer agents. As we come to better understand STAT signaling in cancer, our ability to directly _target the STAT pathway as a means of cancer therapy will be enhanced, thus contributing to a more personalized approach of treating patients.
STAT Signaling
Signal transducer and activator of transcription (STAT) proteins are a family of transcription factors that regulate critical cellular processes, such as proliferation, differentiation and apoptosis.1 When a growth factor or cytokine binds to its receptor, it either activates its intrinsic tyrosine kinase activity or it causes the receptor chains to aggregate, bringing associated tyrosine kinases, usually JAKs, into juxtaposition.2 This activates their kinase activity, which mediates the subsequent tyrosine phosphorylation of the JAKs themselves as well as the cytokine receptor chains. The highly tyrosine phosphorylated receptor-kinase complex then serves as a docking site for proteins, such as STATs, which possess src-homology-2 (SH2) domains that allow binding to specific tyrosine-phosphorylated amino acid sequences.3 The STATs recruited in this way become phosphorylated on unique tyrosine residues necessary for activation,4 then dissociate from the receptor-kinase complex and dimerize via reciprocal phosphotyrosine-SH2 interactions.5 The STAT dimers translocate to the nucleus where they bind to a nine base pair sequence in the regulatory regions of _target genes, thereby modulating their expression.6
STATs may also function as monomers and as non-phosphorylated dimers; however, in most circumstances it is the tyrosine phosphorylated dimer that is the critical mediator of signal transduction of the pathway. In some instances, the activity of the STAT transcription factor can be further modulated by the phosphorylation of the STAT protein on a serine residue.7 The activation of STATs is normally both rapid and transient and is subject to tight regulation. Such regulation includes not only kinase activation, but also inhibitory proteins that mediate the inactivation of STATs and prevent further signaling. These inhibitory regulators include phosphatases, suppressors of cytokine signaling (SOCS), protein inhibitors of activated STATs (PIAS) and nuclear ubiquitin E3 ligases.
STAT-mediated gene expression is involved in many normal physiological processes, such as proliferation, survival and differentiation. In hematopoietic cells in particular, cytokines whose effects are transduced by STATs play a central role in regulating the production of red blood cells, platelets and the full spectrum of white blood cells. Thus, it is not surprising that inappropriate activation of STATs plays a critical role in the formation and maintenance of the full spectrum of hematopoietic cancers, particularly those of the myeloid lineage. This can occur by a variety of mechanisms, including the autocrine or paracrine production of growth factors, activation of kinases by mutations, or loss of negative regulators (Fig. 1).

Figure 1. STAT transcription factors, primarily STAT3 and STAT5, are activated in myeloid malignancies through a variety of mechanisms, including autocrine and paracrine growth factors, mutated receptors and kinases and decreased activity of negative regulators including phosphatases and SOCS proteins. Activated dimers bind to the regulatory region of _target genes, recruit co-activators and modulate transcription of key _target genes.
Acute myeloid leukemia
Deregulated STAT signaling is associated with increased cellular proliferation, disturbed differentiation and arrested apoptosis, which are the hallmarks of leukemogenesis. Constitutive activation of two of the family members, STAT3 and STAT5, either alone or together, has been demonstrated in leukemic cell lines and blasts in a substantial proportion of patients with acute myeloid leukemia (AML).8-12 Constitutive activation of STAT3 and the presence of a truncated isoform, STAT3B, were correlated with a poor clinical outcome.12 Moreover, expression of the STAT3B isoform was more prevalent in relapse as compared with diagnosis.13 Recently, it was suggested that induced phosphorylation of signaling intermediates was more informative for understanding the biology of leukemic cells than the basal phosphorylation state. Using single cell flow cytometry, it was shown that potentiated STAT3 and STAT5 phosphorylation post growth factor stimulation was associated with a negative outcome for patients receiving standard AML chemotherapy.14,15
Several mechanisms have been implicated for the constitutive activation of STATs in leukemias, including autocrine/paracrine stimulation by cytokines16 and the effects of kinases activated through mutations. Some of these mutations include chromosomal translocations generating fusion proteins with constitutive tyrosine kinase activity, such as BCR/ABL, which is a kinase fusion protein in chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL) that leads to the constitutive activation of STAT5. Mutations in FMS related tyrosine kinase 3 (FLT3), either involving internal tandem duplications (ITD) or point mutations in the activating loop of the tyrosine kinase domain, are observed in approximately 30% of AML patients and are associated with poorer prognosis.17 FLT3 ITD mutations cause the constitutive activation of FLT3, leading to aberrant activation of multiple downstream pathways, including STAT5.18-20 The activation of STAT5 by FLT3 ITD is independent of Src and JAK kinases.21 Further supporting the pathogenic role of this mutation, FLT3-ITD expression confers factor independent growth in murine IL-3-dependent cell lines and causes a fatal myeloproliferative disorder in murine bone marrow transplantation models and in FLT3-ITD knock-in mice.22,23
Several small molecule FLT3-tyrosine kinase inhibitors (TKI) have been developed and examined in AML patients as single agents or in combination with chemotherapy. The induction of cytotoxicity by FLT3 inhibitors is closely correlated with deactivation of STAT5, while resistance to FLT3 inhibition is associated with persistent activation of STAT5.24 It is hypothesized that upregulation of FLT3 ligand and the silencing of SOCS expression by methylation of its genetic regulatory elements combine to enhance STAT signaling activity. These data support the use of combination therapy of FLT3 inhibitors with agents _targeting the STAT pathway as treatment for AML patients with FLT3 mutations.25
Chronic myelogenous leukemia
Chronic myelogenous leukemia (CML) is characterized by the presence of the Philadelphia chromosome, the reciprocal translocation of chromosomes 9 and 22 that generate the fusion protein BCR/ABL. This protein functions as a tyrosine kinase and can transform hematopoietic cells.26 STAT5 is constitutively activated by both the 190 kD and 210 kD isoforms of BCR/ABL.27-30 STAT5 activation is correlated with functional effects on cell cycle progression and resistance to apoptosis through increased expression of cyclin D1 and Bcl-xl, respectively31,32 and is essential for leukemic cell survival.33,34 Murine STAT5A-null bone marrow cells were inefficient in generating and maintaining a CML-like disease, suggesting an important role of STAT5 in the pathogenesis of CML.35 STAT5 activation may play a critical role in drug resistance in CML through the induction of P-glycoprotein and the modulation of telomerase activity,36 and high expression of STAT5 accounts for TKI resistance.37,38
Currently, BCR/ABL kinase inhibition by imatinib, and the related kinase inhibitors nilotinib and dasatinib, is considered standard therapy for CML.38,39 Imatinib leads to complete inhibition of STAT5 activation, and this is likely a key part of the effectiveness of this therapeutic approach. However, resistance to imatinib develops in a subset of patients, generally through mutations in BCR/ABL that impair binding of the inhibitors to the ATP-binding site.
Several approaches have been taken to identify _targets other than BCR/ABL for treating CML resistant to kinase inhibitors.40,41 Given the central role of STAT5 in mediating the pathogenic effects of BCR/ABL, this is an appealing _target, as discussed in more detail below.42,43
Another suggested mechanism for imatinib resistance is activation of STAT3 within the bone marrow microenvironment. This novel mechanism suggests the utility of using STAT3 inhibitors to increase the efficacy of BCR-ABL inhibitors.44
Myeloproliferative neoplasms
Myeloproliferative neoplasms (MPN) are a group of clonal disorders that arise from the transformation of hematopoietic stem cells. For many years the molecular pathogenesis of these diseases remained unknown. It was reported that a subset of the patients with polycythemia vera (PV) displayed constitutive STAT3 activation in their peripheral granulocytes.45 By applying a panel of inhibitors, it was also shown that spontaneous erythropoietin-independent differentiation in PV is due to a constitutive activation of signaling pathways including JAK2-STAT5, PI3K and Src.46 STAT5 nuclear translocation and activation was detected in megakaryocytes and in circulating CD34+ cells from the majority of patients with idiopathic myelofibrosis and the spontaneous growth of these cells was abolished by STAT5 or JAK2 inhibition.47
In 2005, several groups reported a single acquired point mutation in JAK2 in the majority of patients with Philadelphia chromosome (Ph)-negative MPN.48-51 This JAK2 mutation is a valine to phenylalanine substitution at position 617 (JAK2 V617F) in the kinase-dead JH2 domain. It is believed that this mutation disrupts the auto-inhibitory effect of the JH2 domain on the JH1 domain, which leads to both constitutive activation and hypersensitivity to the effect of cytokines.52 This mutation is believed to play a critical role in the pathogenesis of these disorders; mice transplanted with bone marrow cells transduced by a retrovirus expressing JAK2 V617F rapidly develop erythrocytosis, progressing to a myelofibrotic state within a few months.52-54 Reflecting the central role of STAT5 in the pathogenesis of these diseases, there is constitutive activation of STAT5 in JAK2 V617F-expressing Ba/F3 cells and a significant increase in phosphorylated STAT5 in the bone marrow and spleens of JAK2 V617F animals.53,54
The ability of JAK2 V617F to induce cytokine-independent activation of the JAK2 and STAT5 pathways and transformation to cytokine independence requires the coexpression of homodimeric Type I cytokine receptors, such as the erythropoietin receptor (EpoR), thrombopoietin receptor (TpoR) or granulocyte colony-stimulating-factor receptor (GCSFR). EpoR mutations that impair erythropoietin-mediated JAK2 or STAT5 activation also impair transformation mediated by the JAK2 V617F kinase, indicating that JAK2 V617F requires a cytokine receptor scaffold for its transforming and signaling activities.55,56 Introduction of a constitutively active form of STAT5 and the overexpression of the STAT5 _target gene Bcl-xl into human erythroid progenitors induces an erythropoietin-independent terminal differentiation and endogenous erythroid colony (EEC) formation, which is a hallmark of PV. STAT5 and Bcl-xl knock-down in human erythroid progenitors inhibits colony-forming unit-erythroid (CFU-E) formation in the presence of erythropoietin. These results suggest that JAK2 V617F may induce EEC via the STAT5-dependent Bcl-xl expression.57
The mutational frequency of JAK2 V617F is estimated at over 95% in PV, 50% in essential thrombocytosis (ET) and primary myelofibrosis (PMF), 20% in certain other MPNs including refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T) and less than 5% in AML or myelodysplastic syndrome.58 Thus, JAK2 V617F plays a critical role in a significant subset of MPNs, suggesting that _targeting this kinase may be a useful treatment strategy.
In addition to STAT5, STAT3 is also activated in MPNs. Immunostaining of bone marrow biopsies show three specific patterns of phosphorylated STAT3 and STAT5 that differed significantly from the normal pattern. Specifically, there is uniformly increased STAT3 and STAT5 phosphorylation in PV; increased STAT3 phosphorylation and reduced STAT5 phosphorylation in ET; and uniformly reduced STAT3 and STAT5 phosphorylation in PMF. Interestingly, in all evaluated MPNs, the STAT5 and STAT3 phosphorylation pattern is not influenced by the presence of the JAK2 V617F mutation.59 By contrast, in another study examining phospho-STAT5 immunohistochemistry of bone marrow biopsies of chronic MPN, all patients with the JAK2 V617F mutation showed abnormal nuclear STAT5 phosphorylation. In the JAK2 wild-type group, STAT5 phosphorylation was observed in about a third of the patients.60,61
In addition to JAK2 V617F, other JAK2 mutations have been described in MPN. Mutations in exon 12 of JAK2 are consistently associated with increased levels of tyrosine phosphorylated JAK2 and STAT5. When transduced into Ba/F3 cells, all four JAK2 exon 12 mutations caused growth factor hypersensitivity and activation of biochemical pathways associated with erythropoietin signaling.62 Interestingly, JAK2 exon 12 mutations did not affect the level of STAT5 phosphorylation or Akt phosphorylation in immunohistochemistry of bone marrow biopsies from a small number of patients,61 demonstrating the complexity of the molecular mechanisms in MPN.
A gain of function mutation in the thrombopoietin receptor (MPL) is found in primary myelofibrosis. This mutation, MPL W515L, as well as other MPL mutations, has a prevalence of 4% in ET and up to 11% in primary myelofibrosis.58,63 Expression of MPLW515L in 32D, UT7 or Ba/F3 cells conferred cytokine independent growth and thrombopoietin hypersensitivity and resulted in constitutive phosphorylation of JAK2, STAT3, STAT5, Akt and ERK. In a murine bone marrow transplant assay, expression of MPLW515L resulted in a fully penetrant myeloproliferative disorder characterized by marked thrombocythemia, splenomegaly due to extramedullary hematopoiesis and increased reticulin fibrosis. Pharmacological reduction of JAK kinase activity inhibited MPLW515L-mediated proliferation and JAK-STAT signaling in vitro.63 In a murine model, JAK2 inhibition improved survival, normalized white blood cell counts and platelet counts and markedly reduced extramedullary hematopoiesis and bone marrow fibrosis. There was a dose-dependent inhibition of STAT signaling, including potent inhibition of STAT3 and STAT5 phosphorylation in primary tissues from MPLW515L mice treated with the JAK inhibitor.64
Several JAK kinase inhibitors are currently in clinical trials. They are effective in alleviating constitutional symptoms and reducing spleen size but they have not been sufficient in inducing histologic or molecular remission. In addition, they can induce side effects including myelosuppression, gastrointestinal disturbances, asymptomatic elevation of liver and pancreatic enzymes, peripheral neuropathy and hyperacute relapse of symptoms during treatment interruption.65 Since JAK mutations in MPN do not always occur in the predominant or ancestral mutant clone, the development of inhibitors to common mediators of diverse signaling pathways in this disease is very desirable. One attractive convergence point of these diverse pathways is the STAT signaling pathway, and thus the development of STAT inhibitors may help improve clinical outcomes for patients with MPN.
It is also worth mentioning that other rarer myeloid malignancies such as systemic mastocytosis (characterized by a D816V mutation in KIT), hypereosinophilic syndrome (characterized by the FIP1L1-PDGFRa tyrosine kinase fusion protein generated by an interstitial deletion on chromosome 4q12) and chronic myeloproliferative diseases (CMPD) with t(5;12) (generating the TEL-PDGFR tyrosine kinase fusion protein) exhibit constitutive activation of STATs, which play a significant role in the pathogenesis of these diseases.66-72 Thus, it is clear that STATs, particularly STAT5 and STAT3, are activated in the full spectrum of myeloid diseases, regardless of the upstream mutational event. These proteins then mediate the transcriptional activation of _target genes that directly drive the phenotype of these cells, including proliferation, survival, self-renewal and resistance to chemotherapy (Fig. 2). This suggests that pharmacological STAT inhibitors might be particularly beneficial for the treatment of patients suffering from these malignancies.
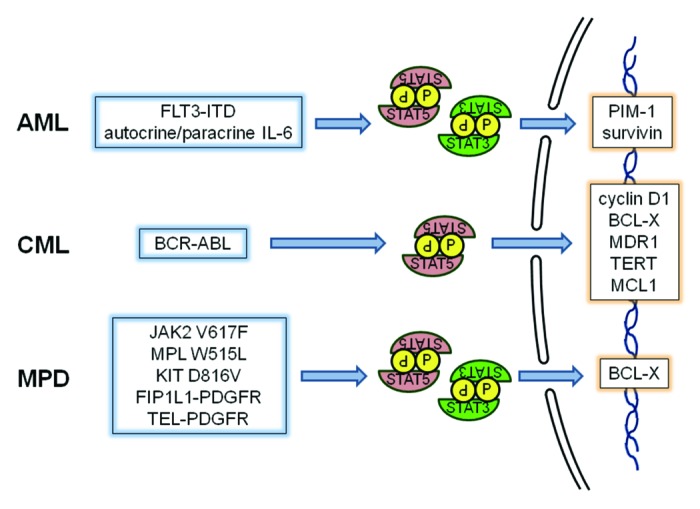
Figure 2. In myeloid leukemias and myeloproliferative neoplasms, a variety of mutations can lead to the activation of tyrosine kinases that can phosphorylate STATs, particularly STAT5 and STAT3. These STATs then drive the transcriptional activation of genes regulating survival, proliferation, self-renewal and other phenotypes characteristic of these diseases.
_targeting the STAT Pathway for the Treatment of Hematologic Malignancies
Since STATs are activated in numerous blood cancers and are essential to the pathogenesis of these tumors, _targeting STATs is an attractive approach for therapeutic intervention. The activation of STATs can occur through the constitutive activity of tyrosine kinases, such as BCR/ABL, FLT3 and JAK2, as well as activation by autocrine and paracrine factors, loss of negative regulators and other mechanisms. Inhibiting tyrosine kinases is an appealing strategy for treating these diseases, in that it addresses the driving mutation in the malignant cell and can shut down several downstream pathways simultaneously (Fig. 3). In fact, the development of imatinib and other BCR/ABL kinase inhibitors represents a triumph of the molecular therapy of cancer. However, there are several limitations to this strategy. First, resistance often emerges to kinase inhibitors. This can occur through further mutations of the kinase, blocking the ability of the drug to bind to the _target.73,74 In addition, activation of other kinases may occur to circumvent the dependence on the inhibited kinase.75 Thus, inhibition of a common downstream mediator of the effects of these activated kinases holds out the promise for increased efficacy even in the setting of additional kinase mutations, the ability to block the effects of other activated kinases and the potential to synergize with kinase inhibitors and other therapies. The large number of tyrosine kinases that can be activated in hematological cancers converges on a small number of transcription factors, which then regulate the transcription of the genes driving the tumor phenotype. Therefore, an appealing strategy is to directly _target key transcription factors, such as STAT3 and STAT5, which may have broad applicability for cancer therapy (Fig. 4).

Figure 3. The activation of STATs in cancer cells can be blocked by modulating _targets resulting in loss of STAT phosphorylation. This includes inhibition of receptors and their ligands, inhibition of activated kinases (both mutated and unmutated) or activation of negative regulators such as phosphatases and SOCS proteins.

Figure 4. In addition to inhibiting kinases, STATs can be _targeted directly by blocking their ability to form activated dimers, translocate into the nucleus, bind DNA or recruit co-activators.
Strategies to developing STAT inhibitors: cell-based screens
The multiple steps through which an unphosphorylated STAT molecule in the cytoplasm proceed to activate gene transcription in the nucleus affords a number of opportunities for _targeted inhibition. One strategy to identify inhibitors of the various steps in the STAT signaling pathway is to establish a cell-based assay in which the transcriptional activity of STATs can be monitored using a reporter, such as luciferase. Coupled with a counter screen to exclude non-specific effects, this approach allows the ability to rapidly screen thousands of compounds to identify specific STAT inhibitors. The open-ended nature of this screen allows for the identification of STAT inhibitors at any step in the signaling pathway, although it can be challenging to deconvolute how a hit derived from this assay specifically blocks STAT function. One compound that has emerged from this approach is pimozide, which inhibits both STAT3 and STAT5 in hematopoietic tumors including CML, AML and MPNs.42,76 Pimozide inhibits STAT3 and STAT5 phosphorylation, but several lines of evidence strongly suggest that it does not inhibit kinases such as BCR/ABL, FLT3 and JAK2. As expected by virtue of its _targeting a downstream mediator, pimozide is effective in models of CML driven by BCR/ABL mutations, such as T315I, that render it resistant to currently available kinase inhibitors.
Pimozide has also displayed anti-leukemic effects in in vivo models. In a mouse model of AML driven by a FLT3 ITD mutation, pimozide results in a notable reduction in tumor burden (Nelson and Frank, manuscript under revision). Pimozide, which is FDA approved for neurological disorders, is known to have a good safety profile in humans. Reflecting this, pimozide is effective at blocking colony formation in vitro from CD34+ cells derived from patients with CML, but has minimal effect on colony formation from CD34+ cells derived from healthy donors. Nonetheless, it is not yet clear if effective anti-tumor doses are achievable in humans. Ultimately, it is possible to test the pharmacodynamic effects of pimozide in AML patients by treating with doses known to be safe in humans, then testing for changes in STAT5 phosphorylation in blasts in the peripheral blood or bone marrow.
Other STAT inhibitors that have been identified by this approach include nifuroxazide, which appears to act through kinase inhibition, and pyrimethamine, whose mechanism of action is still being elucidated.77-79 Thus, cell-based screens represent one useful strategy for developing STAT inhibitors with potential for clinical development.
The role of phosphatases in STAT inhibition
STAT activation represents a balance between phosphorylation by tyrosine kinases and deactivation largely by phosphatases. In some cancers, negative regulators of STATs exhibit low activity through decreased expression, often through promoter methylation.80 Therefore, one method of reducing STAT signaling is by enhancing the activity of these negative regulators. There are several reports of drugs whose effects may be dependent on these negative regulators.81,82 For example, sunitinib and sorafenib are kinase inhibitors that are approved for use in solid tumors, though they may have benefit in hematopoietic tumors as well. However, several recent reports have also suggested a role for phosphatases in the action of these drugs. By inhibiting the activity of phosphatases using sodium vanadate or by siRNA-mediated knockdown, the ability of sorafenib or sunitinib to decrease STAT3 tyrosine phosphorylation was reduced. In addition, the reduction of phosphatase activity inhibited the ability of sunitinib to kill tumor cells in vitro. Therefore, it is possible that the activation of phosphatases is a critical component of the clinical effects of sorafenib, sunitinib and other kinase inhibitors. It is not surprising that phosphatases play an important albeit indirect role in dephosphorylating activated STATs after kinase inhibition. However, these studies on sorafenib and sunitinib suggest a more active role of these drugs on phosphatases, and thus, the dephosphorylation of the STATs may occur through both the inhibition of the kinase and activation of the phosphatase. Thus, the inhibition of STAT phosphorylation by the activation of phosphatases may gain increased importance in the near future.
Inhibition of DNA binding: decoy oligonucleotides
Transcription factors bind to DNA to regulate expression of their _target genes, and thus one method of inhibiting their activity is to prevent them from binding to their _target sequence. One means of doing so is by competing the protein away from binding to the regulatory regions of genes. Synthetic double stranded oligonucleotides containing a STAT binding sequence can act as decoys, such that activated STATs bind to this sequence, rather than to the regulatory regions of their endogenous _target genes.83 The K562 CML cell line depends on the constitutive activation of STAT5, making it a logical model to test this approach. Introducing a decoy oligonucleotide into these cells reduces STAT5 transcriptional activity leading to the reduction in expression of critical STAT5 _target genes and the induction of apoptosis. Importantly, this decoy oligonucleotide has no effect on the myeloid HL-60 cell line, which has no constitutively activated STATs, demonstrating that the effect of the decoy oligonucleotide depends on the presence of activated STAT5.84 Therefore, preventing STAT transcriptional activity through the use of a decoy oligonucleotide may be an effective way of reducing STAT activity in tumor cells.
Inhibition of DNA binding: small molecules
Since double-stranded oligonucleotides may have pharmacological properties limiting their applicability in vivo, alternate approaches to inhibit STAT-DNA binding are also of potential importance. To identify small molecules that directly inhibit STAT3 DNA binding activity, a library of compounds was screened using an in vitro binding assay.85 A platinum (IV) complex, IS3 295, was identified by its ability to inhibit STAT3 DNA binding. Though other platinum-based compounds such as cisplatin bind to DNA, their binding is non-specific. In contrast, IS3 295 inhibits STAT1 and STAT3 homo- and heterodimers from binding DNA, while having no effect on the ability of STAT5 homodimers or the unrelated E2F1 protein to bind to DNA. In contrast, cisplatin has no effect on the DNA binding activity of any of these proteins, demonstrating the specificity of IS3 295 to STAT1 and STAT3. Significantly, IS3 295 did not disrupt the binding of STATs that were already bound to DNA, suggesting that they could only bind to free STAT proteins. This led to the suggestion that IS3 295 directly interacts with the DNA binding domain of STATs, thereby preventing them from binding to DNA. IS3 295 inhibited STAT-mediated gene transcription and it led to apoptosis in cells containing constitutively activated STAT3.
Flavopiridol is a drug that has well-known antineoplastic activity due to its ability to inhibit cyclin-dependent kinases, but intriguing data suggest it might also disrupt STAT3-DNA binding.86 Using a variety of cell-free assays, it has been shown that flavopiridol inhibits the DNA binding activity of STAT3, while not affecting DNA binding of other proteins. In addition, flavopiridol decreases the transcription of Mcl-1, a STAT3 _target gene important in apoptosis regulation. Though flavopiridol affects RNA polymerase II phosphorylation, it does not cause a global reduction in gene transcription, suggesting that flavopiridol has some selectivity to STAT3. Therefore, the data on flavopiridol and IS3 295 suggest that it is possible to inhibit the interaction of STAT3 with DNA and kill tumor cells, and thus _targeted DNA binding inhibitors may have significant therapeutic potential.
Dimerization inhibitors
The activity of STATs is critically dependent on their SH2 domains. These are required for recruitment of STATs to activated receptor-kinase complexes where they become phosphorylated, and for each monomer to bind to the phosphorylated tyrosine of its binding partner, allowing active dimers to form. Though there may be biological effects of STAT monomers, tyrosine phosphorylated STAT dimers are likely the predominant active molecule for transcriptional regulation. Therefore, small molecules that specifically block SH2 domains would likely be useful STAT inhibitors. Using a structure-based virtual screen as well as the interrogation of chemical libraries, several such dimerization inhibitors have been identified.87,88 The first dimerization inhibitor discovered was STA-21. This compound disrupts dimer formation, has no effect on STAT3 phosphorylation and lacks any effect on STAT1 or STAT5.89 Treatment of cells with STA-21 reduced the expression of STAT3 _target genes and induced apoptosis in cancer cell lines containing activated STAT3. Significantly, this compound was used in a small clinical trial.90 Topically applied STA-21 was successfully used to treat the skin lesions of psoriasis, a disease characterized by constitutive STAT3 activity. It is unclear what would be the bioavailability of this compound should it be given systemically for cancer patients.
Other similar approaches have led to both peptide-based and non-peptide small molecules that _target the STAT3 SH2 domain.91-94 For example, C188-9, which selectively blocks STAT3 but not STAT1 phosphorylation, induces apoptosis in AML cell lines.95 Taken together, these studies suggest that STAT dimerization inhibitors may be important approaches to the treatment of cancer.
Concluding Remarks
Advances in our understanding of myeloid diseases has revealed that STAT transcription factors play a key role in activating genes driving the inappropriate proliferation, survival and self-renewal characteristic of these diseases. In addition to providing insights into their pathogenesis, these findings have also opened up new possibilities for _targeted therapies of these diseases. The increasing use of tyrosine kinase inhibitors in clinical practice and in clinical trials has been a major advance in cancer therapy, and many of these drugs exert some or all of their effects through inhibition of STATs. However, directly _targeting STATs is also likely to have a clinical benefit that may complement or exceed that of kinase inhibitors. Even in CML, where the _targeting of BCR/ABL has led to great success, issues such as the emergence of drug resistance or the inability to eradicate the leukemic stem cell may be overcome with direct STAT5 inhibitors. In MPNs and AML, kinase inhibitors _targeting JAK2 or FLT3 have been significantly less effective, perhaps due to co-activation of other pathways. While STATs and other transcription factors had traditionally been viewed as difficult _targets to modulate pharmacologically, it is clear that with the use of a variety of strategies significant progress is being made. Thus, STAT inhibitors, alone or combined with kinase inhibitors and other therapies, may lead to enhanced clinical benefit.
Acknowledgments
Research from our laboratory reported in this manuscript was supported by the NIH (NS050830), the Multiple Myeloma Research Foundation (Norwalk, CT), the Kittredge Foundation (Dana-Farber Cancer Institute), the Brent Leahey Fund (Dana-Farber Cancer Institute), Gabrielle's Angel Foundation (New York, NY) and the Claudia Adams Barr Program in Innovative Basic Cancer Research (Dana-Farber Cancer Institute).
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/20006
References
- 1.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 2.Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K, Silvennoinen O. Signaling through the hematopoietic cytokine receptors. Annu Rev Immunol. 1995;13:369–98. doi: 10.1146/annurev.iy.13.040195.002101. [DOI] [PubMed] [Google Scholar]
- 3.Heim MH, Kerr IM, Stark GR, Darnell JE., Jr Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science. 1995;267:1347–9. doi: 10.1126/science.7871432. [DOI] [PubMed] [Google Scholar]
- 4.Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-γ. Science. 1993;261:1744–6. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- 5.Shuai K, Horvath CM, Huang LHT, Qureshi SA, Cowburn D, Darnell JE., Jr Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76:821–8. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr., Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–39. doi: 10.1016/S0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 7.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 8.Gouilleux-Gruart V, Gouilleux F, Desaint C, Claisse J-F, Capiod J-C, Delobel J, et al. STAT-related transcription factors are constitutively activated in peripheral blood cells from acute leukemia patients. Blood. 1996;87:1692–7. [PubMed] [Google Scholar]
- 9.Xia Z, Baer MR, Block AW, Baumann H, Wetzler M. Expression of signal transducers and activators of transcription proteins in acute myeloid leukemia blasts. Cancer Res. 1998;58:3173–80. [PubMed] [Google Scholar]
- 10.Birkenkamp KU, Geugien M, Lemmink HH, Kruijer W, Vellenga E. Regulation of constitutive STAT5 phosphorylation in acute myeloid leukemia blasts. Leukemia. 2001;15:1923–31. doi: 10.1038/sj.leu.2402317. [DOI] [PubMed] [Google Scholar]
- 11.Spiekermann K, Biethahn S, Wilde S, Hiddemann W, Alves F. Constitutive activation of STAT transcription factors in acute myelogenous leukemia. Eur J Haematol. 2001;67:63–71. [PubMed] [Google Scholar]
- 12.Benekli M, Xia Z, Donohue KA, Ford LA, Pixley LA, Baer MR, et al. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood. 2002;99:252–7. doi: 10.1182/blood.V99.1.252. [DOI] [PubMed] [Google Scholar]
- 13.Xia Z, Sait SNJ, Baer MR, Barcos M, Donohue KA, Lawrence D, et al. Truncated STAT proteins are prevalent at relapse of acute myeloid leukemia. Leuk Res. 2001;25:473–82. doi: 10.1016/S0145-2126(00)00158-2. [DOI] [PubMed] [Google Scholar]
- 14.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud Ò, Gjertsen BT, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 15.Kornblau SM, Minden MD, Rosen DB, Putta S, Cohen A, Covey T, et al. Dynamic single-cell network profiles in acute myelogenous leukemia are associated with patient response to standard induction therapy. Clin Cancer Res. 2010;16:3721–33. doi: 10.1158/1078-0432.CCR-10-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuringa J-J, Wierenga ATJ, Kruijer W, Vellenga E. Constitutive Stat3, Tyr705, and Ser727 phosphorylation in acute myeloid leukemia cells caused by the autocrine secretion of interleukin-6. Blood. 2000;95:3765–70. [PubMed] [Google Scholar]
- 17.Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic _target in AML: still challenging after all these years. Blood. 2010;116:5089–102. doi: 10.1182/blood-2010-04-261867. [DOI] [PubMed] [Google Scholar]
- 18.Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Müller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–14. [PubMed] [Google Scholar]
- 19.Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000;19:624–31. doi: 10.1038/sj.onc.1203354. [DOI] [PubMed] [Google Scholar]
- 20.Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin Cancer Res. 2003;9:2140–50. [PubMed] [Google Scholar]
- 21.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110:370–4. doi: 10.1182/blood-2006-05-024018. [DOI] [PubMed] [Google Scholar]
- 22.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–8. doi: 10.1182/blood.V99.1.310. [DOI] [PubMed] [Google Scholar]
- 23.Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. 2005;105:4792–9. doi: 10.1182/blood-2004-11-4430. [DOI] [PubMed] [Google Scholar]
- 24.Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: the induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood. 2006;108:3494–503. doi: 10.1182/blood-2006-04-015487. [DOI] [PubMed] [Google Scholar]
- 25.Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic _targets in AML. Blood. 2009;113:4052–62. doi: 10.1182/blood-2008-05-156422. [DOI] [PubMed] [Google Scholar]
- 26.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 27.Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183:811–20. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ilaria RL, Jr., Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271:31704–10. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 29.Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–54. [PubMed] [Google Scholar]
- 30.Frank DA, Varticovski L. BCR/abl leads to the constitutive activation of Stat proteins, and shares an epitope with tyrosine phosphorylated Stats. Leukemia. 1996;10:1724–30. [PubMed] [Google Scholar]
- 31.Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000;96:2269–76. [PubMed] [Google Scholar]
- 32.de Groot RP, Raaijmakers JA, Lammers JW, Koenderman L. STAT5-Dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol Cell Biol Res Commun. 2000;3:299–305. doi: 10.1006/mcbr.2000.0231. [DOI] [PubMed] [Google Scholar]
- 33.Sillaber C, Gesbert F, Frank DA, Sattler M, Griffin JD. STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood. 2000;95:2118–25. [PubMed] [Google Scholar]
- 34.Basákiewicz-Masiuk M, Machaliński B. The role of the STAT5 proteins in the proliferation and apoptosis of the CML and AML cells. Eur J Haematol. 2004;72:420–9. doi: 10.1111/j.1600-0609.2004.00242.x. [DOI] [PubMed] [Google Scholar]
- 35.Ye D, Wolff N, Li L, Zhang S, Ilaria RL., Jr STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood. 2006;107:4917–25. doi: 10.1182/blood-2005-10-4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada O, Ozaki K, Furukawa T, Machida M, Wang YH, Motoji T, et al. Activation of STAT5 confers imatinib resistance on leukemic cells through the transcription of TERT and MDR1. Cell Signal. 2011;23:1119–27. doi: 10.1016/j.cellsig.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Hölbl A, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117:3409–20. doi: 10.1182/blood-2009-10-248211. [DOI] [PubMed] [Google Scholar]
- 38.Nam S, Williams A, Vultur A, List A, Bhalla K, Smith D, et al. Dasatinib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol Cancer Ther. 2007;6:1400–5. doi: 10.1158/1535-7163.MCT-06-0446. [DOI] [PubMed] [Google Scholar]
- 39.Kindler T, Breitenbuecher F, Kasper S, Stevens T, Carius B, Gschaidmeier H, et al. In BCR-ABL-positive cells, STAT-5 tyrosine-phosphorylation integrates signals induced by imatinib mesylate and Ara-C. Leukemia. 2003;17:999–1009. doi: 10.1038/sj.leu.2402940. [DOI] [PubMed] [Google Scholar]
- 40.Scherr M, Chaturvedi A, Battmer K, Dallmann I, Schultheis B, Ganser A, et al. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML) Blood. 2006;107:3279–87. doi: 10.1182/blood-2005-08-3087. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Cai D, Brendel C, Barett C, Erben P, Manley PW, et al. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109:2147–55. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- 42.Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117:3421–9. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Zeng J, Shi M, Zhao S, Bai W, Cao W, et al. _targeted blockage of signal transducer and activator of transcription 5 signaling pathway with decoy oligodeoxynucleotides suppresses leukemic K562 cell growth. DNA Cell Biol. 2011;30:71–8. doi: 10.1089/dna.2010.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7:3169–75. doi: 10.1158/1535-7163.MCT-08-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Röder S, Steimle C, Meinhardt G, Pahl HL. STAT3 is constitutively active in some patients with Polycythemia rubra vera. Exp Hematol. 2001;29:694–702. doi: 10.1016/S0301-472X(01)00637-3. [DOI] [PubMed] [Google Scholar]
- 46.Ugo V, Marzac C, Teyssandier I, Larbret F, Lécluse Y, Debili N, et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol. 2004;32:179–87. doi: 10.1016/j.exphem.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Komura E, Chagraoui H, Mansat de Mas V, Blanchet B, de Sepulveda P, Larbret F, et al. Spontaneous STAT5 activation induces growth factor independence in idiopathic myelofibrosis: possible relationship with FKBP51 overexpression. Exp Hematol. 2003;31:622–30. doi: 10.1016/S0301-472X(03)00085-7. [DOI] [PubMed] [Google Scholar]
- 48.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 49.James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 50.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 51.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 52.Delhommeau F, Pisani DF, James C, Casadevall N, Constantinescu S, Vainchenker W. Oncogenic mechanisms in myeloproliferative disorders. Cell Mol Life Sci. 2006;63:2939–53. doi: 10.1007/s00018-006-6272-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–81. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–60. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 55.Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102:18962–7. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funakoshi-Tago M, Tago K, Abe M, Sonoda Y, Kasahara T. STAT5 activation is critical for the transformation mediated by myeloproliferative disorder-associated JAK2 V617F mutant. J Biol Chem. 2010;285:5296–307. doi: 10.1074/jbc.M109.040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garçon L, Rivat C, James C, Lacout C, Camara-Clayette V, Ugo V, et al. Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood. 2006;108:1551–4. doi: 10.1182/blood-2005-10-009514. [DOI] [PubMed] [Google Scholar]
- 58.Tefferi A. Molecular drug _targets in myeloproliferative neoplasms: mutant ABL1, JAK2, MPL, KIT, PDGFRA, PDGFRB and FGFR1. J Cell Mol Med. 2009;13:215–37. doi: 10.1111/j.1582-4934.2008.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teofili L, Martini M, Cenci T, Petrucci G, Torti L, Storti S, et al. Different STAT-3 and STAT-5 phosphorylation discriminates among Ph-negative chronic myeloproliferative diseases and is independent of the V617F JAK-2 mutation. Blood. 2007;110:354–9. doi: 10.1182/blood-2007-01-069237. [DOI] [PubMed] [Google Scholar]
- 60.Aboudola S, Murugesan G, Szpurka H, Ramsingh G, Zhao X, Prescott N, et al. Bone marrow phospho-STAT5 expression in non-CML chronic myeloproliferative disorders correlates with JAK2 V617F mutation and provides evidence of in vivo JAK2 activation. Am J Surg Pathol. 2007;31:233–9. doi: 10.1097/01.pas.0000213338.25111.d3. [DOI] [PubMed] [Google Scholar]
- 61.Grimwade LF, Happerfield L, Tristram C, McIntosh G, Rees M, Bench AJ, et al. Phospho-STAT5 and phospho-Akt expression in chronic myeloproliferative neoplasms. Br J Haematol. 2009;147:495–506. doi: 10.1111/j.1365-2141.2009.07870.x. [DOI] [PubMed] [Google Scholar]
- 62.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koppikar P, Abdel-Wahab O, Hedvat C, Marubayashi S, Patel J, Goel A, et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood. 2010;115:2919–27. doi: 10.1182/blood-2009-04-218842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tefferi A, Pardanani A. JAK inhibitors in myeloproliferative neoplasms: rationale, current data and perspective. Blood Rev. 2011;25:229–37. doi: 10.1016/j.blre.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 66.Ning ZQ, Li J, Arceci RJ. Signal transducer and activator of transcription 3 activation is required for Asp(816) mutant c-Kit-mediated cytokine-independent survival and proliferation in human leukemia cells. Blood. 2001;97:3559–67. doi: 10.1182/blood.V97.11.3559. [DOI] [PubMed] [Google Scholar]
- 67.Harir N, Boudot C, Friedbichler K, Sonneck K, Kondo R, Martin-Lannerée S, et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood. 2008;112:2463–73. doi: 10.1182/blood-2007-09-115477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baumgartner C, Cerny-Reiterer S, Sonneck K, Mayerhofer M, Gleixner KV, Fritz R, et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: subcellular distribution and role of the transforming oncoprotein KIT D816V. Am J Pathol. 2009;175:2416–29. doi: 10.2353/ajpath.2009.080953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic _target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–14. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 70.Buitenhuis M, Verhagen LP, Cools J, Coffer PJ. Molecular mechanisms underlying FIP1L1-PDGFRA-mediated myeloproliferation. Cancer Res. 2007;67:3759–66. doi: 10.1158/0008-5472.CAN-06-4183. [DOI] [PubMed] [Google Scholar]
- 71.Cain JA, Xiang Z, O’Neal J, Kreisel F, Colson A, Luo H, et al. Myeloproliferative disease induced by TEL-PDGFRB displays dynamic range sensitivity to Stat5 gene dosage. Blood. 2007;109:3906–14. doi: 10.1182/blood-2006-07-036335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilbanks AM, Mahajan S, Frank DA, Druker BJ, Gilliland DG, Carroll M. TEL/PDGFbetaR fusion protein activates STAT1 and STAT5: a common mechanism for transformation by tyrosine kinase fusion proteins. Exp Hematol. 2000;28:584–93. doi: 10.1016/S0301-472X(00)00138-7. [DOI] [PubMed] [Google Scholar]
- 73.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 74.Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 75.Ito T, Tanaka H, Kimura A. Establishment and characterization of a novel imatinib-sensitive chronic myeloid leukemia cell line MYL, and an imatinib-resistant subline MYL-R showing overexpression of Lyn. Eur J Haematol. 2007;78:417–31. doi: 10.1111/j.1600-0609.2007.00835.x. [DOI] [PubMed] [Google Scholar]
- 76.Bar-Natan M, Nelson EA, Walker SR, Kuang Y, Distel RJ, Frank DA. Dual inhibition of Jak2 and STAT5 enhances killing of myeloproliferative neoplasia cells. Leukemia. 2012;26:1407–10. doi: 10.1038/leu.2011.338. [DOI] [PubMed] [Google Scholar]
- 77.Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, et al. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008;112:5095–102. doi: 10.1182/blood-2007-12-129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet. 2011;20:4143–54. doi: 10.1093/hmg/ddr338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Onco_target. 2011;2:518–24. doi: 10.18632/onco_target.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watanabe D, Ezoe S, Fujimoto M, Kimura A, Saito Y, Nagai H, et al. Suppressor of cytokine signalling-1 gene silencing in acute myeloid leukaemia and human haematopoietic cell lines. Br J Haematol. 2004;126:726–35. doi: 10.1111/j.1365-2141.2004.05107.x. [DOI] [PubMed] [Google Scholar]
- 81.Yang F, Jove V, Xin H, Hedvat M, Van Meter TE, Yu H. Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways. Mol Cancer Res. 2010;8:35–45. doi: 10.1158/1541-7786.MCR-09-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blechacz BR, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50:1861–70. doi: 10.1002/hep.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boccaccio C, Andò M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–8. doi: 10.1038/34657. [DOI] [PubMed] [Google Scholar]
- 84.Wang X, Zeng J, Shi M, Zhao S, Bai W, Cao W, et al. _targeted blockage of signal transducer and activator of transcription 5 signaling pathway with decoy oligodeoxynucleotides suppresses leukemic K562 cell growth. DNA Cell Biol. 2011;30:71–8. doi: 10.1089/dna.2010.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280:32979–88. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- 86.Lee YK, Isham CR, Kaufman SH, Bible KC. Flavopiridol disrupts STAT3/DNA interactions, attenuates STAT3-directed transcription, and combines with the Jak kinase inhibitor AG490 to achieve cytotoxic synergy. Mol Cancer Ther. 2006;5:138–48. doi: 10.1158/1535-7163.MCT-05-0235. [DOI] [PubMed] [Google Scholar]
- 87.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–42. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 88.Gunning PT, Minden MD, Khoury H, Page BD, Laister RC, Fletcher S, et al. Small molecule Stat5-SH2 domain inhibitors exhibit potent anti-leukemia activity. J Med Chem. 2012;55:1407–55. doi: 10.1021/jm200720n. [DOI] [PubMed] [Google Scholar]
- 89.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyoshi K, Takaishi M, Nakajima K, Ikeda M, Kanda T, Tarutani M, et al. Stat3 as a therapeutic _target for the treatment of psoriasis: a clinical feasibility study with STA-21, a Stat3 inhibitor. J Invest Dermatol. 2011;131:108–17. doi: 10.1038/jid.2010.255. [DOI] [PubMed] [Google Scholar]
- 91.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–42. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 92.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–6. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gunning PT, Glenn MP, Siddiquee KAZ, Katt WP, Masson E, Sebti SM, et al. _targeting protein-protein interactions: suppression of Stat3 dimerization with rationally designed small-molecule, nonpeptidic SH2 domain binders. Chembiochem. 2008;9:2800–3. doi: 10.1002/cbic.200800291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol. 2010;79:1398–409. doi: 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood. 2011;117:5701–9. doi: 10.1182/blood-2010-04-280123. [DOI] [PMC free article] [PubMed] [Google Scholar]